HRness in Breast and Ovarian Cancers

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Tumorigenesis of Breast and Ovarian Epithelial Carcinomas
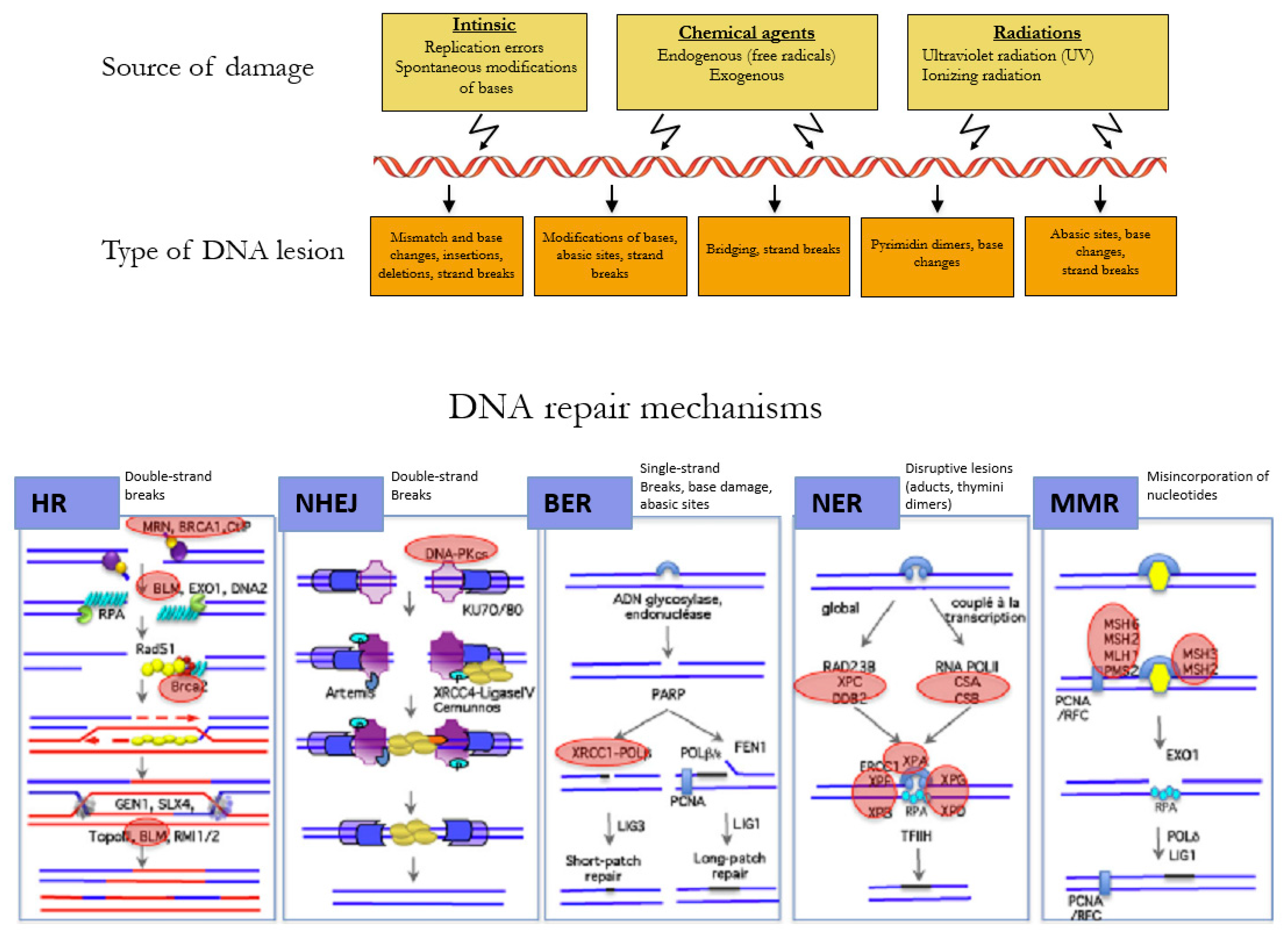
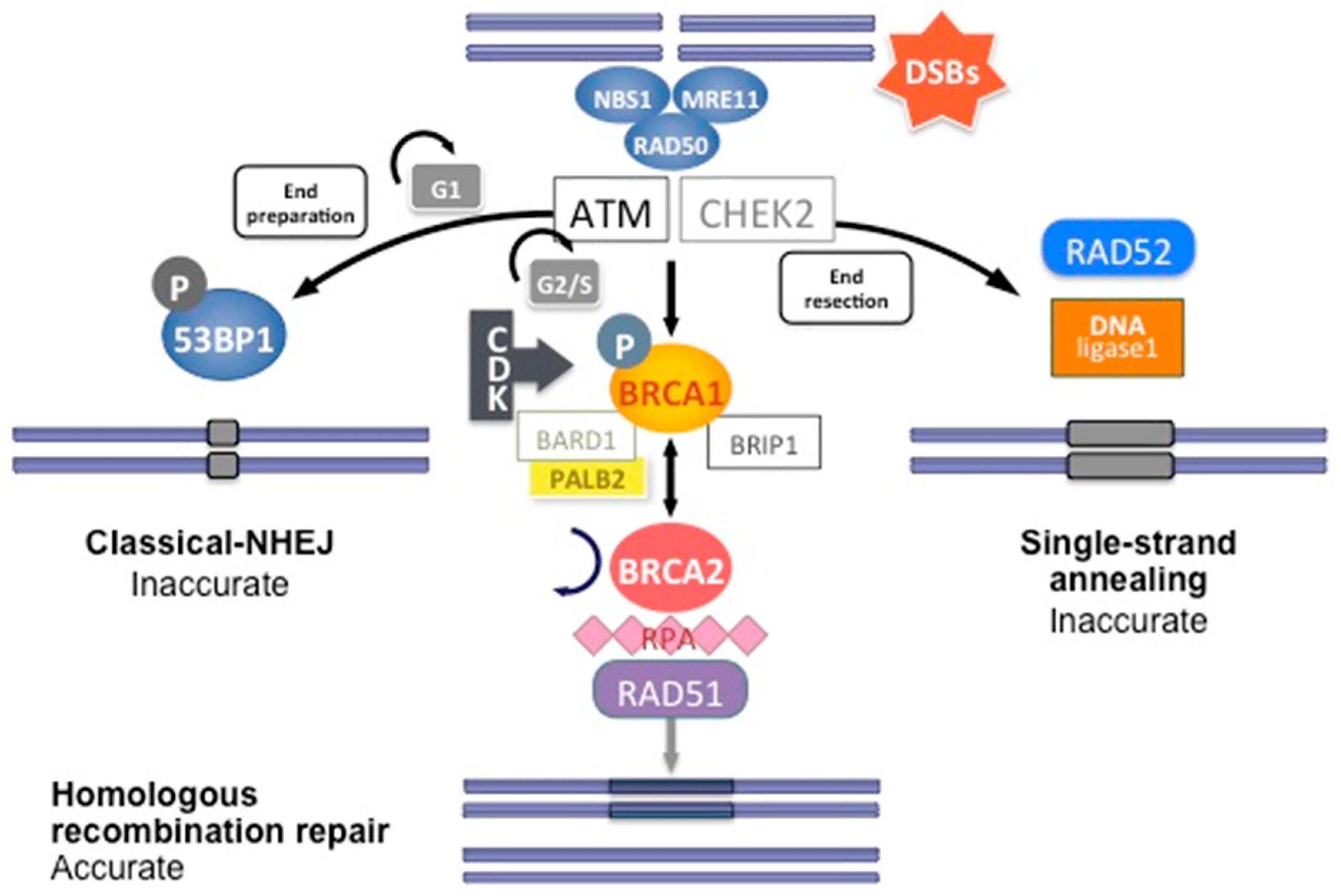
1.1. Maintenance of Genome Integrity
1.1.1. Cell Cycle, DNA Repair, and Apoptosis
1.1.2. Reparation Mechanisms of DNA
1.1.3. Dysfunction of the Repair Pathways
- (1)
- Protein Expression Alteration and Mutation in the Coding Regions
- (2)
- Presence of Missense Variants
- (3)
- Promoter Methylation and miRNA
- (4)
- Transcription and Post-Translational Regulation
1.1.4. Compensatory Mechanisms to Other Reparation Pathways
1.2. Signature
1.3. Temporal Order and Somatic Tumor-Driving Events of BRCA-Associated Tumorigenesis
1.4. Conclusion for Routine Practice
2. Description of Mutations in HR Pathways in Breast and Ovarian Cancers
2.1. Hereditary Breast and Ovarian Cancers
2.1.1. Breast Cancers Related to BRCA1/2 Mutations
2.1.2. Ovarian Cancer Related to BRCA1/2 Mutations
3. Unidentified HR Deficiency Mechanisms in Ovarian and Breast Cancers
3.1. BRCA1/BRCA2 Variants of Uncertain Significance
3.2. Variants in HR Genes Beyond BRCA1⁄2
4. Management
4.1. Risk Reduction Interventions
4.2. Implications for Treatment Response: BRCA1/2 Mutations as Predictive Biomarkers
4.2.1. Sensitivity to Platinum Salts
4.2.2. HR Deficiency and Development of a Targeted Therapy: PARP Inhibitor Treatments
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, M.A.; Geyer, F.C.; Lacroix-Triki, M.; Marchió, C.; Reis-Filho, J.S. Breast cancer precursors revisited: Molecular features and progression pathways. Histopathology 2010, 57, 171–192. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.C.; De, S.; Almendro, V.; Gönen, M.; Park, S.Y.; Blum, J.L.; Herlihy, W.; Ethington, G.; Schnitt, S.J.; Tung, N.; et al. Evolutionary pathways in BRCA1-associated breast tumors. Cancer Discov. 2012, 2, 503–511. [Google Scholar] [CrossRef]
- Bøxrresen-Dale, A.-L. TP53 and breast cancer. Hum. Mutat. 2003, 21, 292–300. [Google Scholar] [CrossRef]
- Olivier, M.; Langerød, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bièche, I.; et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef]
- Pang, J.-M.B.; Savas, P.; Fellowes, A.P.; Mir Arnau, G.; Kader, T.; Vedururu, R.; Hewitt, C.; Takano, E.A.; Byrne, D.J.; Choong, D.Y.; et al. Breast ductal carcinoma in situ carry mutational driver events representative of invasive breast cancer. Mod. Pathol. 2017, 30, 952–963. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef]
- Ducie, J.; Dao, F.; Considine, M.; Olvera, N.; Shaw, P.A.; Kurman, R.J.; Shih, I.-M.; Soslow, R.A.; Cope, L.; Levine, D.A. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun. 2017, 8, 990. [Google Scholar] [CrossRef]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.-L.; Shih, I.-M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma—Evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, M.; Lu, S.; Zhang, Y.; Wang, H. Promoter hypermethylation of FANCF plays an important role in the occurrence of ovarian cancer through disrupting Fanconi anemia-BRCA pathway. Cancer Biol. Ther. 2006, 5, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [PubMed]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkäs, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.-W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Macintyre, G.; Goranova, T.E.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.-A.; Hanif, A.; Wilson, C.; et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Chirackal Manavalan, A.P.; Pilarova, K.; Kluge, M.; Bartholomeeusen, K.; Rajecky, M.; Oppelt, J.; Khirsariya, P.; Paruch, K.; Krejci, L.; Friedel, C.C.; et al. CDK12 controls G1/S progression by regulating RNAPII processivity at core DNA replication genes. EMBO Rep. 2019, 20, e47592. [Google Scholar] [CrossRef]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef]
- Nishitani, H.; Lygerou, Z. Control of DNA replication licensing in a cell cycle. Genes Cells 2002, 7, 523–534. [Google Scholar] [CrossRef]
- Takeda, D.Y.; Dutta, A. DNA replication and progression through S phase. Oncogene 2005, 24, 2827–2843. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Tomlinson, I. Replicative DNA polymerase mutations in cancer. Curr. Opin. Genet. Dev. 2014, 24, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef]
- Dexheimer, T.S. DNA Repair Pathways and Mechanisms. In DNA Repair of Cancer Stem Cells; Mathews, L.A., Cabarcas, S.M., Hurt, E.M., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 19–32. ISBN 978-94-007-4589-6. [Google Scholar]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair (Amst.) 2015, 36, 13–18. [Google Scholar] [CrossRef]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair (Amst.) 2016, 38, 94–101. [Google Scholar] [CrossRef]
