Abstract
Ovarian and breast cancers are currently defined by the main pathways involved in the tumorigenesis. The majority are carcinomas, originating from epithelial cells that are in constant division and subjected to cyclical variations of the estrogen stimulus during the female hormonal cycle, therefore being vulnerable to DNA damage. A portion of breast and ovarian carcinomas arises in the context of DNA repair defects, in which genetic instability is the backdrop for cancer initiation and progression. For these tumors, DNA repair deficiency is now increasingly recognized as a target for therapeutics. In hereditary breast/ovarian cancers (HBOC), tumors with BRCA1/2 mutations present an impairment of DNA repair by homologous recombination (HR). For many years, BRCA1/2 mutations were only screened on germline DNA, but now they are also searched at the tumor level to personalize treatment. The reason of the inactivation of this pathway remains uncertain for most cases, even in the presence of a HR-deficient signature. Evidence indicates that identifying the mechanism of HR inactivation should improve both genetic counseling and therapeutic response, since they can be useful as new biomarkers of response.
1. Tumorigenesis of Breast and Ovarian Epithelial Carcinomas
Breast carcinomas arise in the terminal duct lobular units of the collecting ducts (the functional unit of the breast), as a consequence of a continuum of lesions and molecular alterations from normal glands to malignant tumors. The two main localizations of invasive cancers are inside the milk duct for ductal carcinomas and inside the milk glands (lobules) for lobular carcinomas.
The cell origin and the molecular alterations that drive breast carcinogenesis differ among the subtypes. The work of Perou and Sorlie allowed the subdivision of breast cancers in five subtypes, distinguished by differences in their gene expression patterns with distinct clinical behaviors: basal, erb-b2 receptor tyrosine kinase 2 (ERBB/HER2), luminal A, luminal B, and normal breast-like [1,2].
Evidence suggests that at the molecular level, these different molecular subtypes evolve along two different pathways of progression: (1) low-grade-like pathway, characterized by gain of 1q, loss of 16q, infrequent amplification of 17q12, gene expression signature associated with estrogen receptor (ER) phenotype, diploid or near-diploid karyotypes, and low tumor grade, including luminal A and part of luminal B tumors, and (2) the high-grade-like pathway, characterized by loss of 13q, gain of chromosomal region 11q13, amplification of 17q12 (region of ERBB2/HER2 gene), and expression of signature genes involved in the cell cycle and cellular proliferation, including intermediate-high grade tumors such as HER2-positive and triple-negative breast cancer (TNBC or breast cancer negative for ER, progesterone receptor (PR), and ERBB2/HER2 gene amplification) [3]. For breast cancer, the inactivation pathway can be based either on the inactivation of TP53, or the inactivation of PTEN [4]. Somatic mutations in TP53 are present in 20–40% of breast cancers [5,6], whereas mutations in PTEN are less frequent (1–5% of breast cancers). Studies have shown that TP53 mutations can occur in ductal carcinoma in situ (DCIS) before the development of invasive breast cancer, which implies an important role for TP53 inactivation early in breast cancer [7]. While these mutations are almost null in low-grade DCIS, they are reported at a frequency of 30–40% in high-grade DCIS. TP53 and PTEN alterations mainly occur in ER-negative cancers, whereas ER-positive have more GATA3, PIK3CA, and CCND1 alterations [8].
Ovarian carcinomas correspond to 90% of ovarian cancers and comprise different subtypes of disease with specific morphologies and molecular patterns. It has been hypothesized that high-grade serous ovarian carcinomas (HGSOC) originate from pre-malignant lesions in the tubas (serous tubal intraepithelial carcinoma) instead of the ovary itself, since both share the same morphological and molecular features, which involves mutations in the TP53 gene as an early event [9,10]. Atypical lesions within the fimbriated end of the fallopian tube (serous tubal intraepithelial carcinomas (STIC)) display similar morphology and TP53 signatures as HGSOC, suggesting that the neoplastic process may originate at these tubal lesions and shed into the ovary, where they aggressively progress [11]. Compelling data suggests the same origin for low-grade serous carcinomas, but that they progress from benign serous cystadenoma to borderline serous tumors and then on to low-grade carcinomas.
Integrated genomic analysis led to the shift that ovarian cancer was not just one disease, but rather several distinct diseases presenting different histological and molecular features. HGSOC are characterized by nearly universal TP53 abnormalities, also detected in endometrioid and other high-grade diseases. This subtype also presents high genomic instability, somatic DNA copy-number changes, and whole genome duplications. Homologous recombination deficiency (HRD) is present in about 50% of HGSOC. Overall, TP53 mutations occur in 96% of cases, BRCA1 and BRCA2 mutations in 22% of cases (15–20% of these are germline), and additional somatic mutations in six other genes are identified in 2–6% of cases (NF1, RB1, CDK12, FAT3, CSMD3, and GABRA6). Recent molecular analysis, which was based on the profile of RNA and microRNA expression, stratified HGSOC into four different prognostic subtypes (C1-mesenchymal, C2-immune, C3-differentiated, C4-proliferative) and seven copy-number signatures. However, contrary to breast cancer, the molecular stratification is not yet validated for accurate prediction of drug sensitivity and/or resistance to treatment [12,13,14,15,16,17].
Breast and ovarian cancers are mostly carcinomas, originated from epithelial cells that are in constant division and subjected to cyclical variations of the estrogen stimulus during the female hormonal cycle, therefore being vulnerable to DNA damage. A portion of breast and ovarian carcinomas arises in the context of DNA repair defects, in which genetic instability is the backdrop for cancer initiation and progression. This is particularly relevant for triple-negative breast cancers and high-grade serous ovarian cancers, for which DNA repair deficiency is increasingly recognized as a target for therapeutics.
1.1. Maintenance of Genome Integrity
1.1.1. Cell Cycle, DNA Repair, and Apoptosis
The cell cycle is divided into 4 phases: G1 (preparation of the DNA replication), S (DNA replication), G2 (preparation of the mitosis), and M (Mitosis). When a cell is out of the cell cycle, it is in the G0 phase. The cell cycle is controlled by different cyclin-dependent kinases (CDKs). Each CDK is specifically linked to a cyclin which is crucial for its kinase activity. The different CDK-cyclin dimers modulate the progression of cells through the cell cycle and each CDK-cyclin complex is specific of one or several phase(s) of the cell cycle. The CDK2-cycline E dimer modulates the G1/S transition, and the dimer CDK1-cycline A modulates the G2 and S phases. The CDK1-cycline B dimer modulates the M phase [18]. During the G1 phase, CDK4-cyclin D and CDK6-cyclin D phosphorylate the protein RB. This phosphorylation inhibits the RB-E2F association. Once liberated from RB, the transcriptional activator E2F activates the transcription of genes indispensable for the DNA replication or S phase [19]. The transition to each phase of the cell cycle is controlled by proteins such as CDK12 [18]. The regulation of CDK-cyclin complexes are mainly assured through phosphorylation and dephosphorylation cycles [20]. During S phase, the quantity of DNA is doubled with the replication forks. The double-stranded DNA is separated into single-stranded DNA, allowing the recruitment of replication protein A (RPA) and then the loading of the replicative DNA polymerases and proliferating cell nuclear antigen (PCNA) sliding clamps [21]. Three DNA polymerases—Pol α, Pol δ, and Pol ɛ—are essential for DNA replication. After Pol α initiates DNA synthesis, Pol δ takes over on the lagging and Pol ɛ takes over on the leading strand, performing the bulk of replication with very high fidelity [22]. The main actors are the polymerases Pol ε (coded by the POLE gene) and Pol δ (coded by the POLD1 gene). Dysfunction of these proteins generates intrinsic DNA errors [23].
Cell cycle, DNA repair, and apoptosis are tightly connected to maintaining the genome integrity of cells and tissues in development and in constant division (Figure 1). The cell cycle, once initiated, could face DNA damage. If so, cells stop their progression through the cell cycle at specific points (cell cycle checkpoints) to correctly repair the errors in the DNA. This is achieved through the interaction existing between the CDKs and the proteins sensitive to DNA alterations. After the DNA repair, the cells can restart their progression through the cell cycle. However, if the DNA damages are too important, the cells undergo apoptosis. There are three cell cycle checkpoints: G1/S, S, and G2/M checkpoints [24]. Moreover, three canonical S-phase "checkpoint pathways” are involved in the maintenance of DNA integrity: the replication checkpoint detects the stalled replication fork, the S-M checkpoint blocks mitosis until the entire genome has been successfully duplicated, and the intra-S phase checkpoint, whose main regulators are ATR and ATM kinases, is sensitive to double-strand-breaks (DSBs) [25]. This system will detect DNA errors, leading to the stop of the cell cycle and the direct reparation of these errors.
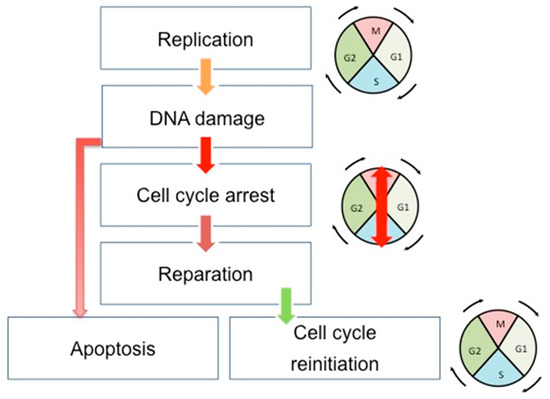
Figure 1.
Schematic view of the correlation between cell cycle, DNA repair, and apoptosis pathways. During the cell cycle, if the DNA error is correctly repaired, the cells can restart their progression through the cell cycle. However, if the DNA damages are too important, the cells undergo apoptosis. Double-headed arrows indicate the evolution of the cell cycle phases: M (Mitosis), G1, S (Synthesis), and G2.
1.1.2. Reparation Mechanisms of DNA
The DNA of a cell can suffer between 10,000 to 100,000 damages a day caused by environmental agents and endogenous processes, such as transcription and replication of the DNA (Figure 2) [26]. The choice of the repair mechanism used by the cell to repair its DNA is determined by the type of lesion and the position of the cell in the cell cycle. Single-strand breaks are repaired by base excision repair (BER) [27], bulkier single-strand lesions that distort the DNA helical structure are repaired by Nucleotide Excision Repair (NER) [28], and misincorporation of nucleotides resulting in mismatches in the DNA sequence are repaired by Mismatch Repair (MMR) [29].
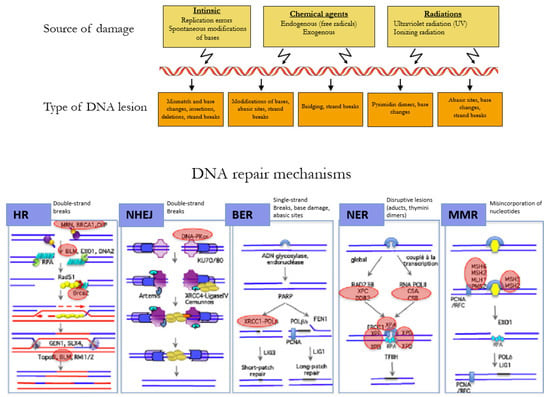
Figure 2.
Mechanisms of DNA repair according to the type of lesion. DNA double-strand breaks are repaired by the error-free pathway Homologous Recombination (HR) or the error-prone pathway Non-Homologous End Joining (NHEJ). Single-strand breaks are repaired by base excision repair (BER), bulkier single-strand lesions that distort the DNA helical structure are repaired by Nucleotide Excision Repair (NER), and misincorporation of nucleotides resulting in mismatches in the DNA sequence are repaired by Mismatch Repair (MMR). Adapted with permission from Rass et al. [30].
DNA double-strand breaks (DSBs) are carried out by two major mechanisms that differ in their fidelity and template requirement: Non-Homologous End Joining (NHEJ) and Homologous Recombination (HR). NHEJ is an error-prone pathway that does not use a template for DNA repair but simply ligates the broken DNA ends together, which leads to an accumulation of errors such as little insertions or deletions [31]. However, HR repair is a highly conserved mechanism that enables the accurate repair of DNA double-strand breaks by using the intact sister chromatid as a template for repair, thereby maintaining the sequence integrity (Figure 3). As it requires a template for repair, it occurs mainly during the late S and G2 phases of the cell cycle. BRCA1 and BRCA2 are key components of this pathway that involves the coordinated interaction of BRCA1 and BRCA2 with other DNA repair proteins such as ATM, CHEK2, BARD1, BRIP1/BACH1, MRE11, RAD50, NBN/NBS1, RAD51C, RAD51D, and PALB2. In cells that are deficient of BRCA1⁄2, the repair of DNA double-strand breaks relies on the error-prone NHEJ pathway [32].