- Rass, E.; Grabarz, A.; Bertrand, P.; Lopez, B.-S. [Double strand break repair, one mechanism can hide another: Alternative non-homologous end joining]. Cancer Radiother. 2012, 16, 1–10. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Connor, A.A.; Gallinger, S. Characterization, Detection, and Treatment Approaches for Homologous Recombination Deficiency in Cancer. Trends Mol. Med. 2017, 23, 1121–1137. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef]
- Brzovic, P.S.; Keeffe, J.R.; Nishikawa, H.; Miyamoto, K.; Fox, D.; Fukuda, M.; Ohta, T.; Klevit, R. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. USA 2003, 100, 5646–5651. [Google Scholar] [CrossRef]
- Joukov, V.; Chen, J.; Fox, E.A.; Green, J.B.A.; Livingston, D.M. Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc. Natl. Acad. Sci. USA 2001, 98, 12078–12083. [Google Scholar] [CrossRef]
- Yu, X.; Chini, C.C.S.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-protein binding domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef]
- Yu, X.; Fu, S.; Lai, M.; Baer, R.; Chen, J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006, 20, 1721–1726. [Google Scholar] [CrossRef]
- Takaoka, M.; Miki, Y. BRCA1 gene: Function and deficiency. Int. J. Clin. Oncol. 2018, 23, 36–44. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2017, 18, 1264. [Google Scholar] [CrossRef] [PubMed]
- Hughes-Davies, L.; Huntsman, D.; Ruas, M.; Fuks, F.; Bye, J.; Chin, S.-F.; Milner, J.; Brown, L.A.; Hsu, F.; Gilks, B.; et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell 2003, 115, 523–535. [Google Scholar] [CrossRef]
- Bork, P.; Blomberg, N.; Nilges, M. Internal repeats in the BRCA2 protein sequence. Nat. Genet. 1996, 13, 22–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Chen, C.F.; Chen, Y.; Xiao, J.; Sharp, Z.D.; Lee, W.H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. USA 1998, 95, 5287–5292. [Google Scholar] [CrossRef]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002, 420, 287–293. [Google Scholar] [CrossRef]
- Martinez, J.S.; Baldeyron, C.; Carreira, A. Molding BRCA2 function through its interacting partners. Cell Cycle 2015, 14, 3389–3395. [Google Scholar] [CrossRef]
- von Nicolai, C.; Ehlén, Å.; Martin, C.; Zhang, X.; Carreira, A. A second DNA binding site in human BRCA2 promotes homologous recombination. Nat. Commun. 2016, 7, 12813. [Google Scholar] [CrossRef]
- Yang, H.; Jeffrey, P.D.; Miller, J.; Kinnucan, E.; Sun, Y.; Thoma, N.H.; Zheng, N.; Chen, P.-L.; Lee, W.-H.; Pavletich, N.P. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 2002, 297, 1837–1848. [Google Scholar] [CrossRef]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.H.; West, S.C. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fujiwara, Y.; Yamamoto, S.; Shibuya, H. A meiosis-specific BRCA2 binding protein recruits recombinases to DNA double-strand breaks to ensure homologous recombination. Nat. Commun. 2019, 10, 722. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, J. DNA Damage-Induced Cell Cycle Checkpoint Control Requires CtIP, a Phosphorylation-Dependent Binding Partner of BRCA1 C-Terminal Domains. Mol. Cell. Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Chai, Y.L.; Shyam, E.; Reddy, P.; Rao, V.N. Induction of apoptosis by the tumor suppressor protein BRCA1. Oncogene 1996, 13, 1–7. [Google Scholar]
- Scully, R.; Anderson, S.F.; Chao, D.M.; Wei, W.; Ye, L.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar] [CrossRef]
- Bochar, D.A.; Wang, L.; Beniya, H.; Kinev, A.; Xue, Y.; Lane, W.S.; Wang, W.; Kashanchi, F.; Shiekhattar, R. BRCA1 is associated with a human SWI/SNF-related complex: Linking chromatin remodeling to breast cancer. Cell 2000, 102, 257–265. [Google Scholar] [CrossRef]
- Marmorstein, L.Y.; Ouchi, T.; Aaronson, S.A. The BRCA2 gene product functionally interacts with p53 and RAD51. Proc. Natl. Acad. Sci. USA 1998, 95, 13869–13874. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Tumour Suppressor Mechanisms in the Control of Chromosome Stability: Insights from <em>BRCA2</em>. Mol. Cells 2014, 37, 95–99. [Google Scholar] [CrossRef]
- Pelttari, L.M.; Heikkinen, T.; Thompson, D.; Kallioniemi, A.; Schleutker, J.; Holli, K.; Blomqvist, C.; Aittomäki, K.; Bützow, R.; Nevanlinna, H. RAD51C is a susceptibility gene for ovarian cancer. Hum. Mol. Genet. 2011, 20, 3278–3288. [Google Scholar] [CrossRef]
- Pelttari, L.M.; Kiiski, J.; Nurminen, R.; Kallioniemi, A.; Schleutker, J.; Gylfe, A.; Aaltonen, L.A.; Leminen, A.; Heikkilä, P.; Blomqvist, C.; et al. A Finnish founder mutation in RAD51D: Analysis in breast, ovarian, prostate, and colorectal cancer. J. Med. Genet. 2012, 49, 429–432. [Google Scholar] [CrossRef]