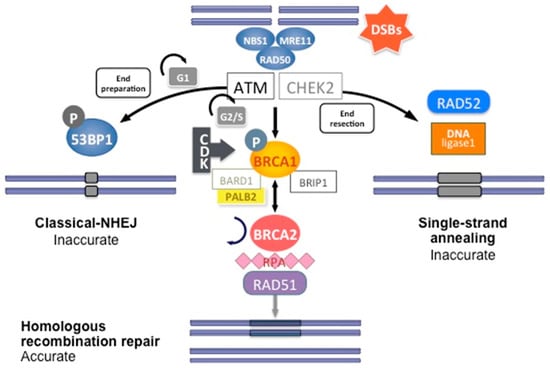
Figure 3.
DNA double-strand break repair by homologous recombination. Coordinated interaction of BRCA1 and BRCA2 with other HR proteins is required to repair double-strand breaks.
The reparation mechanisms of NHEJ and HR are not involved equally during the cell cycle. In G0 and M phases, CDK activity is low and no sister chromatid is available, favoring NHEJ pathways. NHEJ is mainly present during the G1 phase, whereas HR is mainly present during the S phase. In the M phase, both HR and NHEJ repair are extremely reduced, and DSB that arise during mitosis are repaired by single-strand annealing (a DNA damage repair mechanism that uses homologous repeats to bridge DSB ends, causing a deletion rearrangement between the repeats), resulting in large-scale chromosomal rearrangements. CDK activity, which increases in the S and G2 phases of the cell cycle, also favors BRCA1 activation and DNA repair by HR.
In short, HR DNA repair begins after recognition of the 5′ends of the double-strand DNA break by the MRN complex (MRE11-RAD50-NBS1) (Figure 3). This complex recruits Ataxia telangiectasia mutated (ATM), a protein kinase. ATM subsequently phosphorylates downstream proteins, particularly BRCA1, as well as CHEK1 and CHEK2, which are two serine/threonine-protein kinases inducing cell cycle arrest at the G1/S and G2/M cell cycle checkpoints, allowing DNA damages repair [24]. The phosphorylation of BRCA1 by ATM induces its recruitment to DNA damage sites and its binding to the BRCA1-associated RING domain (BARD1), a E3 ubiquitin-protein ligase essential for BRCA1 stability [24]. CDK activity, which increases in the S and G2 phases of the cell cycle, also favors BRCA1 activation and DNA repair by HR. BRCA1 activation then allows extensive 5′ end resection to produce 3′ single-stranded DNA and the induction of the RAD51 loading to the single-stranded DNA by the BRCA1/BRCA2/PALB2 complex. PALB2 recruits BRCA2 and RAD51 to DNA break sites by enhancing BRCA1–BRCA2 interaction and binding DNA with high affinity for D loop [24]. DNA is then repaired using the homologous region of the chromatid as a replicative template.
BRCA1 and BRCA2 are therefore essential proteins involved in HR. Their dysfunction leads to genomic instability, which is a hallmark of cancer [33]. The BRCA1 gene is located on chromosome 17 (17q21) and encompasses 24 exons. It was originally mapped in 1990 and subsequently cloned in 1994 [34]. This gene encodes a 1863-amino acid protein that contains at the N-terminus, a nuclear export signal (NES), and a zinc-binding RING domain (Exons 2–7, aa 8–96, Figure 4) [35,36]. The RING domain heterodimerizes with BARD1 to form an E3 ubiquitin ligase. The main function of BRCA1⁄BARD1 complex is its E3 ubiquitin ligase activity (post damage) at double-strand break sites, which results in the ubiquitination of other proteins involved in DNA damage repair, such as CtIP and H2AX [37,38,39]. Ubiquitinated CtIP binds to chromatin to manage G2⁄M checkpoint control. Ubiquitinated H2AX remodels chromatin so that it becomes accessible for DNA repair machinery. In its carboxyl-terminus, there are tandem repeats of two BRCA1 carboxyl-terminal (BRCT) domains (Exons 16–24, aa 1646–1855). Each comprises of about 100 amino acids and engages in forming functional macromolecules complexes with partner proteins (CtIP, BACH1, Abraxas). More central, BRCA1 has two nuclear localization signals (NLS): one DNA binding domain and one Serine-glutamine (SQ) cluster domain containing several threonine and serine residues which can become phosphorylated [40]. BRCA1 also interacts with BRCA2 via the bridging protein PALB2 (partner and localizer of BRCA2) through BRCA1 coiled-coil domain during RAD51 recruitment to double-strand breaks [41].
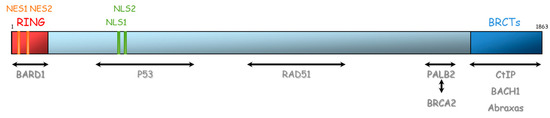
Figure 4.
Representation of BRCA1 protein and its domains of interaction with other proteins. NES: Nuclear export signal, NLS: nuclear localization signal, BRCT: BRCA1 carboxyl-terminal, BARD1: BRCA1-associated RING domain.
The BRCA2 gene is located on chromosome 13 (13q12.3) and encompasses 27 exons (26 coding exons). BRCA2 encodes a 3418-amino acid protein that also contains motifs that mediate its interaction with partner proteins (Figure 5). Currently, three BRCA2 regions have been described as particularly important for HR function: (1) N-terminal PALB2-binding site (aa 10–40), that physically links BRCA1 and BRCA2 and is critical for maintenance of the DNA repair function of these proteins [41,42,43,44]. The same region has also been implicated in binding to EMSY [45]. (2) BRC repeat (aa 900–2000) corresponds to eight consecutive motifs of about 35 residues located in the central region of the protein (in the exon 11), with a well-described function of interaction with RAD51 and other partners [46,47,48], and (3) C-terminal region (aa 2459–3190), composed of three oligosaccharide binding folds (or OB folds), a helical domain, and a tower domain that together constitute the DNA binding region [49,50,51]. Another RAD51 binding region (aa 3265–3330) was shown in the C-terminal region [52]. Recently, Carreira et al. showed a new DNA interaction with the N-terminal [50] and Shibuya et al. showed an interaction with MEILB2/HSF2BP with aa 2117–2339 [53].
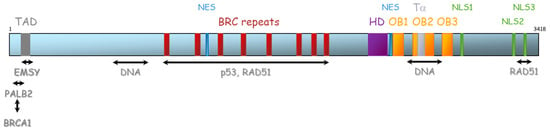
Figure 5.
Representation of BRCA2 protein and its domains of interaction with protein partners. TAD: transactivation domain, NES: nuclear export domain, HD: helical domain, OB: oligosaccharide binding, Tα: Tower alpha, NLS: Nuclear localization signal.
In addition to the maintenance of genomic integrity, BRCA1 and BRCA2 have other cellular functions whose failure might also be related to carcinogenesis. BRCA1 is involved in checkpoint regulation during cell cycle, which is a strategy that transiently inhibits DNA synthesis allowing for the repair of DNA lesions [54]. BRCA1 participates in the maintenance of centrosome number during late S and G2⁄M phases of the cell cycle and in the regulation of apoptosis [55]. BRCA1 regulates its expression at the RNA transcription level and through participation on chromatin remodeling [56,57]. Contrary to BRCA1, the role of BRCA2 in transcriptional and cell cycle regulation is less certain but some studies support such roles [58]. BRCA2 has been shown to play a role in a number of mitotic processes, including the spindle assembly checkpoint, cytokinesis, and daughter cell abscission [53,59].
1.1.3. Dysfunction of the Repair Pathways
- (1)
- Protein Expression Alteration and Mutation in the Coding Regions
Most of the genes implicated in DNA repair have been classified as tumor suppressor genes. Their dysfunction can be related to the absence of the protein or to the inactivation of functional domains. The main cause of dysfunction is the absence of protein due to mutations in the coding region. Mutations in genes linked to HR (HR genes) have been reported in breast and ovarian cancers [15,60,61] and in recent years, several studies have evaluated the consequences of the absence of BRCA1/2 expressions. However, presently, there is no appropriate antibody to routinely test the absence of the BRCA1 or BRCA2 protein in tumors presenting BRCA1/2 mutations by immunohistochemistry. Despite the lack of such antibody, some studies showed a correlation between BRCA1 mutation status and protein expression for ovarian carcinomas [62,63]. For BRCA2, data seems to be more heterogeneous and difficult to interpret [64]. No correlation was found for BRCA2 expression in prostate cancer [65]. However, there are a few examples of interest for performing immunohistochemistry to evaluate BRCA1 protein [62] and nuclear expression for BRCA2-associated tumor [66]. The functional RAD51 foci assay (which showed to be highly discriminative of HRD [67]) could emerge to identify any HR deficiency.
- (2)
- Presence of Missense Variants
A genetic variant can be defined as an alteration in the most common DNA nucleotide sequence (of reference sequence). They can be inferred as pathogenic since they result in predicted truncating or null proteins, and/or are frequent enough in breast–ovarian cancer families that their risk of disease can be estimated directly [68].
The presence of missense variant in functional domains can hurdle the pathway of reparation. Pathogenic missense variants generally have impact on domains directly implied in the DNA reparation activity, such as the BRCT and RING domains of the BRCA1 gene.
- (3)
- Promoter Methylation and miRNA
The deregulation of the protein expression can be related to level of expression without any mutation in the coding sequence. In addition to mutations in BRCA1⁄2 genes, the presence of genetic instability may be a consequence of mutations or epigenetic silencing of BRCA1⁄2 or other HR genes. The main possibility is related to promoter methylation. Aberrant BRCA1 promoter methylation is seen in 5%–30% of ovarian cancers [69,70] and in 11%–14% of sporadic breast cancer. It is more frequent (~30%) among TNBC [69,71]. Promoter hypermethylation in ovarian and TNBC samples have also been described in other HR genes, such as PALB2, ATM, RAD50, RAD51C, and FANCF [72,73]. In contrast, BRCA2 promoter hypermethylation is a very rare event and certainly not well defined in breast and ovarian tumorigenesis. Additionally, BRCA post-transcriptional downregulation through miRNA has been described in breast and ovarian carcinogenesis, which could also explain cases sharing BRCA histopathological features with no mutation identified [74].
- (4)
- Transcription and Post-Translational Regulation
The regulation of the transcription of certain genes involved in DNA repair has been well described. The role of CDK12 has been described as an activator of the HR genes promoters [75]. The protein EMSY is more inclined to have a negative impact on the expression, which explains the role of the amplification of its gene [76]. Moreover, steroid hormones may affect BRCA1 expression indirectly, by altering the proliferative status of the cell rather than acting directly on DNA sequences. So far, no estrogen receptor (ER) site has been identified on BRCA1 regulatory regions [77].
Another level of alteration could happen in the post-translational regulation. Some post-translational alterations, such as phosphorylation, can be clearly responsible for activation or inactivation of a pathway [78]. The mechanism implying ubiquitin modification is also known to limit the activity of some proteins by accelerating the intracytoplasmic destruction of them. This mechanism has been described with HR proteins [79].
1.1.4. Compensatory Mechanisms to Other Reparation Pathways
Since there are several DNA repair pathways, some compensatory mechanisms have been observed [80]. For example, if the HR pathway is inactivated, the BER and the alternative NHEJ DNA repair will try to compensate for it imperfectly [30]. Moreover, since HR requires a full coordination of different proteins, the impact on the pathway will probably differ according to the protein affected.
1.2. Signature
The inefficiency of the DNA repair mechanism generates a scenario of genetic instability. Mutational signatures are then designed to identify the homologous recombination-deficient (HRD) phenotype and characterize a larger population which can benefit from DNA damaging agents, extending beyond BRCA mutant tumors. Every signature serves as an imprint of a distinct DNA damage and repair process operative in the tumor at some point during tumoral development.
This includes signatures based on the evaluation of the following: (1) Copy number alteration [81] (CNA) profiles, which are determined by the identification of DNA gains or losses in the tumor. CNA can be evaluated by comparative genomic hybridization array (aCGH), multiplex ligation-dependent probe amplification (MLPA), or single-nucleotide polymorphism (SNP) arrays. (2) Loss of heterozygosity (LOH) score, evaluated by the imbalance in the ratio of parental alleles from the normal 1:1 [82]. (3) Telomeric allelic imbalance, which calculates the allelic imbalance extending from the site of DNA damage to the telomere [83,84]. (4) Large-scale transitions that consist of chromosome breaks (translocations, inversions, or deletions) of at least 10 Mb between adjacent regions [85], and (5) mRNA and miRNA expression [86]. (6) Mutational signature based on the experience of the TCGA (The Cancer Genome Atlas Program)—signature 3 [87]. Finally, two scores were developed combining different methods to improve the sensitivity to identify the BRCAness phenotype: myChoice HRD test (Myriad Genetics) combines measures of LOH, TAI, and LST [88], and Foundation Medicine HR score combines measures of BRCA1⁄2 mutation status and percentage of LOH [89]. The threshold of composite scores have been described and validated in prospective clinical trials [90].