- Garg, K.; Levine, D.A.; Olvera, N.; Dao, F.; Bisogna, M.; Secord, A.A.; Berchuck, A.; Cerami, E.; Schultz, N.; Soslow, R.A. BRCA1 immunohistochemistry in a molecularly characterized cohort of ovarian high-grade serous carcinomas. Am. J. Surg. Pathol. 2013, 37, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Meisel, J.L.; Hyman, D.M.; Garg, K.; Zhou, Q.; Dao, F.; Bisogna, M.; Gao, J.; Schultz, N.D.; Grisham, R.N.; Phillips, M.; et al. The performance of BRCA1 immunohistochemistry for detecting germline, somatic, and epigenetic BRCA1 loss in high-grade serous ovarian cancer. Ann. Oncol. 2014, 25, 2372–2378. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.A.; Candido Dos Reis, F.J. Immunohistochemistry for the detection of BRCA1 and BRCA2 proteins in patients with ovarian cancer: A systematic review. J. Clin. Pathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nientiedt, C.; Heller, M.; Endris, V.; Volckmar, A.-L.; Zschäbitz, S.; Tapia-Laliena, M.A.; Duensing, A.; Jäger, D.; Schirmacher, P.; Sültmann, H.; et al. Mutations in BRCA2 and taxane resistance in prostate cancer. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Honrado, E.; Osorio, A.; Palacios, J.; Milne, R.L.; Sánchez, L.; Díez, O.; Cazorla, A.; Syrjakoski, K.; Huntsman, D.; Heikkilä, P.; et al. Immunohistochemical Expression of DNA Repair Proteins in Familial Breast Cancer Differentiate BRCA2-Associated Tumors. JCO 2005, 23, 7503–7511. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Healey, S.; Devereau, A.; Hogervorst, F.B.; Monteiro, A.N.; Nathanson, K.L.; Radice, P.; Stoppa-Lyonnet, D.; Tavtigian, S.; Wappenschmidt, B.; et al. ENIGMA—Evidence-based Network for the Interpretation of Germline Mutant Alleles: An international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 2012, 33, 2–7. [Google Scholar] [CrossRef]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Geisler, J.P.; Hatterman-Zogg, M.A.; Rathe, J.A.; Buller, R.E. Frequency of BRCA1 dysfunction in ovarian cancer. J. Natl. Cancer Inst. 2002, 94, 61–67. [Google Scholar] [CrossRef]
- Lips, E.H.; Mulder, L.; Oonk, A.; van der Kolk, L.E.; Hogervorst, F.B.L.; Imholz, A.L.T.; Wesseling, J.; Rodenhuis, S.; Nederlof, P.M. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br. J. Cancer 2013, 108, 2172–2177. [Google Scholar] [CrossRef]
- Bernards, S.S.; Pennington, K.P.; Harrell, M.I.; Agnew, K.J.; Garcia, R.L.; Norquist, B.M.; Swisher, E.M. Clinical characteristics and outcomes of patients with BRCA1 or RAD51C methylated versus mutated ovarian carcinoma. Gynecol. Oncol. 2018, 148, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Dite, G.S.; Makalic, E.; Schmidt, D.F.; Giles, G.G.; Hopper, J.L.; Southey, M.C. Tumour morphology of early-onset breast cancers predicts breast cancer risk for first-degree relatives: The Australian Breast Cancer Family Registry. Breast Cancer Res. 2012, 14, R122. [Google Scholar] [CrossRef] [PubMed]
- Moskwa, P.; Buffa, F.M.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.J.; Beech, J.; Kulshrestha, R.; Abdelmohsen, K.; Weinstock, D.M.; et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 2011, 41, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Paculová, H.; Kohoutek, J. The emerging roles of CDK12 in tumorigenesis. Cell Div. 2017, 12, 7. [Google Scholar] [CrossRef]
- Hou, J.; Wang, Z.; Yang, L.; Guo, X.; Yang, G. The function of EMSY in cancer development. Tumor Biol. 2014, 35, 5061–5066. [Google Scholar] [CrossRef]
- Gudas, J.M.; Nguyen, H.; Li, T.; Cowan, K.H. Hormone-dependent regulation of BRCA1 in human breast cancer cells. Cancer Res. 1995, 55, 4561–4565. [Google Scholar]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef]
- Lee, N.S.; Kim, S.; Jung, Y.W.; Kim, H. Eukaryotic DNA damage responses: Homologous recombination factors and ubiquitin modification. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2018, 809, 88–98. [Google Scholar] [CrossRef]
- Vanderstichele, A.; Busschaert, P.; Olbrecht, S.; Lambrechts, D.; Vergote, I. Genomic signatures as predictive biomarkers of homologous recombination deficiency in ovarian cancer. Eur. J. Cancer 2017, 86, 5–14. [Google Scholar] [CrossRef]
- Gross, E.; van Tinteren, H.; Li, Z.; Raab, S.; Meul, C.; Avril, S.; Laddach, N.; Aubele, M.; Propping, C.; Gkazepis, A.; et al. Identification of BRCA1-like triple-negative breast cancers by quantitative multiplex-ligation-dependent probe amplification (MLPA) analysis of BRCA1-associated chromosomal regions: A validation study. BMC Cancer 2016, 16, 811. [Google Scholar] [CrossRef]
- Wang, Z.C.; Birkbak, N.J.; Culhane, A.C.; Drapkin, R.; Fatima, A.; Tian, R.; Schwede, M.; Alsop, K.; Daniels, K.E.; Piao, H.; et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin. Cancer Res. 2012, 18, 5806–5815. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Karlan, B.Y.; Taniguchi, T.; Fountzilas, E.; Francoeur, N.; Levine, D.A.; Cannistra, S.A. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J. Clin. Oncol. 2010, 28, 3555–3561. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- von Wahlde, M.-K.; Timms, K.M.; Chagpar, A.; Wali, V.B.; Jiang, T.; Bossuyt, V.; Saglam, O.; Reid, J.; Gutin, A.; Neff, C.; et al. Intratumor Heterogeneity of Homologous Recombination Deficiency in Primary Breast Cancer. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2017, 8, 319. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Nilsson, M.P.; Olsson, E.; George, A.M.; Chen, Y.; Kvist, A.; Törngren, T.; Vallon-Christersson, J.; Hegardt, C.; Häkkinen, J.; et al. Targeted sequencing of BRCA1 and BRCA2 across a large unselected breast cancer cohort suggests that one-third of mutations are somatic. Ann. Oncol. 2016, 27, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Fraga, M.F.; Guo, M.; Garcia-Foncillas, J.; Hedenfalk, I.; Godwin, A.K.; Trojan, J.; Vaurs-Barrière, C.; Bignon, Y.J.; Ramus, S.; et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum. Mol. Genet. 2001, 10, 3001–3007. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, A.M.; Spearman, A.D.; Tseng, S.Y.; Sweet, K.; Toland, A.E. Methylation not a frequent “second hit” in tumors with germline BRCA mutations. Fam. Cancer 2009, 8, 339–346. [Google Scholar] [CrossRef]
- Van Heetvelde, M.; Van Bockstal, M.; Poppe, B.; Lambein, K.; Rosseel, T.; Atanesyan, L.; Deforce, D.; Van Den Berghe, I.; De Leeneer, K.; Van Dorpe, J.; et al. Accurate detection and quantification of epigenetic and genetic second hits in BRCA1 and BRCA2-associated hereditary breast and ovarian cancer reveals multiple co-acting second hits. Cancer Lett. 2018, 425, 125–133. [Google Scholar] [CrossRef]
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D.; et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef]
- Tan, S.L.W.; Chadha, S.; Liu, Y.; Gabasova, E.; Perera, D.; Ahmed, K.; Constantinou, S.; Renaudin, X.; Lee, M.; Aebersold, R.; et al. A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 2017, 169, 1105–1118. [Google Scholar] [CrossRef]
- Colas, C.; Golmard, L.; de Pauw, A.; Caputo, S.M.; Stoppa-Lyonnet, D. “Decoding hereditary breast cancer” benefits and questions from multigene panel testing. Breast 2019, 45, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.-H.; et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Ware, M.D.; Buisson, M.; Zetoune, A.B.; Stoppa-Lyonnet, D.; Sinilnikova, O.M.; Mazoyer, S. Does the nonsense-mediated mRNA decay mechanism prevent the synthesis of truncated BRCA1, CHK2, and p53 proteins? Hum. Mutat. 2008, 29, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, E.; Jesson, B.; Briaux, A.; Nogues, C.; Chabaud, V.; Demange, L.; Sokolowska, J.; Coulet, F.; Barouk-Simonet, E.; Bignon, Y.J.; et al. Rare germline large rearrangements in the BRCA1/2 genes and eight candidate genes in 472 patients with breast cancer predisposition. Breast Cancer Res. Treat. 2012, 133, 1179–1190. [Google Scholar] [CrossRef]
- Welcsh, P.L.; King, M.C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef]
- Lesueur, F.; Mebirouk, N.; Jiao, Y.; Barjhoux, L.; Belotti, M.; Laurent, M.; Léone, M.; Houdayer, C.; Bressac-de Paillerets, B.; Vaur, D.; et al. GEMO, a National Resource to Study Genetic Modifiers of Breast and Ovarian Cancer Risk in BRCA1 and BRCA2 Pathogenic Variant Carriers. Front. Oncol. 2018, 8, 490. [Google Scholar] [CrossRef]
- Castéra, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.-R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016. [Google Scholar] [CrossRef]
- Balmaña, J.; Digiovanni, L.; Gaddam, P.; Walsh, M.F.; Joseph, V.; Stadler, Z.K.; Nathanson, K.L.; Garber, J.E.; Couch, F.J.; Offit, K.; et al. Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing. J. Clin. Oncol. 2016, 34, 4071–4078. [Google Scholar] [CrossRef]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Timms, K.M.; Liu, S.; Chen, H.; Litton, J.K.; Potter, J.; Lanchbury, J.S.; Stemke-Hale, K.; Hennessy, B.T.; Arun, B.K.; et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res. 2011, 17, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Southey, M.C.; Ramus, S.J.; Dowty, J.G.; Smith, L.D.; Tesoriero, A.A.; Wong, E.E.M.; Dite, G.S.; Jenkins, M.A.; Byrnes, G.B.; Winship, I.; et al. Morphological predictors of BRCA1 germline mutations in young women with breast cancer. Br. J. Cancer 2011, 104, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Bégin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485. [Google Scholar] [CrossRef]