HRD tumors represent up to 50% of HGSOC and more than 20% of basal breast cancers, but a BRCA mutation is identified in only 20% of them [14,91]. According to the results of recent trials, even if the tumor is sporadic, the identification of an HRD phenotype helps in personalizing therapy. The comprehension of breast- and ovarian-associated carcinogenesis has evolved from solely mutation identification in candidate genes onwards to the integration of a large volume of genomics and transcriptome data, revealing recurrently altered pathways and signatures of mutational processes. All methods described above (individually or in combination) were able to discriminate HR-deficient tumors which were correlated with responsiveness to platinum and PARPi (Poly (ADP-ribose) polymerase (PARP) inhibitors), and resulted in improved outcomes.
1.3. Temporal Order and Somatic Tumor-Driving Events of BRCA-Associated Tumorigenesis
In relation to tumorigenesis specifically related to BRCA1 and BRCA2, data suggest that BRCA bi-allelic inactivation renders the cell vulnerable to genomic instability, being the background for successive mutations that culminate in cancer development. Conforming to this theory, Von Waldhe et al. recently demonstrated concordance between HRD scores across different regions of the same BRCA-associated breast cancer, indicating that HRD affects the entire primary tumor and corresponds to a founding event [92].
As typical tumor suppressor genes, the inactivation of the second allele of BRCA1⁄2 is presumed to be a rate-limiting step [93]. LOH is the most common second hit event of breast and ovarian BRCA1⁄2-associated carcinogenesis. It is a consequence of large deletions, genomic rearrangements, incorrect mitosis, or deficient DNA repair. It has been reported in 90% (breast) and 91% (ovarian) of BRCA1-associated cancers and in 54% (breast) and 84% (ovarian) of BRCA2-associated cancers [94]. Alternative second-hit mechanisms, such as somatic inactivating point mutations, have been described in a small minority of BRCA1-associated breast and ovarian cancers [95,96]. Furthermore, hypermethylation of the BRCA1 promoter has also been responsible for the silencing of the wild-type allele, but only in a minority of the cases [97,98].
Van Heetvelde et al. described the panorama of second-hit events in breast and ovarian cancers from patients harboring germline BRCA1⁄2 mutations. Copy neutral LOH was the most prevalent mechanism of wild-type (WT) allele inactivation (detected in 69% of breast cancers and 67% in ovarian cancers). However, most intriguing was that only a minority of tumors (35% breast and 47% ovarian cancers) presented loss of the WT allele in all cancerous cells but in the majority of the cases, different mechanisms of WT allele inactivation were present in the same tumor [99]. Moreover, somatic intragenic deletions and methylated subclones were found in combination with partial LOH.
It has been suggested that heterozygous mutations affecting BRCA1 and BRCA2 might be enough for carcinogenesis, even when the remaining WT allele remains expressed. Recent genomic studies have showed that a significant fraction of cancers arising in BRCA mutation carriers retain a functional WT allele. Jonsson et al. observed this in 25% of breast and ovarian cancers with BRCA1⁄2 mutations [100]. In line with this, Maxwell et al. observed the retention of the WT allele in 46% breast and 16% ovarian BRCA2-associated cancers. However, it was less frequent for BRCA1 breast (7%) and ovarian (10%) cancers [94]. The prevalence of 8% was globally estimated in a large panorama of tumors and clearly more frequent in lung cancer (up to 20%) [100]. Furthermore, some lines of evidence suggest that the presence of heterozygous truncating BRCA1 and BRCA2 mutations may render cells vulnerable to haploinsufficiency, when exposed to replication stress [101].
1.4. Conclusion for Routine Practice
Currently, the molecular testing with the highest level of evidence is based on the screening of the coding sequences of BRCA1 and BRCA2 genes as the main drivers of HR pathway inactivation. This analysis is most often performed through NGS (Next-Generation Sequencing) in laboratories with access to updated databases. The clinical utility of the screening of other HR genes is still under investigation and more information on their implication is still needed for therapeutic and prevention decisions. Mutations in some of these genes, such as PALB2, RAD51C, and RAD51D have also been linked to hereditary predisposition to cancer [102]. There is currently no other option for detection of the HR deficiency, not even with immunohistochemistry. The HR signatures look promising, but interpretation of data and the clinical utility is still challenging.
2. Description of Mutations in HR Pathways in Breast and Ovarian Cancers
In 2018, the CIMBA (Consortium of Investigators of Modifiers of BRCA1/2) consortium presented an inventory of the current state of BRCA1 and BRCA2 mutations. There are 1650 unique BRCA1 and 1731 unique BRCA2 mutations distributed within these genes [103]. Different types of mutations have been reported: frameshift, nonsense, missense, and splice. Frameshift are the most common type, followed by nonsense mutations. The most common effect of the mutations was premature translation termination and the majority of mutant mRNAs were predicted to undergo nonsense-mediated mRNA decay (NMD) [104]. In the 2000s, large rearrangement (deletion or duplication of one or more exons) were also highlighted [105]. Despite having the same spectrum of mutations, the frequency distribution by mutation type, effect, or function differed significantly (p < 0.05) between BRCA1 and BRCA2 mutation carriers in the CIMBA cohort [103]. These differences are largely because genomic rearrangements and missense mutations account for a much higher proportion of alterations in BRCA1 when compared to BRCA2, as previously described [106,107].
Mutations in HR genes beyond BRCA1⁄2 have been reported in breast and ovarian cancers [15,60,61]. As expected, BRCA1/BRCA2 genes were the most commonly altered genes, followed by several genes, including CHEK2, PALB2, RAD51C, and RAD51D [108]. Some were preferentially affected by germline alterations (e.g., BRCA1/2, CHEK2, FANCM, PALB2), whereas others (e.g., ATM, BAP1, CDK12) were preferentially affected by somatic events [109].
The prevalence of germline HR genes alterations in patients with breast cancer is about 10%. After BRCA1⁄2, the main HR genes affected are CHEK2, ATM, BRIP1, PALB2, PTEN, NBN, RAD51C, RAD51D, MSH6, and PMS2 [110]. For ovarian cancer, HR mutations are identified in more than 25% of the cases [95,109]. Beyond BRCA1⁄2, the main HR genes affected are RAD51D, BRIP1, RAD51C, CHEK2, PALB2, and BARD1.
Analysis of TCGA data confirmed the prevalence of HR pathway alterations in 10% and 25% of breast and ovarian cancers, respectively. It was demonstrated that bi-allelic alterations in HR genes are mutually exclusive of each other [87,109]. Analysis of ∼1000 samples confirmed the same pattern of HRD in breast cancer samples of germline (nonsense and frameshift) PALB2 mutations carriers, while it was not observed in ATM or CHEK2 [109].
2.1. Hereditary Breast and Ovarian Cancers
2.1.1. Breast Cancers Related to BRCA1/2 Mutations
Breast cancers associated with gBRCA1⁄2 mutations or pathogenic variants represent 3%–5% of cases [111]. The percentage of somatic BRCA1⁄2 (sBRCA1⁄2) mutations in breast cancer is not well established. However, two studies found that 3% of unselected cases present sBRCA1⁄2 mutations [8,96].
About 70% of breast tumors arising in BRCA1 mutation carriers are “triple negative” [112]. On the other hand, only 10% to 20% of TNBCs carry a BRCA1 mutation [113,114]. BRCA1-associated tumors generally present a higher mitotic rate and are peculiarly higher-grade tumors, presenting greatly increased mitotic count, pushing margins, lymphocytic infiltrate, trabecular growth pattern, and necrosis [115,116,117]. These tumors generally express myoepithelial cell-type cytokeratins (CK5⁄6, CK14, and CK17) and present a basal-like gene expression profile [116]. A previous study showed that reduced expression of CK8⁄18 could help discriminate the basal tumors of BRCA1 carriers from those sporadic tumors [118].
BRCA2 breast carcinomas are most closely like sporadic tumors, generally expressing the estrogen receptor (77%) and are in the minority triple negative [112,119]. RNA tumor profiling demonstrated that BRCA2 tumors are mainly of the luminal B subtype and are more likely than non-BRCA2 tumors to be ER-positive and of high grade, with pushing margins [120,121].
Mavaddat et al. evaluated the histopathological characteristics of the largest cohort of breast cancer patients harboring BRCA1/2 germline mutations. This included 4325 patients with BRCA1 mutations and 2568 patients with BRCA2 mutations [112]. Breast tumors were mostly invasive ductal carcinomas (occurring in milk ducts) for both BRCA1 (80%) and BRCA2 (83%) carriers. Lobular carcinoma (occurring in breast lobules) was the second most common subtype for BRCA2 carriers (8.4%), and medullary carcinoma (a subtype of invasive ductal carcinoma) for BRCA1 carriers (9.4%). The frequency of TNBC was 69% for BRCA1 and 16% for BRCA2. HER2-positive was 13% for BRCA1 and 10% for BRCA2 tumors. BRCA1 tumors were a majority grade 3 (77%), while for BRCA2, half were grade 3. For BRCA1 carriers, the grade of the tumor decreased with increasing age, as well as the proportion of estrogen receptor-negative tumors. In contrast, the grade and the proportion of ER-negative tumors increased with age for BRCA2. Such findings are in agreement with previous studies with a smaller number of participants [122,123]. Pathological data was available for 702 BRCA1 and 302 BRCA2 mutation carriers in the same cohort that developed a contralateral breast cancer [112]. The median interval for a second breast cancer was 5.2 years. Interestingly, ER/PR status of the first breast tumor was predictive of the ER/PR of the second cancer for both BRCA1 and BRCA2 carriers, suggesting that the second tumor arises in the same genetic and environmental background with the same pathology.
Concerning the prognosis of BRCA-associated breast cancers, recently, the POSH study showed no difference in survival for patients carrying a BRCA mutation when compared to those with sporadic breast cancer [124]. However, in the TNBC subgroup, BRCA carriers had a better survival than non-carriers, which may be related to better sensitivity to chemotherapy. This survival advantage of the TN BRCA mutant subgroup was also confirmed in a recent meta-analysis [125].
2.1.2. Ovarian Cancer Related to BRCA1/2 Mutations
Ovarian epithelial cancers associated with BRCA1⁄2 mutations represent ~22% of the cases, with 15% germline and ~7% somatic mutations [14]. In the absence of BRCA1 or BRCA2 protein function, the preferential use of error-prone DNA repair mechanisms leads to genomic instability, a peculiar feature of breast and ovarian cancers arising from BRCA mutations that may favor carcinogenesis.
The majority of ovarian tumors related to BRCA1 and BRCA2 constitutional mutations are serous carcinomas (67%), followed by endometrioid (12%), clear-cell (2%), and mucinous carcinomas (1%) [112]. Tumors in BRCA1⁄2 carriers are more likely than tumors in age-matched controls to be invasive serous adenocarcinomas and unlikely to be borderline or mucinous tumors. They are of higher grade, with a higher percentage of solid components and are more likely to stain strongly to TP53 [126]. There are no significant differences in ovarian cancer morphology or grade between BRCA1 and BRCA2 tumors [112]. However, BRCA1 carriers present a higher ovarian cancer lifetime risk than BRCA2. The cumulative ovarian cancer risk to age 80 years is around 44% and 17% for BRCA1 and BRCA2 carriers, respectively.
Evidence suggests that genomic instability is present in both hereditary and sporadic cancers but occurring in different stages of cancer development and with different molecular basis. While in hereditary cancers, genetic instability probably precedes the acquisition of mutations in oncogenes and tumor suppressor genes, and therefore precedes the acquisition of other hallmarks of cancer [127], studies suggest that the first hallmark acquired in sporadic cancers may be increased activation of growth signaling, secondary to mutations in oncogenes or anti-oncogenes.
According to the mutator hypothesis, genomic instability in hereditary cancers is related to mutations in caretaker genes (genes involved in maintaining genomic stability) that happens during early carcinogenesis. Classical caretaker genes are DNA repair genes, including BRCA1⁄2, and mitosis checkpoint genes. Chromosomal abnormalities are present from the stage of precancerous lesions and participate in cancer development by increasing of the spontaneous mutation rate [128]. The observation that only a part of chromosomal abnormalities is seen in all tumor cells is in line with the hypothesis that tumor cells originate from a single genetic unstable cell which continues to accumulate mutations during cancer development. The results of high-throughput sequencing studies showed that mutations in caretaker genes were infrequent in sporadic cancers [129,130,131,132]. However, that inactivation of caretaker genes can also be purely sporadic and define a specific subtype with comparable features of tumors carrying germline mutations. In sporadic cancers, the genetic instability is probably related to an oncogenic-induced collapse of DNA replication forks.