- Lakhani, S.R.; Van De Vijver, M.J.; Jacquemier, J.; Anderson, T.J.; Osin, P.P.; McGuffog, L.; Easton, D.F. The pathology of familial breast cancer: Predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J. Clin. Oncol. 2002, 20, 2310–2318. [Google Scholar] [CrossRef]
- Mulligan, A.M.; Pinnaduwage, D.; Bane, A.L.; Bull, S.B.; O’Malley, F.P.; Andrulis, I.L. CK8/18 expression, the basal phenotype, and family history in identifying BRCA1-associated breast cancer in the Ontario site of the breast cancer family registry. Cancer 2011, 117, 1350–1359. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Couch, F.J.; Parsons, M.T.; McGuffog, L.; Barrowdale, D.; Bolla, M.K.; Wang, Q.; Healey, S.; Schmutzler, R.; Wappenschmidt, B.; et al. Refined histopathological predictors of BRCA1 and BRCA2 mutation status: A large-scale analysis of breast cancer characteristics from the BCAC, CIMBA, and ENIGMA consortia. Breast Cancer Res. 2014, 16, 3419. [Google Scholar] [CrossRef]
- Larsen, M.J.; Kruse, T.A.; Tan, Q.; Lænkholm, A.-V.; Bak, M.; Lykkesfeldt, A.E.; Sørensen, K.P.; Hansen, T.V.O.; Ejlertsen, B.; Gerdes, A.-M.; et al. Classifications within molecular subtypes enables identification of BRCA1/BRCA2 mutation carriers by RNA tumor profiling. PLoS ONE 2013, 8, e64268. [Google Scholar] [CrossRef]
- Bane, A.L.; Beck, J.C.; Bleiweiss, I.; Buys, S.S.; Catalano, E.; Daly, M.B.; Giles, G.; Godwin, A.K.; Hibshoosh, H.; Hopper, J.L.; et al. BRCA2 mutation-associated breast cancers exhibit a distinguishing phenotype based on morphology and molecular profiles from tissue microarrays. Am. J. Surg. Pathol. 2007, 31, 121–128. [Google Scholar] [CrossRef]
- Tung, N.; Wang, Y.; Collins, L.C.; Kaplan, J.; Li, H.; Gelman, R.; Comander, A.H.; Gallagher, B.; Fetten, K.; Krag, K.; et al. Estrogen receptor positive breast cancers in BRCA1 mutation carriers: Clinical risk factors and pathologic features. Breast Cancer Res. 2010, 12, R12. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Metcalfe, K.; Sun, P.; Hanna, W.M.; Lynch, H.T.; Ghadirian, P.; Tung, N.; Olopade, O.I.; Weber, B.L.; McLennan, J.; et al. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: The influence of age, grade, and histological type. Clin. Cancer Res. 2004, 10, 2029–2034. [Google Scholar] [CrossRef] [PubMed]
- Copson, E.R.; Maishman, T.C.; Tapper, W.J.; Cutress, R.I.; Greville-Heygate, S.; Altman, D.G.; Eccles, B.; Gerty, S.; Durcan, L.T.; Jones, L.; et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): A prospective cohort study. Lancet Oncol. 2018, 19, 169–180. [Google Scholar] [CrossRef]
- Baretta, Z.; Mocellin, S.; Goldin, E.; Olopade, O.I.; Huo, D. Effect of BRCA germline mutations on breast cancer prognosis: A systematic review and meta-analysis. Medicine (Baltimore) 2016, 95, e4975. [Google Scholar] [CrossRef]
- Lakhani, S.R.; Manek, S.; Penault-Llorca, F.; Flanagan, A.; Arnout, L.; Merrett, S.; McGuffog, L.; Steele, D.; Devilee, P.; Klijn, J.G.M.; et al. Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clin. Cancer Res. 2004, 10, 2473–2481. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Loeb, L.A. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991, 51, 3075–3079. [Google Scholar]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Santana Dos Santos, E.; Lallemand, F.; Burke, L.; Stoppa-Lyonnet, D.; Brown, M.; Caputo, S.M.; Rouleau, E. Non-Coding Variants in BRCA1 and BRCA2 Genes: Potential Impact on Breast and Ovarian Cancer Predisposition. Cancers (Basel) 2018, 10, 453. [Google Scholar] [CrossRef] [PubMed]
- Caputo, S.; Benboudjema, L.; Sinilnikova, O.; Rouleau, E.; Béroud, C.; Lidereau, R. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res. 2012, 40, D992–D1002. [Google Scholar] [CrossRef] [PubMed]
- Béroud, C.; Letovsky, S.I.; Braastad, C.D.; Caputo, S.M.; Beaudoux, O.; Bignon, Y.J.; Bressac-De Paillerets, B.; Bronner, M.; Buell, C.M.; Collod-Béroud, G.; et al. BRCA Share: A Collection of Clinical BRCA Gene Variants. Hum. Mutat. 2016. [Google Scholar] [CrossRef]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Whiley, P.J.; Guidugli, L.; Walker, L.C.; Healey, S.; Thompson, B.A.; Lakhani, S.R.; Da Silva, L.M.; kConFab Investigators; Tavtigian, S.V.; Goldgar, D.E.; et al. Splicing and multifactorial analysis of intronic BRCA1 and BRCA2 sequence variants identifies clinically significant splicing aberrations up to 12 nucleotides from the intron/exon boundary. Hum. Mutat. 2011, 32, 678–687. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Whiley, P.J.; Thompson, B.; Feng, B.; Healey, S.; Brown, M.A.; Pettigrew, C.; kConFab; Van Asperen, C.J.; Ausems, M.G.E.M.; et al. BRCA1 R1699Q variant displaying ambiguous functional abrogation confers intermediate breast and ovarian cancer risk. J. Med. Genet. 2012, 49. [Google Scholar] [CrossRef]