3. Unidentified HR Deficiency Mechanisms in Ovarian and Breast Cancers
3.1. BRCA1/BRCA2 Variants of Uncertain Significance
Ten percent of individuals undergoing genetic BRCA1/2 screening receive test results reporting variants of uncertain clinical significance (VUS). These sequence variations are either in-frame deletions/insertions, missense, silent variants, or variants in intronic and regulatory regions that may influence splicing or translation. They present an unknown functional effect on BRCA1 and BRCA2 and cannot currently be classified as either pathogenic or of low clinical significance. A large number of missense variants and virtually all non-coding deep intronic or promoter variants remain of unknown significance (VUS) since they cause subtle changes in protein structure (for missense variants) or in the amount of produced protein (for non-coding variants), being generally difficult to reliably determine their pathogenicity merely from clinical genetic information [133]. A VUS finding should not be considered clinically useful and should not be taken into account for clinical decisions until further evidence emerges to shift interpretation. Medical advice should be solely based on family and personal medical presentation. But in some cases, they are managed inappropriately as pathogenic mutation leading to psychological distress and inappropriate interventions in patients [103]. Even though individual VUS are rare, the identification of a VUS is not a rare event and has a tendency to increase with concomitant sequencing of several genes in NGS panels. Information about VUS is collected in different databases [134,135,136,137]. Attempts to evaluate the clinical significance of these variants include frequency analysis in case-control studies, personal and familial history, co-segregation of the variant with disease in affected families, co-occurrence in trans with deleterious mutations, in silico prediction models, and functional and tumoral data.
BRCA1/2 VUS classification is particularly challenging. This is why, in 2009, an international consortium was created, which allowed for the classification of a certain number of variants [68,138,139,140,141,142,143,144,145]. This consortium has recently extended the scope of this study for other HBOC genes.
A number of in silico tools are available to help understand if a given intronic or exonic variant leads to an improper exon and intron recognition on messenger RNA and results in the generation of an aberrant transcript of the mutated gene, as well as whether a specific amino acid change may impact protein function [146,147,148,149,150,151,152,153,154,155,156,157]. Functional assays are complementary to in silico prediction, also being used to evaluate the impact of VUS on RNA splicing and/or on protein function [143,153,158,159,160,161,162,163,164,165].
3.2. Variants in HR Genes Beyond BRCA1⁄2
Since the discovery of BRCA1 and BRCA2 genes 25 years ago, several other breast cancer susceptibility genes have been identified. With the current popularization of next-generation sequencing, the single-gene strategy is used in selected circumstances. Most services are now sequencing BRCA1⁄2, along with other genes related to breast and ovarian cancer hereditary predisposition in the context of a HBOC gene panel. These high to moderate penetrance mutations may also contribute to hereditary predisposition, such as TP53, PTEN, STK11, CDH1, ATM, BRIP1, PALB2, and RAD51 isoforms (RAD51C, D, B). However, altogether these mutations only explain ~5% of the unsolved cases [108] and VUS are also identified in these new genes. Some of these genes have been found to increase the risk of cancer similar to BRCA1⁄2, sharing the same care guidelines for cases where a BRCA1⁄2 mutation has been identified. BRCA1⁄2 HBOC can be distinguished from these other disorders based on the spectrum of tumors present in the family and with the aid of germline genetic screening. PALB2 variants have now been determined to be of high penetrance [15]. ATM, CHEK2, and BARD1 genes are considered to have a moderate increase in risk [166]. Numerous other genes are suspected to be related to the risk of breast cancer, such as NF1, RAD51C, RAD51D, BRIP1, NBN, MRE11A, FANCM, RECQL, MLH1, MSH2, MSH6, and PMS2. However, they still need confirmation given their low penetrance and divergent results between studies. These previously cited genes are usually included in HBOC panels [102,166]. Still, some of the commercial panels mix genes related to different hereditary cancer syndromes, adding the challenge of interpreting the clinical risk of mutations related to other syndromes when they are identified in HBOC patients. This leads to the increase of uninterpretable results, since the number of variants of uncertain significance increases when multiple genes are tested. For multi-gene panel testing, a mutation is identified in ~30% of HBOC patients, most commonly in BRCA1 and BRCA2 [167]. It is thus worth mentioning that despite technology advances and recent democratization of access to genetic screening, the predisposition mechanism remains undefined for about two-thirds of families meeting the clinical criteria for HBOC.
4. Management
4.1. Risk Reduction Interventions
The importance of identifying at-risk individuals lies in providing appropriate screening, surveillance, and risk reduction interventions. The individualized approach should include discussion about the risks and benefits of risk-reduction surgeries, taking into account patient′s age, priorities, previous cancer history, comorbidities, and cancer-related anxiety. Prospective studies demonstrated that for BRCA carriers without a personal history of cancer, bilateral risk-reducing mastectomy (RRM) is associated with 90% or more decreased risk of breast cancer with a residual risk of 1%–2% [168,169]. But the decision to undergo RRM and the ideal time can be influenced by life events, being uncertain for some women. For individuals with HBOC who choose not to undergo risk-reducing surgery, proper follow up with intensive cancer screening has an impact on early detection of cancer with increased cure rate. It is important to mention that for patients with a strong familial breast cancer risk, even if a mutation is not identified, appropriate follow-up and awareness training with monthly self-breast examination should begin at 18 years with clinical breast examination recommended at 25 years, and from then on, every 6 months. Between 25 and 29 years, radiographic screening is suggested. From 30 to 75 years, annual MRI (magnetic resonance imaging) and mammography is recommended. A recent prospective randomized study that performed paired MRI and mammography in women with high risk for breast cancer confirmed the benefit of adding MRI to the screening of this population. This study showed that 61% of the tumors would not have been diagnosed by just a mammography, and it also demonstrated that MRI allowed the diagnosis of cancer at an earlier stage. [170]. For BRCA1⁄2 mutation carriers older than 50 years, the addition of MRI to mammography improves screening sensitivity by a magnitude similar to that observed in younger women [171].
Ovarian cancer risk should also be taken into account, but screening for ovarian cancer is more challenging due to the low sensitivity of the exams. It is advised that risk-reducing salpingo-oophorectomy (RRSO) be offered between age 35 and 40 years for women with BRCA1 mutations who have completed childbearing. For BRCA2 carriers, it can be delayed until the age of 45 years, since only 1% of this population presents ovarian cancer by age 50. Nevertheless, health considerations related to premature surgical menopause, including an increased risk of osteoporosis and cardiovascular disease, should be discussed with women considering surgery. RRSO is the only evidenced-based strategy to prevent ovarian and fallopian tubes cancer. It is associated with an 80% reduction of ovarian cancer risk, a 50% reduction of breast cancer risk in premenopausal women, and of breast and ovarian-cancer specific mortality [172]. Annual screening with CA125 and transvaginal ultrasound may be considered for women who refuse prophylactic surgery, with limited sensitivity (less than 50%) and positive predictive value (less than 17%) [173]. Studies show that both are ineffective in detecting tumors during the very early stage to influence prognosis. The PROSE (Prevention and Observation of Surgical Endpoints) study evaluated the effect of risk-reducing salpingo-oophorectomy on mortality and confirmed that the surgical group had lower all-cause mortality (HR 0.40; 95% CI 0.26–0.61), breast cancer-specific mortality (HR 0.44; 95% CI 0.26–0.76), and ovarian cancer-specific mortality (HR 0.21; 95% CI 0.06–0.80) [168]. Some questions about the extent of surgery remain unanswered, such as whether adding hysterectomy to the procedure has survival benefits, and even if just performing salpingectomy alone would be sufficient for risk reduction. The latter is based on the pathophysiology of ovarian cancer and its likely origin is in situ lesions located in fallopian tubes. Recent data has suggested that women with BRCA1⁄2 mutations present an increased risk for uterine serous carcinoma, which generated an extensive discussion whether hysterectomy should be performed at the time of prophylactic surgery [174,175]. However, current standard guidelines do not include hysterectomy as part of risk reducing surgery and the decision to perform a concurrent hysterectomy should be individualized [176,177].
Since germline BRCA2 mutations are associated with a five-to-eight increase in the risk of developing prostate cancers, which are more aggressive and with a shorter survival rate, male carriers should begin prostate screening at age 45. Additionally, BRCA2 mutations are present in 7% of pancreatic cancers irrespective of familial history, and accounting for ~10% of hereditary pancreatic cancers. Therefore, an individualized screening should be advised, preferably in the context of a clinical screening protocol because there is no consensus for pancreatic cancer screening in most institutions so far.
In addition to intensified screening and risk reduction surgeries, some pharmacological measures have proven to positively impact the management of BRCA carriers. Chemoprevention with Tamoxifen may be offered for breast cancer primary prevention of BRCA2 carriers, since 75% of BRCA2-associated breast cancer are ER-positive [178]. However, for BRCA1 carriers, the current use of tamoxifen is less studied, and data is inadequate to support the use of tamoxifen, since they present mainly TNBC. Beyond Tamoxifen, observational studies have shown that oral contraceptives reduce the risk of ovarian cancer by 30% and 40% in the general and BRCA population, respectively. The concern about theoretical increased risk of breast cancer was not confirmed in studies of women with HBOC syndrome. However, data from randomized controlled trials is lacking and therefore, the use of oral contraceptives for prevention of ovarian cancer in women who have not undergone risk-reducing salpingo-oophorectomy is controversial [179,180].
4.2. Implications for Treatment Response: BRCA1/2 Mutations as Predictive Biomarkers
4.2.1. Sensitivity to Platinum Salts
Platinum salts, such as cisplatin and carboplatin, are effective breast and ovarian cancer treatments. They act as DNA cross-linking agents forming intra-strand crosslinks and are especially active in cells lacking HR function. Although their clinical effectiveness as first-line chemotherapy for breast cancers has been confirmed (overall response rate (ORR) of 50% for cisplatin and 30% for carboplatin), studies have shown that they have only modest activity in previously treated metastatic breast cancers [181,182]. Tutt et al. were able to demonstrate that the presence of a germline BRCA mutation was predictive of a greater benefit in the metastatic scenario. The trial included 376 unselected TNBC patients after first-line treatment failure who were randomized to receive either carboplatin or docetaxel. While there was no difference between ORR to carboplatin and ORR to Docetaxel in the overall population (ORR 31.4% × 34%), subjects with a BRCA1⁄2 germline mutation had a significantly better response to carboplatin than to docetaxel, doubling the overall response rate (ORR 68% × 33.3%, p = 0.03). However, the highest platinum sensitivity was limited to BRCA mutation carriers. Such benefit was neither observed for subjects with a high HRD score, nor for tumors presenting BRCA1 promoter [183]. These results were consistent with previous results from a smaller phase 2 trial in metastatic TNBC in which platinum agents were active, especially in the presence of BRCA1⁄2 mutations but not in the presence of BRCA1 promoter methylation [184].
The activity of platinum salts was also evaluated in early breast cancer, with proven benefits in the neoadjuvant scenario for the TNBC subtype, regardless of BRCA1 status [185]. Data from a retrospective study support the use of platinum salts in the neoadjuvant treatment of women with a BRCA mutation. Expressive response rates have been observed for BRCA1 mutation carriers treated with cisplatin monotherapy compared with standard regimens based on anthracycline and taxanes (pathological complete response (pCR)= 83% × 8–22%) [186]. However, usefulness is still questionable of BRCA1⁄2 mutations as predictive biomarkers of platinum response in the neoadjuvant scenario. Some authors advocate that gBRCA1⁄2 mutation carriers have a higher likelihood of achieving pCR thanks to a higher sensitivity to cytotoxic agents in general, regardless of the addition of platinum salts [187]. GeparSixto was a phase II study which confirmed the benefit of adding carboplatin to neoadjuvant chemotherapy with increase of pathological complete response rate (53.2% × 36.9%, p = 0.005), an advantage translated in a superior disease-free survival rate at 3 years. The secondary analysis of GeparSixto trial, performed to evaluate if BRCA1⁄2 status was predictive of response to chemotherapy, could not confirm this hypothesis. It found that the addition of carboplatin did not increase pCR rate in mutation carriers (65.4% × 66.7% in treated versus untreated, respectively). Surprisingly, in the wild-type population, neoadjuvant carboplatin significantly increased it (55% × 36.4% OR 2.14, 95% CI 1.28–3.58, p = 0.004). Additionally, gBRCA1⁄2 mutation carriers experienced a better disease free survival (DFS), which was not significantly improved by the addition of carboplatin (82.5% in carboplatin treated × 86.3% untreated patients).
Little data is available in the adjuvant setting for platinum salts in gBRCA1⁄2-associated breast cancers. In 2014, Dwadasi et al. randomized TNBC patients who had residual disease after neoadjuvant chemotherapy based on anthracycline and taxanes, receiving four additional cycles of adjuvant cisplatin, with and without the PARPi rucaparib [188]. The primary end point was similar in both arms and was not different between patients with BRCA-associated and sporadic tumors (85% × 79%, respectively). Yet it is noteworthy that there was no relapse in any of the eight patients with gBRCA mutation.
4.2.2. HR Deficiency and Development of a Targeted Therapy: PARP Inhibitor Treatments
The rationale to use Poly (ADP-ribose) polymerase (PARP) inhibitors to treat tumors harboring BRCA1⁄2 mutations is based on the principle of synthetic lethality, a concept in which if only one of the two genes is mutated, then it is compatible with viability, while a mutation in both leads to cellular death [189,190].
PARPs are a large family of multifunctional enzymes that play a key role in the repair of single-strand breaks (SSB) through base excision repair. PARP1 is best characterized of the 17 members of the PARP protein family [191]. PARP1 is the major target of PARPi. The inhibition of PARP impairs the repair of SSBs through disruption of the BER pathway and PARP1 trapping that happens through inhibition of auto-PARylation and/or PARP release from DNA. These events lead to accumulation of SSB, which lead to DSBs at the replication fork and thus to the death of homologous recombination-deficient cells such as BRCA1⁄2 mutants in a process named “synthetic lethality.” This concept has moved from the field of genetics to medical oncology, opening new perspectives for treating tumors containing the BRCAness HR-deficient phenotype.
The first trial evaluating the efficacy of PARPi (olaparib) in breast cancer was published in 2009 [192]. This phase I trial included 60 heavily pretreated women, 3 carrying a BRCA mutation. One out of these 3 patients presented a complete response for 60 months. The second had stable disease for 7 months [192]. These results led to the approval of 2 phase II trials, including women with gBRCA1⁄2 mutations with advanced breast cancer who presented response rates ranging from 12% to 41% [193,194]. Recently, a prospective phase III trial compared olaparib to standard-of-care chemotherapy in patients with metastatic breast cancer harboring a gBRCA1⁄2 mutation. The PFS was significantly longer in the olaparib group (7 × 4.2 months HR 0.58, 95%CI 0.43–0.80, p < 0.001), as well as improvement to quality of life. No significant benefit in overall survival has been proven yet [195].
More recent studies have investigated the benefit of adding platinum salts in comparison and in combination with PARPi for the treatment of BRCA-related early breast cancer. Telli et al. reported a pCR of 36% in a single-arm phase II study that evaluated the combination of iniparib, gemcitabine, and carboplatin for the neoadjuvant treatment of BRCA mutation. The study confirmed that a high loss of heterozygosity score was a predictor of better response [196]. Next, the combination of a PARPi (Veliparib) with carboplatin in addition to standard neoadjuvant chemotherapy with Docetaxel was evaluated in the BrighTNess trial, a phase III randomized study that included stage II–III TNBC. In this trial, the addition of carboplatin and veliparib increased pCR rate in both gBRCA1⁄2 mutation carriers (57%) and wild-type patients (53%), but with no significant differences in patients who received only carboplatin [197].
Just like for early breast cancer, several studies are now comparing platinum salts to PARPi and evaluating them in combination for advanced breast cancer. A recently published phase II trial evaluated the efficacy of adding the PARPi veliparib to chemotherapy regimens (carboplatin and paclitaxel or temozolamide) in patients with gBRCA1⁄2 mutated metastatic breast cancer. A numerical but not statistically significant increase in progression free survival (PFS) and overall survival (OS) was observed with the addition of veliparib to the platinum-based regimen carboplatin and paclitaxel [198].
Beyond breast cancer, PARPis have been widely tested for ovarian cancer treatment in different settings. High-grade serous carcinoma (HGSOC), the most common subtype of ovarian cancer, is characterized by nearly universal TP53 mutations (96%) and high genomic instability. As stated before, one half of HGSOC displays defects in the HR DNA repair pathway, with mutations identified in BRCA1⁄2 in ~22% of the cases with ~15% germline and ~7% of tumoral mutations [14]. Mutations in other HR genes are less common and are present in about 3% of the cases. Sporadic tumors also display HR defects as BRCA mutants (the BRCAness phenotype), and consequently higher response rate to platinum-based chemotherapy and PARPi. Most patients with advanced-stage ovarian carcinoma are initially treated with platinum-based chemotherapy, but the majority of them will ultimately relapse. Longer treatment-free intervals and improved overall survival rates observed in this group are related to their inability to repair DNA damage. Based on this rationale, two phase I studies tested the safety and benefit of olaparib for treatment of ovarian cancer harboring gBRCA1⁄2 mutations [192,199]. In the first study, Fong et al. enrolled 60 solid tumor patients, in which ovarian tumors led with 21 cases. Of the 21 ovarian tumors, all of which received at least one line of chemotherapy, 16 had gBRCA1⁄2 mutations. Response was only documented in these 16 patients harboring gBRCA1⁄2 mutations in both platinum-sensitive (61.5%) and platinum-resistant (41.7%) cohorts [199]. This study supported the anti-tumor activity of PARP inhibition for the treatment of ovarian cancer. Subsequently, in the expansion phase, only ovarian cancer carriers of BRCA1 or BRCA2 mutations were enrolled. Of the 50 patients, 20 (40%) presented partial or complete response and 3 (6%) presented disease stabilization. The authors further confirmed a significant association between the clinical benefit rate and platinum-free interval.
Subsequent phase II studies confirmed the efficacy of olaparib as monotherapy for the treatment of metastatic HGSOC patients harboring gBRCA1⁄2 mutations, with ORR ranging from 33%–41% and a median response duration of 8.8 months [200].
Next, the trials focused on the use of olaparib in the maintenance scenario for platinum-sensitive relapsed ovarian cancer. Ledermann et al. confirmed the improvement of PFS by olaparib initially in a retrospective pre-planned analysis of a phase II trial, and subsequently in a prospective trial (8.4 months versus 4.8 months; HR 0.35; 95% CI 0.25–0.49; p < 0.001). The benefit was even greater in the presence of BRCA1⁄2 germline or somatic mutations [201]. Also, SOLO 2 phase III trial met its primary end point, with more improved PFS with olaparib than with placebo (19.1 months versus 5.5 months, HR 0.30; 95% CI 0.22–0.41) [202]. Following these results, olaparib was also tested in newly diagnosed ovarian cancer patients, after administration of platinum-based adjuvant chemotherapy. In the adjuvant scenario, olaparib significantly reduced the risk of disease progression or death by 70% [203].
Therefore, olaparib was initially approved by the Food and Drug Administration (FDA) in 2014 for the maintenance treatment of BRCA1⁄2-mutated ovarian cancer. The approval was extended in 2018 to all platinum-sensitive patients, regardless of BRCA1⁄2 status, because it was realized that the benefit extended to all HRD tumors. Following the SOLO1 trial, olaparib was also approved in first-line maintenance for BRCA-mutated (BRCAm) advanced ovarian cancer. Currently, two other PARPi, niraparib and rucaparib, have been approved by the FDA for the treatment of ovarian cancer. Other PARPi, such as veliparib and talozaparib, are under development and testing, based on the rationale described above.
The phase III NOVA study confirmed the benefit of niraparib in the maintenance setting of platinum-sensitive HGSOC. The authors stratified the analysis by the presence of BRCA1⁄2 mutation, and in the wild-type group, by the presence of HR deficiency. The benefit of niraparib was more pronounced among patients with gBRCA1⁄2 mutation (PFS 21 versus 5.5 months, HR 0.27; 95% CI 0.17–0.41). However, it was not negligible among gBRCA1⁄2 wild-type patients with HR-deficient tumors (12.9 versus 3.8 months, HR 0.38; 95% CI 0.24–0.59). These results led to FDA approval of niraparib in the maintenance setting, regardless of BRCA1⁄2 status. Additionally, niraparib antitumor activity was also documented for late-line treatment of ovarian cancer patients, with greater benefit among HRD-positive tumors, regardless of the relation to a BRCA1⁄2 mutation [204].
Ultimately, rucaparib was also approved by the FDA for maintenance treatment of ovarian cancer, based on the results of ARIEL2 and ARIEL3 trials [89,205]. As for niraparib, a preplanned analysis of PFS, according to a tumor genomic profiling test for homologous recombination and loss of heterozygosis analysis, confirmed that the benefit of the PARPi was bigger but not restricted to BRCA mutant tumors. The PFS was 16.6 months and 13.4 months in patients with BRCAm and homologous recombination-deficient ovarian carcinomas, respectively (versus 5.4 months for patients who received placebo; p < 0.0001).
In line with these findings, it is clear that the population with potential benefit from PARPi is likely wider than germline BRCA mutation-associated disease. However, it is known that a portion of the patients even carrying the mutation will present primary or secondary resistance to the treatment. For this reason, biomarkers are in development to broaden the selection of patients, with the potential clinical benefit from these agents.
5. Conclusions
A portion of breast and ovarian cancers can be defined by a common pathway involved in the tumorigenesis with an impairment of DNA repair by homologous recombination (HR). However, the reason of the inactivation of this pathway remains unclear for most cases, even in the presence of an HR-deficient signature. It is becoming clear that alternative mechanisms (beyond BRCA1 and BRCA2 mutations) can also be used as new biomarkers of therapeutic response. Elucidating them can help improve both genetic counseling and therapeutic response.
Author Contributions
Conceptualization, E.R., E.S.d.S. and S.M.C.; methodology, E.R. and S.M.C.; validation, E.R., E.S.d.S., A.P., F.L. and S.M.C.; formal analysis E.R., E.S.d.S., A.P., F.L. and S.M.C.; resources, E.R. and S.M.C.; data curation, E.R., E.S.d.S., F.L. and S.M.C.; writing—original draft preparation, E.R., E.S.d.S., F.L. and S.M.C.; review and editing, E.R., E.S.d.S., A.P., F.L. and S.M.C.; visualization, all authors.; supervision, E.R. and S.M.C.; project administration, E.R. and S.M.C.; funding acquisition, E.R. and S.M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the French National Cancer Institute (INCa) [PRT-K14-134 to E. SdS, SC and AP], USA [grant number BCRF-16-096, 2017–2018 to AP].
Conflicts of Interest
E Rouleau—Board participation and travel expenses: AstraZeneca, Roche, BMS. The other authors declare no conflict of interest.