- de la Hoya, M.; Soukarieh, O.; López-Perolio, I.; Vega, A.; Walker, L.C.; van Ierland, Y.; Baralle, D.; Santamariña, M.; Lattimore, V.; Wijnen, J.; et al. Combined genetic and splicing analysis of BRCA1 c.[594-2A>C; 641A>G] highlights the relevance of naturally occurring in-frame transcripts for developing disease gene variant classification algorithms. Hum. Mol. Genet. 2016, 25, 2256–2268. [Google Scholar] [CrossRef]
- Dos Santos, E.S.; Caputo, S.M.; Castera, L.; Gendrot, M.; Briaux, A.; Breault, M.; Krieger, S.; Rogan, P.K.; Mucaki, E.J.; Burke, L.J.; et al. Assessment of the functional impact of germline BRCA1/2 variants located in non-coding regions in families with breast and/or ovarian cancer predisposition. Breast Cancer Res. Treat. 2017. [Google Scholar] [CrossRef]
- Burke, L.J.; Sevcik, J.; Gambino, G.; Tudini, E.; Mucaki, E.J.; Shirley, B.C.; Whiley, P.; Parsons, M.T.; De Leeneer, K.; Gutiérrez-Enríquez, S.; et al. BRCA1 and BRCA2 5’ non-coding region variants identified in breast cancer patients alter promoter activity and protein binding. Hum. Mutat. 2018. [Google Scholar] [CrossRef] [PubMed]
- Caputo, S.M.; Léone, M.; Damiola, F.; Ehlen, A.; Carreira, A.; Gaidrat, P.; Martins, A.; Brandão, R.D.; Peixoto, A.; Vega, A.; et al. Full in-frame exon 3 skipping of BRCA2 confers high risk of breast and/or ovarian cancer. Oncotarget 2018, 9, 17334–17348. [Google Scholar] [CrossRef] [PubMed]
- Moghadasi, S.; Meeks, H.D.; Vreeswijk, M.P.; Janssen, L.A.; Borg, Å.; Ehrencrona, H.; Paulsson-Karlsson, Y.; Wappenschmidt, B.; Engel, C.; Gehrig, A.; et al. The BRCA1 c. 5096G>A p.Arg1699Gln (R1699Q) intermediate risk variant: Breast and ovarian cancer risk estimation and recommendations for clinical management from the ENIGMA consortium. J. Med. Genet. 2018, 55, 15–20. [Google Scholar] [CrossRef]
- Parsons, M.T.; Tudini, E.; Li, H.; Hahnen, E.; Wappenschmidt, B.; Feliubadaló, L.; Aalfs, C.M.; Agata, S.; Aittomäki, K.; Alducci, E.; et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum. Mutat. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef]
- Ramensky, V.; Bork, P.; Sunyaev, S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002, 30, 3894–3900. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Greenblatt, M.S.; Lesueur, F.; Byrnes, G.B. IARC Unclassified Genetic Variants Working Group In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 2008, 29, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Houdayer, C.; Caux-Moncoutier, V.; Krieger, S.; Barrois, M.; Bonnet, F.; Bourdon, V.; Bronner, M.; Buisson, M.; Coulet, F.; Gaildrat, P.; et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum. Mutat. 2012, 33, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Soukarieh, O.; Gaildrat, P.; Hamieh, M.; Drouet, A.; Baert-Desurmont, S.; Frébourg, T.; Tosi, M.; Martins, A. Exonic Splicing Mutations Are More Prevalent than Currently Estimated and Can Be Predicted by Using In Silico Tools. PLoS Genet. 2016, 12, e1005756. [Google Scholar] [CrossRef]
- Vallée, M.P.; Sera, T.L.D.; Nix, D.A.; Paquette, A.M.; Parsons, M.T.; Bell, R.; Hoffman, A.; Hogervorst, F.B.L.; Goldgar, D.E.; Spurdle, A.B.; et al. Adding In Silico Assessment of Potential Splice Aberration to the Integrated Evaluation of BRCA Gene Unclassified Variants. Hum. Mutat. 2016. [Google Scholar] [CrossRef] [PubMed]
- Leman, R.; Gaildrat, P.; Gac, G.L.; Ka, C.; Fichou, Y.; Audrezet, M.-P.; Caux-Moncoutier, V.; Caputo, S.M.; Boutry-Kryza, N.; Léone, M.; et al. Novel diagnostic tool for prediction of variant spliceogenicity derived from a set of 395 combined in silico/in vitro studies: An international collaborative effort. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Leman, R.; Tubeuf, H.; Raad, S.; Tournier, I.; Derambure, C.; Lanos, R.; Gaildrat, P.; Castelain, G.; Hauchard, J.; Killian, A.; et al. Assessment of branch point prediction tools to predict physiological branch points and their alteration by variants. BMC Genom. 2020, 21, 86. [Google Scholar] [CrossRef]
- Muller, D.; Rouleau, E.; Schultz, I.; Caputo, S.; Lefol, C.; Bièche, I.; Caron, O.; Noguès, C.; Limacher, J.M.; Demange, L.; et al. An entire exon 3 germ-line rearrangement in the BRCA2 gene: Pathogenic relevance of exon 3 deletion in breast cancer predisposition. BMC Med. Genet. 2011, 12, 121. [Google Scholar] [CrossRef]
- Gaildrat, P.; Krieger, S.; Giacomo, D.D.; Abdat, J.; Révillion, F.; Caputo, S.; Vaur, D.; Jamard, E.; Bohers, E.; Ledemeney, D.; et al. Multiple sequence variants of BRCA2 exon 7 alter splicing regulation. J. Med. Genet. 2012, 49, 609–617. [Google Scholar] [CrossRef]
- Millot, G.; Carvalho, M.A.; Caputo, S.M.; Vreeswijk, M.P.G.; Brown, M.A.; Webb, M.; Rouleau, E.; Neuhausen, S.L.; Hansen, T.V.O.; Galli, A.; et al. A Guide for Functional Analysis of BRCA1 Variants of Uncertain Significance (VUS). Hum. Mutat. 2012, 33, 1526–1537. [Google Scholar] [CrossRef]
- Guidugli, L.; Carreira, A.; Caputo, S.M.; Ehlen, A.; Galli, A.; Monteiro, A.N.A.; Neuhausen, S.L.; Hansen, T.V.O.; Couch, F.J.; Vreeswijk, M.P.G. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum. Mutat. 2014, 35, 151–164. [Google Scholar] [CrossRef]
- Jhuraney, A.; Velkova, A.; Johnson, R.C.; Kessing, B.; Carvalho, R.S.; Whiley, P.; Spurdle, A.B.; Vreeswijk, M.P.G.; Caputo, S.M.; Millot, G.A.; et al. BRCA1 Circos: A visualisation resource for functional analysis of missense variants. J. Med. Genet. 2015, 52, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Petitalot, A.; Dardillac, E.; Jacquet, E.; Nhiri, N.; Guirouilh-Barbat, J.; Julien, P.; Bouazzaoui, I.; Bonte, D.; Feunteun, J.; Schnell, J.A.; et al. Combining Homologous Recombination and Phosphopeptide-binding Data to Predict the Impact of BRCA1 BRCT Variants on Cancer Risk. Mol. Cancer Res. 2019, 17, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Meulemans, L.; Mesman, R.L.S.; Caputo, S.M.; Krieger, S.; Guillaud-Bataille, M.; Caux-Moncoutier, V.; Léone, M.; Boutry-Kryza, N.; Sokolowska, J.; Révillion, F.; et al. Skipping nonsense to maintain function: The paradigm of BRCA2 exon 12. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef]
- Couch, F.J.; Nathanson, K.L.; Offit, K. Two decades after BRCA: Setting paradigms in personalized cancer care and prevention. Science 2014, 343, 1466–1470. [Google Scholar] [CrossRef]
- Domchek, S.M.; Friebel, T.M.; Singer, C.F.; Evans, D.G.; Lynch, H.T.; Isaacs, C.; Garber, J.E.; Neuhausen, S.L.; Matloff, E.; Eeles, R.; et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010, 304, 967–975. [Google Scholar] [CrossRef]
- Carbine, N.E.; Lostumbo, L.; Wallace, J.; Ko, H. Risk-reducing mastectomy for the prevention of primary breast cancer. Cochrane Database Syst. Rev. 2018, 4, CD002748. [Google Scholar] [CrossRef]
- Saadatmand, S.; Geuzinge, H.A.; Rutgers, E.J.T.; Mann, R.M.; de Roy van Zuidewijn, D.B.W.; Zonderland, H.M.; Tollenaar, R.A.E.M.; Lobbes, M.B.I.; Ausems, M.G.E.M.; van ’t Riet, M.; et al. MRI versus mammography for breast cancer screening in women with familial risk (FaMRIsc): A multicentre, randomised, controlled trial. Lancet Oncol. 2019, 20, 1136–1147. [Google Scholar] [CrossRef]
- Phi, X.-A.; Houssami, N.; Obdeijn, I.-M.; Warner, E.; Sardanelli, F.; Leach, M.O.; Riedl, C.C.; Trop, I.; Tilanus-Linthorst, M.M.A.; Mandel, R.; et al. Magnetic resonance imaging improves breast screening sensitivity in BRCA mutation carriers age ≥ 50 years: Evidence from an individual patient data meta-analysis. J. Clin. Oncol. 2015, 33, 349–356. [Google Scholar] [CrossRef]
- Hartmann, L.C.; Lindor, N.M. The Role of Risk-Reducing Surgery in Hereditary Breast and Ovarian Cancer. N. Engl. J. Med. 2016, 374, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Stirling, D.; Evans, D.G.R.; Pichert, G.; Shenton, A.; Kirk, E.N.; Rimmer, S.; Steel, C.M.; Lawson, S.; Busby-Earle, R.M.C.; Walker, J.; et al. Screening for familial ovarian cancer: Failure of current protocols to detect ovarian cancer at an early stage according to the international Federation of gynecology and obstetrics system. J. Clin. Oncol. 2005, 23, 5588–5596. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.A.; Pike, M.C.; Jotwani, A.R.; Friebel, T.M.; Soslow, R.A.; Levine, D.A.; Nathanson, K.L.; Konner, J.A.; Arnold, A.G.; Bogomolniy, F.; et al. Uterine Cancer After Risk-Reducing Salpingo-oophorectomy Without Hysterectomy in Women With BRCA Mutations. JAMA Oncol. 2016, 2, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Havrilesky, L.J.; Moss, H.A.; Chino, J.; Myers, E.R.; Kauff, N.D. Mortality reduction and cost-effectiveness of performing hysterectomy at the time of risk-reducing salpingo-oophorectomy for prophylaxis against serous/serous-like uterine cancers in BRCA1 mutation carriers. Gynecol. Oncol. 2017, 145, 549–554. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians and Gynecologists; ACOG Committee on Practice Bulletins—Gynecology; ACOG Committee on Genetics; Society of Gynecologic Oncologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet. Gynecol. 2009, 113, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pilarski, R.; Berry, M.; Buys, S.S.; Farmer, M.; Friedman, S.; Garber, J.E.; Kauff, N.D.; Khan, S.; Klein, C.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J. Natl. Compr. Canc. Netw. 2017, 15, 9–20. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Wieand, S.; Hale, K.; Lee, M.; Walsh, T.; Owens, K.; Tait, J.; Ford, L.; Dunn, B.K.; Costantino, J.; et al. Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention Trial. JAMA 2001, 286, 2251–2256. [Google Scholar] [CrossRef]