References
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, M.A.; Geyer, F.C.; Lacroix-Triki, M.; Marchió, C.; Reis-Filho, J.S. Breast cancer precursors revisited: Molecular features and progression pathways. Histopathology 2010, 57, 171–192. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.C.; De, S.; Almendro, V.; Gönen, M.; Park, S.Y.; Blum, J.L.; Herlihy, W.; Ethington, G.; Schnitt, S.J.; Tung, N.; et al. Evolutionary pathways in BRCA1-associated breast tumors. Cancer Discov. 2012, 2, 503–511. [Google Scholar] [CrossRef]
- Bøxrresen-Dale, A.-L. TP53 and breast cancer. Hum. Mutat. 2003, 21, 292–300. [Google Scholar] [CrossRef]
- Olivier, M.; Langerød, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bièche, I.; et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef]
- Pang, J.-M.B.; Savas, P.; Fellowes, A.P.; Mir Arnau, G.; Kader, T.; Vedururu, R.; Hewitt, C.; Takano, E.A.; Byrne, D.J.; Choong, D.Y.; et al. Breast ductal carcinoma in situ carry mutational driver events representative of invasive breast cancer. Mod. Pathol. 2017, 30, 952–963. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef]
- Ducie, J.; Dao, F.; Considine, M.; Olvera, N.; Shaw, P.A.; Kurman, R.J.; Shih, I.-M.; Soslow, R.A.; Cope, L.; Levine, D.A. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun. 2017, 8, 990. [Google Scholar] [CrossRef]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.-L.; Shih, I.-M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma—Evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, M.; Lu, S.; Zhang, Y.; Wang, H. Promoter hypermethylation of FANCF plays an important role in the occurrence of ovarian cancer through disrupting Fanconi anemia-BRCA pathway. Cancer Biol. Ther. 2006, 5, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [PubMed]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkäs, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.-W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Macintyre, G.; Goranova, T.E.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.-A.; Hanif, A.; Wilson, C.; et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Chirackal Manavalan, A.P.; Pilarova, K.; Kluge, M.; Bartholomeeusen, K.; Rajecky, M.; Oppelt, J.; Khirsariya, P.; Paruch, K.; Krejci, L.; Friedel, C.C.; et al. CDK12 controls G1/S progression by regulating RNAPII processivity at core DNA replication genes. EMBO Rep. 2019, 20, e47592. [Google Scholar] [CrossRef]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef]
- Nishitani, H.; Lygerou, Z. Control of DNA replication licensing in a cell cycle. Genes Cells 2002, 7, 523–534. [Google Scholar] [CrossRef]
- Takeda, D.Y.; Dutta, A. DNA replication and progression through S phase. Oncogene 2005, 24, 2827–2843. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Tomlinson, I. Replicative DNA polymerase mutations in cancer. Curr. Opin. Genet. Dev. 2014, 24, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef]
- Dexheimer, T.S. DNA Repair Pathways and Mechanisms. In DNA Repair of Cancer Stem Cells; Mathews, L.A., Cabarcas, S.M., Hurt, E.M., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 19–32. ISBN 978-94-007-4589-6. [Google Scholar]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair (Amst.) 2015, 36, 13–18. [Google Scholar] [CrossRef]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair (Amst.) 2016, 38, 94–101. [Google Scholar] [CrossRef]
- Rass, E.; Grabarz, A.; Bertrand, P.; Lopez, B.-S. [Double strand break repair, one mechanism can hide another: Alternative non-homologous end joining]. Cancer Radiother. 2012, 16, 1–10. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Connor, A.A.; Gallinger, S. Characterization, Detection, and Treatment Approaches for Homologous Recombination Deficiency in Cancer. Trends Mol. Med. 2017, 23, 1121–1137. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef]
- Brzovic, P.S.; Keeffe, J.R.; Nishikawa, H.; Miyamoto, K.; Fox, D.; Fukuda, M.; Ohta, T.; Klevit, R. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. USA 2003, 100, 5646–5651. [Google Scholar] [CrossRef]
- Joukov, V.; Chen, J.; Fox, E.A.; Green, J.B.A.; Livingston, D.M. Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc. Natl. Acad. Sci. USA 2001, 98, 12078–12083. [Google Scholar] [CrossRef]
- Yu, X.; Chini, C.C.S.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-protein binding domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef]
- Yu, X.; Fu, S.; Lai, M.; Baer, R.; Chen, J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006, 20, 1721–1726. [Google Scholar] [CrossRef]
- Takaoka, M.; Miki, Y. BRCA1 gene: Function and deficiency. Int. J. Clin. Oncol. 2018, 23, 36–44. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2017, 18, 1264. [Google Scholar] [CrossRef] [PubMed]
- Hughes-Davies, L.; Huntsman, D.; Ruas, M.; Fuks, F.; Bye, J.; Chin, S.-F.; Milner, J.; Brown, L.A.; Hsu, F.; Gilks, B.; et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell 2003, 115, 523–535. [Google Scholar] [CrossRef]
- Bork, P.; Blomberg, N.; Nilges, M. Internal repeats in the BRCA2 protein sequence. Nat. Genet. 1996, 13, 22–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Chen, C.F.; Chen, Y.; Xiao, J.; Sharp, Z.D.; Lee, W.H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. USA 1998, 95, 5287–5292. [Google Scholar] [CrossRef]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002, 420, 287–293. [Google Scholar] [CrossRef]
- Martinez, J.S.; Baldeyron, C.; Carreira, A. Molding BRCA2 function through its interacting partners. Cell Cycle 2015, 14, 3389–3395. [Google Scholar] [CrossRef]
- von Nicolai, C.; Ehlén, Å.; Martin, C.; Zhang, X.; Carreira, A. A second DNA binding site in human BRCA2 promotes homologous recombination. Nat. Commun. 2016, 7, 12813. [Google Scholar] [CrossRef]
- Yang, H.; Jeffrey, P.D.; Miller, J.; Kinnucan, E.; Sun, Y.; Thoma, N.H.; Zheng, N.; Chen, P.-L.; Lee, W.-H.; Pavletich, N.P. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 2002, 297, 1837–1848. [Google Scholar] [CrossRef]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.H.; West, S.C. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fujiwara, Y.; Yamamoto, S.; Shibuya, H. A meiosis-specific BRCA2 binding protein recruits recombinases to DNA double-strand breaks to ensure homologous recombination. Nat. Commun. 2019, 10, 722. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, J. DNA Damage-Induced Cell Cycle Checkpoint Control Requires CtIP, a Phosphorylation-Dependent Binding Partner of BRCA1 C-Terminal Domains. Mol. Cell. Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Chai, Y.L.; Shyam, E.; Reddy, P.; Rao, V.N. Induction of apoptosis by the tumor suppressor protein BRCA1. Oncogene 1996, 13, 1–7. [Google Scholar]
- Scully, R.; Anderson, S.F.; Chao, D.M.; Wei, W.; Ye, L.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar] [CrossRef]
- Bochar, D.A.; Wang, L.; Beniya, H.; Kinev, A.; Xue, Y.; Lane, W.S.; Wang, W.; Kashanchi, F.; Shiekhattar, R. BRCA1 is associated with a human SWI/SNF-related complex: Linking chromatin remodeling to breast cancer. Cell 2000, 102, 257–265. [Google Scholar] [CrossRef]
- Marmorstein, L.Y.; Ouchi, T.; Aaronson, S.A. The BRCA2 gene product functionally interacts with p53 and RAD51. Proc. Natl. Acad. Sci. USA 1998, 95, 13869–13874. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Tumour Suppressor Mechanisms in the Control of Chromosome Stability: Insights from <em>BRCA2</em>. Mol. Cells 2014, 37, 95–99. [Google Scholar] [CrossRef]
- Pelttari, L.M.; Heikkinen, T.; Thompson, D.; Kallioniemi, A.; Schleutker, J.; Holli, K.; Blomqvist, C.; Aittomäki, K.; Bützow, R.; Nevanlinna, H. RAD51C is a susceptibility gene for ovarian cancer. Hum. Mol. Genet. 2011, 20, 3278–3288. [Google Scholar] [CrossRef]
- Pelttari, L.M.; Kiiski, J.; Nurminen, R.; Kallioniemi, A.; Schleutker, J.; Gylfe, A.; Aaltonen, L.A.; Leminen, A.; Heikkilä, P.; Blomqvist, C.; et al. A Finnish founder mutation in RAD51D: Analysis in breast, ovarian, prostate, and colorectal cancer. J. Med. Genet. 2012, 49, 429–432. [Google Scholar] [CrossRef]
- Garg, K.; Levine, D.A.; Olvera, N.; Dao, F.; Bisogna, M.; Secord, A.A.; Berchuck, A.; Cerami, E.; Schultz, N.; Soslow, R.A. BRCA1 immunohistochemistry in a molecularly characterized cohort of ovarian high-grade serous carcinomas. Am. J. Surg. Pathol. 2013, 37, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Meisel, J.L.; Hyman, D.M.; Garg, K.; Zhou, Q.; Dao, F.; Bisogna, M.; Gao, J.; Schultz, N.D.; Grisham, R.N.; Phillips, M.; et al. The performance of BRCA1 immunohistochemistry for detecting germline, somatic, and epigenetic BRCA1 loss in high-grade serous ovarian cancer. Ann. Oncol. 2014, 25, 2372–2378. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.A.; Candido Dos Reis, F.J. Immunohistochemistry for the detection of BRCA1 and BRCA2 proteins in patients with ovarian cancer: A systematic review. J. Clin. Pathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nientiedt, C.; Heller, M.; Endris, V.; Volckmar, A.-L.; Zschäbitz, S.; Tapia-Laliena, M.A.; Duensing, A.; Jäger, D.; Schirmacher, P.; Sültmann, H.; et al. Mutations in BRCA2 and taxane resistance in prostate cancer. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Honrado, E.; Osorio, A.; Palacios, J.; Milne, R.L.; Sánchez, L.; Díez, O.; Cazorla, A.; Syrjakoski, K.; Huntsman, D.; Heikkilä, P.; et al. Immunohistochemical Expression of DNA Repair Proteins in Familial Breast Cancer Differentiate BRCA2-Associated Tumors. JCO 2005, 23, 7503–7511. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Healey, S.; Devereau, A.; Hogervorst, F.B.; Monteiro, A.N.; Nathanson, K.L.; Radice, P.; Stoppa-Lyonnet, D.; Tavtigian, S.; Wappenschmidt, B.; et al. ENIGMA—Evidence-based Network for the Interpretation of Germline Mutant Alleles: An international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 2012, 33, 2–7. [Google Scholar] [CrossRef]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Geisler, J.P.; Hatterman-Zogg, M.A.; Rathe, J.A.; Buller, R.E. Frequency of BRCA1 dysfunction in ovarian cancer. J. Natl. Cancer Inst. 2002, 94, 61–67. [Google Scholar] [CrossRef]
- Lips, E.H.; Mulder, L.; Oonk, A.; van der Kolk, L.E.; Hogervorst, F.B.L.; Imholz, A.L.T.; Wesseling, J.; Rodenhuis, S.; Nederlof, P.M. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br. J. Cancer 2013, 108, 2172–2177. [Google Scholar] [CrossRef]
- Bernards, S.S.; Pennington, K.P.; Harrell, M.I.; Agnew, K.J.; Garcia, R.L.; Norquist, B.M.; Swisher, E.M. Clinical characteristics and outcomes of patients with BRCA1 or RAD51C methylated versus mutated ovarian carcinoma. Gynecol. Oncol. 2018, 148, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Dite, G.S.; Makalic, E.; Schmidt, D.F.; Giles, G.G.; Hopper, J.L.; Southey, M.C. Tumour morphology of early-onset breast cancers predicts breast cancer risk for first-degree relatives: The Australian Breast Cancer Family Registry. Breast Cancer Res. 2012, 14, R122. [Google Scholar] [CrossRef] [PubMed]
- Moskwa, P.; Buffa, F.M.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.J.; Beech, J.; Kulshrestha, R.; Abdelmohsen, K.; Weinstock, D.M.; et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 2011, 41, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Paculová, H.; Kohoutek, J. The emerging roles of CDK12 in tumorigenesis. Cell Div. 2017, 12, 7. [Google Scholar] [CrossRef]
- Hou, J.; Wang, Z.; Yang, L.; Guo, X.; Yang, G. The function of EMSY in cancer development. Tumor Biol. 2014, 35, 5061–5066. [Google Scholar] [CrossRef]
- Gudas, J.M.; Nguyen, H.; Li, T.; Cowan, K.H. Hormone-dependent regulation of BRCA1 in human breast cancer cells. Cancer Res. 1995, 55, 4561–4565. [Google Scholar]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef]