- Havrilesky, L.J.; Gierisch, J.M.; Moorman, P.G.; Coeytaux, R.R.; Urrutia, R.P.; Lowery, W.J.; Dinan, M.; McBroom, A.J.; Wing, L.; Musty, M.D.; et al. Oral contraceptive use for the primary prevention of ovarian cancer. Evid. Rep. Technol. Assess (Full Rep.) 2013, 1–514. [Google Scholar]
- Moorman, P.G.; Havrilesky, L.J.; Gierisch, J.M.; Coeytaux, R.R.; Lowery, W.J.; Peragallo Urrutia, R.; Dinan, M.; McBroom, A.J.; Hasselblad, V.; Sanders, G.D.; et al. Oral contraceptives and risk of ovarian cancer and breast cancer among high-risk women: A systematic review and meta-analysis. J. Clin. Oncol. 2013, 31, 4188–4198. [Google Scholar] [CrossRef]
- Martín, M. Platinum compounds in the treatment of advanced breast cancer. Clin. Breast Cancer 2001, 2, 190–208. [Google Scholar] [CrossRef]
- Sledge, G.W.; Loehrer, P.J.; Roth, B.J.; Einhorn, L.H. Cisplatin as first-line therapy for metastatic breast cancer. J. Clin. Oncol. 1988, 6, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.A.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2015, 33, 1902–1909. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Schneeweiss, A.; Loibl, S.; Salat, C.; Denkert, C.; Rezai, M.; Blohmer, J.U.; Jackisch, C.; Paepke, S.; Gerber, B.; et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): A randomised phase 2 trial. Lancet Oncol. 2014, 15, 747–756. [Google Scholar] [CrossRef]
- Byrski, T.; Gronwald, J.; Huzarski, T.; Grzybowska, E.; Budryk, M.; Stawicka, M.; Mierzwa, T.; Szwiec, M.; Wisniowski, R.; Siolek, M.; et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J. Clin. Oncol. 2010, 28, 375–379. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Wang, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Xie, Y. Prevalence of BRCA1 mutations and responses to neoadjuvant chemotherapy among BRCA1 carriers and non-carriers with triple-negative breast cancer. Ann. Oncol. 2015, 26, 523–528. [Google Scholar] [CrossRef]
- Dwadasi, S.; Tong, Y.; Walsh, T.; Danso, M.A.; Ma, C.X.; Silverman, P.; King, M.-C.; Perkins, S.M.; Badve, S.S.; Miller, K. Cisplatin with or without rucaparib after preoperative chemotherapy in patients with triple-negative breast cancer (TNBC): Hoosier Oncology Group BRE09-146. JCO 2014, 32, 1019. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.-H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Jensen, K.C.; Vinayak, S.; Kurian, A.W.; Lipson, J.A.; Flaherty, P.J.; Timms, K.; Abkevich, V.; Schackmann, E.A.; Wapnir, I.L.; et al. Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA1/2 Mutation-Associated Breast Cancer With Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. J. Clin. Oncol. 2015, 33, 1895–1901. [Google Scholar] [CrossRef]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Han, H.S.; Diéras, V.; Robson, M.; Palácová, M.; Marcom, P.K.; Jager, A.; Bondarenko, I.; Citrin, D.; Campone, M.; Telli, M.L.; et al. Veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in patients with BRCA1/2 locally recurrent/metastatic breast cancer: Randomized phase II study. Ann. Oncol. 2018, 29, 154–161. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Kaye, S.B.; Lubinski, J.; Matulonis, U.; Ang, J.E.; Gourley, C.; Karlan, B.Y.; Amnon, A.; Bell-McGuinn, K.M.; Chen, L.-M.; Friedlander, M.; et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin. Oncol. 2012, 30, 372–379. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana dos Santos, E.; Lallemand, F.; Petitalot, A.; Caputo, S.M.; Rouleau, E. HRness in Breast and Ovarian Cancers. Int. J. Mol. Sci. 2020, 21, 3850. https://doi.org/10.3390/ijms21113850
Santana dos Santos E, Lallemand F, Petitalot A, Caputo SM, Rouleau E. HRness in Breast and Ovarian Cancers. International Journal of Molecular Sciences. 2020; 21(11):3850. https://doi.org/10.3390/ijms21113850
Chicago/Turabian StyleSantana dos Santos, Elizabeth, François Lallemand, Ambre Petitalot, Sandrine M. Caputo, and Etienne Rouleau. 2020. "HRness in Breast and Ovarian Cancers" International Journal of Molecular Sciences 21, no. 11: 3850. https://doi.org/10.3390/ijms21113850
APA StyleSantana dos Santos, E., Lallemand, F., Petitalot, A., Caputo, S. M., & Rouleau, E. (2020). HRness in Breast and Ovarian Cancers. International Journal of Molecular Sciences, 21(11), 3850. https://doi.org/10.3390/ijms21113850