- Lee, N.S.; Kim, S.; Jung, Y.W.; Kim, H. Eukaryotic DNA damage responses: Homologous recombination factors and ubiquitin modification. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2018, 809, 88–98. [Google Scholar] [CrossRef]
- Vanderstichele, A.; Busschaert, P.; Olbrecht, S.; Lambrechts, D.; Vergote, I. Genomic signatures as predictive biomarkers of homologous recombination deficiency in ovarian cancer. Eur. J. Cancer 2017, 86, 5–14. [Google Scholar] [CrossRef]
- Gross, E.; van Tinteren, H.; Li, Z.; Raab, S.; Meul, C.; Avril, S.; Laddach, N.; Aubele, M.; Propping, C.; Gkazepis, A.; et al. Identification of BRCA1-like triple-negative breast cancers by quantitative multiplex-ligation-dependent probe amplification (MLPA) analysis of BRCA1-associated chromosomal regions: A validation study. BMC Cancer 2016, 16, 811. [Google Scholar] [CrossRef]
- Wang, Z.C.; Birkbak, N.J.; Culhane, A.C.; Drapkin, R.; Fatima, A.; Tian, R.; Schwede, M.; Alsop, K.; Daniels, K.E.; Piao, H.; et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin. Cancer Res. 2012, 18, 5806–5815. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Karlan, B.Y.; Taniguchi, T.; Fountzilas, E.; Francoeur, N.; Levine, D.A.; Cannistra, S.A. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J. Clin. Oncol. 2010, 28, 3555–3561. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- von Wahlde, M.-K.; Timms, K.M.; Chagpar, A.; Wali, V.B.; Jiang, T.; Bossuyt, V.; Saglam, O.; Reid, J.; Gutin, A.; Neff, C.; et al. Intratumor Heterogeneity of Homologous Recombination Deficiency in Primary Breast Cancer. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2017, 8, 319. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Nilsson, M.P.; Olsson, E.; George, A.M.; Chen, Y.; Kvist, A.; Törngren, T.; Vallon-Christersson, J.; Hegardt, C.; Häkkinen, J.; et al. Targeted sequencing of BRCA1 and BRCA2 across a large unselected breast cancer cohort suggests that one-third of mutations are somatic. Ann. Oncol. 2016, 27, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Fraga, M.F.; Guo, M.; Garcia-Foncillas, J.; Hedenfalk, I.; Godwin, A.K.; Trojan, J.; Vaurs-Barrière, C.; Bignon, Y.J.; Ramus, S.; et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum. Mol. Genet. 2001, 10, 3001–3007. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, A.M.; Spearman, A.D.; Tseng, S.Y.; Sweet, K.; Toland, A.E. Methylation not a frequent “second hit” in tumors with germline BRCA mutations. Fam. Cancer 2009, 8, 339–346. [Google Scholar] [CrossRef]
- Van Heetvelde, M.; Van Bockstal, M.; Poppe, B.; Lambein, K.; Rosseel, T.; Atanesyan, L.; Deforce, D.; Van Den Berghe, I.; De Leeneer, K.; Van Dorpe, J.; et al. Accurate detection and quantification of epigenetic and genetic second hits in BRCA1 and BRCA2-associated hereditary breast and ovarian cancer reveals multiple co-acting second hits. Cancer Lett. 2018, 425, 125–133. [Google Scholar] [CrossRef]
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D.; et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef]
- Tan, S.L.W.; Chadha, S.; Liu, Y.; Gabasova, E.; Perera, D.; Ahmed, K.; Constantinou, S.; Renaudin, X.; Lee, M.; Aebersold, R.; et al. A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 2017, 169, 1105–1118. [Google Scholar] [CrossRef]
- Colas, C.; Golmard, L.; de Pauw, A.; Caputo, S.M.; Stoppa-Lyonnet, D. “Decoding hereditary breast cancer” benefits and questions from multigene panel testing. Breast 2019, 45, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.-H.; et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Ware, M.D.; Buisson, M.; Zetoune, A.B.; Stoppa-Lyonnet, D.; Sinilnikova, O.M.; Mazoyer, S. Does the nonsense-mediated mRNA decay mechanism prevent the synthesis of truncated BRCA1, CHK2, and p53 proteins? Hum. Mutat. 2008, 29, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, E.; Jesson, B.; Briaux, A.; Nogues, C.; Chabaud, V.; Demange, L.; Sokolowska, J.; Coulet, F.; Barouk-Simonet, E.; Bignon, Y.J.; et al. Rare germline large rearrangements in the BRCA1/2 genes and eight candidate genes in 472 patients with breast cancer predisposition. Breast Cancer Res. Treat. 2012, 133, 1179–1190. [Google Scholar] [CrossRef]
- Welcsh, P.L.; King, M.C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef]
- Lesueur, F.; Mebirouk, N.; Jiao, Y.; Barjhoux, L.; Belotti, M.; Laurent, M.; Léone, M.; Houdayer, C.; Bressac-de Paillerets, B.; Vaur, D.; et al. GEMO, a National Resource to Study Genetic Modifiers of Breast and Ovarian Cancer Risk in BRCA1 and BRCA2 Pathogenic Variant Carriers. Front. Oncol. 2018, 8, 490. [Google Scholar] [CrossRef]
- Castéra, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.-R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016. [Google Scholar] [CrossRef]
- Balmaña, J.; Digiovanni, L.; Gaddam, P.; Walsh, M.F.; Joseph, V.; Stadler, Z.K.; Nathanson, K.L.; Garber, J.E.; Couch, F.J.; Offit, K.; et al. Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing. J. Clin. Oncol. 2016, 34, 4071–4078. [Google Scholar] [CrossRef]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Timms, K.M.; Liu, S.; Chen, H.; Litton, J.K.; Potter, J.; Lanchbury, J.S.; Stemke-Hale, K.; Hennessy, B.T.; Arun, B.K.; et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res. 2011, 17, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Southey, M.C.; Ramus, S.J.; Dowty, J.G.; Smith, L.D.; Tesoriero, A.A.; Wong, E.E.M.; Dite, G.S.; Jenkins, M.A.; Byrnes, G.B.; Winship, I.; et al. Morphological predictors of BRCA1 germline mutations in young women with breast cancer. Br. J. Cancer 2011, 104, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Bégin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485. [Google Scholar] [CrossRef]
- Lakhani, S.R.; Van De Vijver, M.J.; Jacquemier, J.; Anderson, T.J.; Osin, P.P.; McGuffog, L.; Easton, D.F. The pathology of familial breast cancer: Predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J. Clin. Oncol. 2002, 20, 2310–2318. [Google Scholar] [CrossRef]
- Mulligan, A.M.; Pinnaduwage, D.; Bane, A.L.; Bull, S.B.; O’Malley, F.P.; Andrulis, I.L. CK8/18 expression, the basal phenotype, and family history in identifying BRCA1-associated breast cancer in the Ontario site of the breast cancer family registry. Cancer 2011, 117, 1350–1359. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Couch, F.J.; Parsons, M.T.; McGuffog, L.; Barrowdale, D.; Bolla, M.K.; Wang, Q.; Healey, S.; Schmutzler, R.; Wappenschmidt, B.; et al. Refined histopathological predictors of BRCA1 and BRCA2 mutation status: A large-scale analysis of breast cancer characteristics from the BCAC, CIMBA, and ENIGMA consortia. Breast Cancer Res. 2014, 16, 3419. [Google Scholar] [CrossRef]
- Larsen, M.J.; Kruse, T.A.; Tan, Q.; Lænkholm, A.-V.; Bak, M.; Lykkesfeldt, A.E.; Sørensen, K.P.; Hansen, T.V.O.; Ejlertsen, B.; Gerdes, A.-M.; et al. Classifications within molecular subtypes enables identification of BRCA1/BRCA2 mutation carriers by RNA tumor profiling. PLoS ONE 2013, 8, e64268. [Google Scholar] [CrossRef]
- Bane, A.L.; Beck, J.C.; Bleiweiss, I.; Buys, S.S.; Catalano, E.; Daly, M.B.; Giles, G.; Godwin, A.K.; Hibshoosh, H.; Hopper, J.L.; et al. BRCA2 mutation-associated breast cancers exhibit a distinguishing phenotype based on morphology and molecular profiles from tissue microarrays. Am. J. Surg. Pathol. 2007, 31, 121–128. [Google Scholar] [CrossRef]
- Tung, N.; Wang, Y.; Collins, L.C.; Kaplan, J.; Li, H.; Gelman, R.; Comander, A.H.; Gallagher, B.; Fetten, K.; Krag, K.; et al. Estrogen receptor positive breast cancers in BRCA1 mutation carriers: Clinical risk factors and pathologic features. Breast Cancer Res. 2010, 12, R12. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Metcalfe, K.; Sun, P.; Hanna, W.M.; Lynch, H.T.; Ghadirian, P.; Tung, N.; Olopade, O.I.; Weber, B.L.; McLennan, J.; et al. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: The influence of age, grade, and histological type. Clin. Cancer Res. 2004, 10, 2029–2034. [Google Scholar] [CrossRef] [PubMed]
- Copson, E.R.; Maishman, T.C.; Tapper, W.J.; Cutress, R.I.; Greville-Heygate, S.; Altman, D.G.; Eccles, B.; Gerty, S.; Durcan, L.T.; Jones, L.; et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): A prospective cohort study. Lancet Oncol. 2018, 19, 169–180. [Google Scholar] [CrossRef]
- Baretta, Z.; Mocellin, S.; Goldin, E.; Olopade, O.I.; Huo, D. Effect of BRCA germline mutations on breast cancer prognosis: A systematic review and meta-analysis. Medicine (Baltimore) 2016, 95, e4975. [Google Scholar] [CrossRef]
- Lakhani, S.R.; Manek, S.; Penault-Llorca, F.; Flanagan, A.; Arnout, L.; Merrett, S.; McGuffog, L.; Steele, D.; Devilee, P.; Klijn, J.G.M.; et al. Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clin. Cancer Res. 2004, 10, 2473–2481. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Loeb, L.A. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991, 51, 3075–3079. [Google Scholar]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Santana Dos Santos, E.; Lallemand, F.; Burke, L.; Stoppa-Lyonnet, D.; Brown, M.; Caputo, S.M.; Rouleau, E. Non-Coding Variants in BRCA1 and BRCA2 Genes: Potential Impact on Breast and Ovarian Cancer Predisposition. Cancers (Basel) 2018, 10, 453. [Google Scholar] [CrossRef] [PubMed]
- Caputo, S.; Benboudjema, L.; Sinilnikova, O.; Rouleau, E.; Béroud, C.; Lidereau, R. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res. 2012, 40, D992–D1002. [Google Scholar] [CrossRef] [PubMed]
- Béroud, C.; Letovsky, S.I.; Braastad, C.D.; Caputo, S.M.; Beaudoux, O.; Bignon, Y.J.; Bressac-De Paillerets, B.; Bronner, M.; Buell, C.M.; Collod-Béroud, G.; et al. BRCA Share: A Collection of Clinical BRCA Gene Variants. Hum. Mutat. 2016. [Google Scholar] [CrossRef]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Whiley, P.J.; Guidugli, L.; Walker, L.C.; Healey, S.; Thompson, B.A.; Lakhani, S.R.; Da Silva, L.M.; kConFab Investigators; Tavtigian, S.V.; Goldgar, D.E.; et al. Splicing and multifactorial analysis of intronic BRCA1 and BRCA2 sequence variants identifies clinically significant splicing aberrations up to 12 nucleotides from the intron/exon boundary. Hum. Mutat. 2011, 32, 678–687. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Whiley, P.J.; Thompson, B.; Feng, B.; Healey, S.; Brown, M.A.; Pettigrew, C.; kConFab; Van Asperen, C.J.; Ausems, M.G.E.M.; et al. BRCA1 R1699Q variant displaying ambiguous functional abrogation confers intermediate breast and ovarian cancer risk. J. Med. Genet. 2012, 49. [Google Scholar] [CrossRef]
- de la Hoya, M.; Soukarieh, O.; López-Perolio, I.; Vega, A.; Walker, L.C.; van Ierland, Y.; Baralle, D.; Santamariña, M.; Lattimore, V.; Wijnen, J.; et al. Combined genetic and splicing analysis of BRCA1 c.[594-2A>C; 641A>G] highlights the relevance of naturally occurring in-frame transcripts for developing disease gene variant classification algorithms. Hum. Mol. Genet. 2016, 25, 2256–2268. [Google Scholar] [CrossRef]
- Dos Santos, E.S.; Caputo, S.M.; Castera, L.; Gendrot, M.; Briaux, A.; Breault, M.; Krieger, S.; Rogan, P.K.; Mucaki, E.J.; Burke, L.J.; et al. Assessment of the functional impact of germline BRCA1/2 variants located in non-coding regions in families with breast and/or ovarian cancer predisposition. Breast Cancer Res. Treat. 2017. [Google Scholar] [CrossRef]
- Burke, L.J.; Sevcik, J.; Gambino, G.; Tudini, E.; Mucaki, E.J.; Shirley, B.C.; Whiley, P.; Parsons, M.T.; De Leeneer, K.; Gutiérrez-Enríquez, S.; et al. BRCA1 and BRCA2 5’ non-coding region variants identified in breast cancer patients alter promoter activity and protein binding. Hum. Mutat. 2018. [Google Scholar] [CrossRef] [PubMed]
- Caputo, S.M.; Léone, M.; Damiola, F.; Ehlen, A.; Carreira, A.; Gaidrat, P.; Martins, A.; Brandão, R.D.; Peixoto, A.; Vega, A.; et al. Full in-frame exon 3 skipping of BRCA2 confers high risk of breast and/or ovarian cancer. Oncotarget 2018, 9, 17334–17348. [Google Scholar] [CrossRef] [PubMed]
- Moghadasi, S.; Meeks, H.D.; Vreeswijk, M.P.; Janssen, L.A.; Borg, Å.; Ehrencrona, H.; Paulsson-Karlsson, Y.; Wappenschmidt, B.; Engel, C.; Gehrig, A.; et al. The BRCA1 c. 5096G>A p.Arg1699Gln (R1699Q) intermediate risk variant: Breast and ovarian cancer risk estimation and recommendations for clinical management from the ENIGMA consortium. J. Med. Genet. 2018, 55, 15–20. [Google Scholar] [CrossRef]
- Parsons, M.T.; Tudini, E.; Li, H.; Hahnen, E.; Wappenschmidt, B.; Feliubadaló, L.; Aalfs, C.M.; Agata, S.; Aittomäki, K.; Alducci, E.; et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum. Mutat. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef]
- Ramensky, V.; Bork, P.; Sunyaev, S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002, 30, 3894–3900. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Greenblatt, M.S.; Lesueur, F.; Byrnes, G.B. IARC Unclassified Genetic Variants Working Group In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 2008, 29, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Houdayer, C.; Caux-Moncoutier, V.; Krieger, S.; Barrois, M.; Bonnet, F.; Bourdon, V.; Bronner, M.; Buisson, M.; Coulet, F.; Gaildrat, P.; et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum. Mutat. 2012, 33, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Soukarieh, O.; Gaildrat, P.; Hamieh, M.; Drouet, A.; Baert-Desurmont, S.; Frébourg, T.; Tosi, M.; Martins, A. Exonic Splicing Mutations Are More Prevalent than Currently Estimated and Can Be Predicted by Using In Silico Tools. PLoS Genet. 2016, 12, e1005756. [Google Scholar] [CrossRef]
- Vallée, M.P.; Sera, T.L.D.; Nix, D.A.; Paquette, A.M.; Parsons, M.T.; Bell, R.; Hoffman, A.; Hogervorst, F.B.L.; Goldgar, D.E.; Spurdle, A.B.; et al. Adding In Silico Assessment of Potential Splice Aberration to the Integrated Evaluation of BRCA Gene Unclassified Variants. Hum. Mutat. 2016. [Google Scholar] [CrossRef] [PubMed]
- Leman, R.; Gaildrat, P.; Gac, G.L.; Ka, C.; Fichou, Y.; Audrezet, M.-P.; Caux-Moncoutier, V.; Caputo, S.M.; Boutry-Kryza, N.; Léone, M.; et al. Novel diagnostic tool for prediction of variant spliceogenicity derived from a set of 395 combined in silico/in vitro studies: An international collaborative effort. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Leman, R.; Tubeuf, H.; Raad, S.; Tournier, I.; Derambure, C.; Lanos, R.; Gaildrat, P.; Castelain, G.; Hauchard, J.; Killian, A.; et al. Assessment of branch point prediction tools to predict physiological branch points and their alteration by variants. BMC Genom. 2020, 21, 86. [Google Scholar] [CrossRef]
- Muller, D.; Rouleau, E.; Schultz, I.; Caputo, S.; Lefol, C.; Bièche, I.; Caron, O.; Noguès, C.; Limacher, J.M.; Demange, L.; et al. An entire exon 3 germ-line rearrangement in the BRCA2 gene: Pathogenic relevance of exon 3 deletion in breast cancer predisposition. BMC Med. Genet. 2011, 12, 121. [Google Scholar] [CrossRef]
- Gaildrat, P.; Krieger, S.; Giacomo, D.D.; Abdat, J.; Révillion, F.; Caputo, S.; Vaur, D.; Jamard, E.; Bohers, E.; Ledemeney, D.; et al. Multiple sequence variants of BRCA2 exon 7 alter splicing regulation. J. Med. Genet. 2012, 49, 609–617. [Google Scholar] [CrossRef]
- Millot, G.; Carvalho, M.A.; Caputo, S.M.; Vreeswijk, M.P.G.; Brown, M.A.; Webb, M.; Rouleau, E.; Neuhausen, S.L.; Hansen, T.V.O.; Galli, A.; et al. A Guide for Functional Analysis of BRCA1 Variants of Uncertain Significance (VUS). Hum. Mutat. 2012, 33, 1526–1537. [Google Scholar] [CrossRef]
- Guidugli, L.; Carreira, A.; Caputo, S.M.; Ehlen, A.; Galli, A.; Monteiro, A.N.A.; Neuhausen, S.L.; Hansen, T.V.O.; Couch, F.J.; Vreeswijk, M.P.G. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum. Mutat. 2014, 35, 151–164. [Google Scholar] [CrossRef]
- Jhuraney, A.; Velkova, A.; Johnson, R.C.; Kessing, B.; Carvalho, R.S.; Whiley, P.; Spurdle, A.B.; Vreeswijk, M.P.G.; Caputo, S.M.; Millot, G.A.; et al. BRCA1 Circos: A visualisation resource for functional analysis of missense variants. J. Med. Genet. 2015, 52, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Petitalot, A.; Dardillac, E.; Jacquet, E.; Nhiri, N.; Guirouilh-Barbat, J.; Julien, P.; Bouazzaoui, I.; Bonte, D.; Feunteun, J.; Schnell, J.A.; et al. Combining Homologous Recombination and Phosphopeptide-binding Data to Predict the Impact of BRCA1 BRCT Variants on Cancer Risk. Mol. Cancer Res. 2019, 17, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Meulemans, L.; Mesman, R.L.S.; Caputo, S.M.; Krieger, S.; Guillaud-Bataille, M.; Caux-Moncoutier, V.; Léone, M.; Boutry-Kryza, N.; Sokolowska, J.; Révillion, F.; et al. Skipping nonsense to maintain function: The paradigm of BRCA2 exon 12. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef]
- Couch, F.J.; Nathanson, K.L.; Offit, K. Two decades after BRCA: Setting paradigms in personalized cancer care and prevention. Science 2014, 343, 1466–1470. [Google Scholar] [CrossRef]
- Domchek, S.M.; Friebel, T.M.; Singer, C.F.; Evans, D.G.; Lynch, H.T.; Isaacs, C.; Garber, J.E.; Neuhausen, S.L.; Matloff, E.; Eeles, R.; et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010, 304, 967–975. [Google Scholar] [CrossRef]
- Carbine, N.E.; Lostumbo, L.; Wallace, J.; Ko, H. Risk-reducing mastectomy for the prevention of primary breast cancer. Cochrane Database Syst. Rev. 2018, 4, CD002748. [Google Scholar] [CrossRef]
- Saadatmand, S.; Geuzinge, H.A.; Rutgers, E.J.T.; Mann, R.M.; de Roy van Zuidewijn, D.B.W.; Zonderland, H.M.; Tollenaar, R.A.E.M.; Lobbes, M.B.I.; Ausems, M.G.E.M.; van ’t Riet, M.; et al. MRI versus mammography for breast cancer screening in women with familial risk (FaMRIsc): A multicentre, randomised, controlled trial. Lancet Oncol. 2019, 20, 1136–1147. [Google Scholar] [CrossRef]
- Phi, X.-A.; Houssami, N.; Obdeijn, I.-M.; Warner, E.; Sardanelli, F.; Leach, M.O.; Riedl, C.C.; Trop, I.; Tilanus-Linthorst, M.M.A.; Mandel, R.; et al. Magnetic resonance imaging improves breast screening sensitivity in BRCA mutation carriers age ≥ 50 years: Evidence from an individual patient data meta-analysis. J. Clin. Oncol. 2015, 33, 349–356. [Google Scholar] [CrossRef]
- Hartmann, L.C.; Lindor, N.M. The Role of Risk-Reducing Surgery in Hereditary Breast and Ovarian Cancer. N. Engl. J. Med. 2016, 374, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Stirling, D.; Evans, D.G.R.; Pichert, G.; Shenton, A.; Kirk, E.N.; Rimmer, S.; Steel, C.M.; Lawson, S.; Busby-Earle, R.M.C.; Walker, J.; et al. Screening for familial ovarian cancer: Failure of current protocols to detect ovarian cancer at an early stage according to the international Federation of gynecology and obstetrics system. J. Clin. Oncol. 2005, 23, 5588–5596. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.A.; Pike, M.C.; Jotwani, A.R.; Friebel, T.M.; Soslow, R.A.; Levine, D.A.; Nathanson, K.L.; Konner, J.A.; Arnold, A.G.; Bogomolniy, F.; et al. Uterine Cancer After Risk-Reducing Salpingo-oophorectomy Without Hysterectomy in Women With BRCA Mutations. JAMA Oncol. 2016, 2, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Havrilesky, L.J.; Moss, H.A.; Chino, J.; Myers, E.R.; Kauff, N.D. Mortality reduction and cost-effectiveness of performing hysterectomy at the time of risk-reducing salpingo-oophorectomy for prophylaxis against serous/serous-like uterine cancers in BRCA1 mutation carriers. Gynecol. Oncol. 2017, 145, 549–554. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians and Gynecologists; ACOG Committee on Practice Bulletins—Gynecology; ACOG Committee on Genetics; Society of Gynecologic Oncologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet. Gynecol. 2009, 113, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pilarski, R.; Berry, M.; Buys, S.S.; Farmer, M.; Friedman, S.; Garber, J.E.; Kauff, N.D.; Khan, S.; Klein, C.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J. Natl. Compr. Canc. Netw. 2017, 15, 9–20. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Wieand, S.; Hale, K.; Lee, M.; Walsh, T.; Owens, K.; Tait, J.; Ford, L.; Dunn, B.K.; Costantino, J.; et al. Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention Trial. JAMA 2001, 286, 2251–2256. [Google Scholar] [CrossRef]
- Havrilesky, L.J.; Gierisch, J.M.; Moorman, P.G.; Coeytaux, R.R.; Urrutia, R.P.; Lowery, W.J.; Dinan, M.; McBroom, A.J.; Wing, L.; Musty, M.D.; et al. Oral contraceptive use for the primary prevention of ovarian cancer. Evid. Rep. Technol. Assess (Full Rep.) 2013, 1–514. [Google Scholar]
- Moorman, P.G.; Havrilesky, L.J.; Gierisch, J.M.; Coeytaux, R.R.; Lowery, W.J.; Peragallo Urrutia, R.; Dinan, M.; McBroom, A.J.; Hasselblad, V.; Sanders, G.D.; et al. Oral contraceptives and risk of ovarian cancer and breast cancer among high-risk women: A systematic review and meta-analysis. J. Clin. Oncol. 2013, 31, 4188–4198. [Google Scholar] [CrossRef]
- Martín, M. Platinum compounds in the treatment of advanced breast cancer. Clin. Breast Cancer 2001, 2, 190–208. [Google Scholar] [CrossRef]
- Sledge, G.W.; Loehrer, P.J.; Roth, B.J.; Einhorn, L.H. Cisplatin as first-line therapy for metastatic breast cancer. J. Clin. Oncol. 1988, 6, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.A.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2015, 33, 1902–1909. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Schneeweiss, A.; Loibl, S.; Salat, C.; Denkert, C.; Rezai, M.; Blohmer, J.U.; Jackisch, C.; Paepke, S.; Gerber, B.; et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): A randomised phase 2 trial. Lancet Oncol. 2014, 15, 747–756. [Google Scholar] [CrossRef]
- Byrski, T.; Gronwald, J.; Huzarski, T.; Grzybowska, E.; Budryk, M.; Stawicka, M.; Mierzwa, T.; Szwiec, M.; Wisniowski, R.; Siolek, M.; et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J. Clin. Oncol. 2010, 28, 375–379. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Wang, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Xie, Y. Prevalence of BRCA1 mutations and responses to neoadjuvant chemotherapy among BRCA1 carriers and non-carriers with triple-negative breast cancer. Ann. Oncol. 2015, 26, 523–528. [Google Scholar] [CrossRef]
- Dwadasi, S.; Tong, Y.; Walsh, T.; Danso, M.A.; Ma, C.X.; Silverman, P.; King, M.-C.; Perkins, S.M.; Badve, S.S.; Miller, K. Cisplatin with or without rucaparib after preoperative chemotherapy in patients with triple-negative breast cancer (TNBC): Hoosier Oncology Group BRE09-146. JCO 2014, 32, 1019. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.-H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Jensen, K.C.; Vinayak, S.; Kurian, A.W.; Lipson, J.A.; Flaherty, P.J.; Timms, K.; Abkevich, V.; Schackmann, E.A.; Wapnir, I.L.; et al. Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA1/2 Mutation-Associated Breast Cancer With Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. J. Clin. Oncol. 2015, 33, 1895–1901. [Google Scholar] [CrossRef]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Han, H.S.; Diéras, V.; Robson, M.; Palácová, M.; Marcom, P.K.; Jager, A.; Bondarenko, I.; Citrin, D.; Campone, M.; Telli, M.L.; et al. Veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in patients with BRCA1/2 locally recurrent/metastatic breast cancer: Randomized phase II study. Ann. Oncol. 2018, 29, 154–161. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Kaye, S.B.; Lubinski, J.; Matulonis, U.; Ang, J.E.; Gourley, C.; Karlan, B.Y.; Amnon, A.; Bell-McGuinn, K.M.; Chen, L.-M.; Friedlander, M.; et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin. Oncol. 2012, 30, 372–379. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).