Association of A Novel Splice Site Mutation in P/Q-Type Calcium Channels with Childhood Epilepsy and Late-Onset Slowly Progressive Non-Episodic Cerebellar Ataxia


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
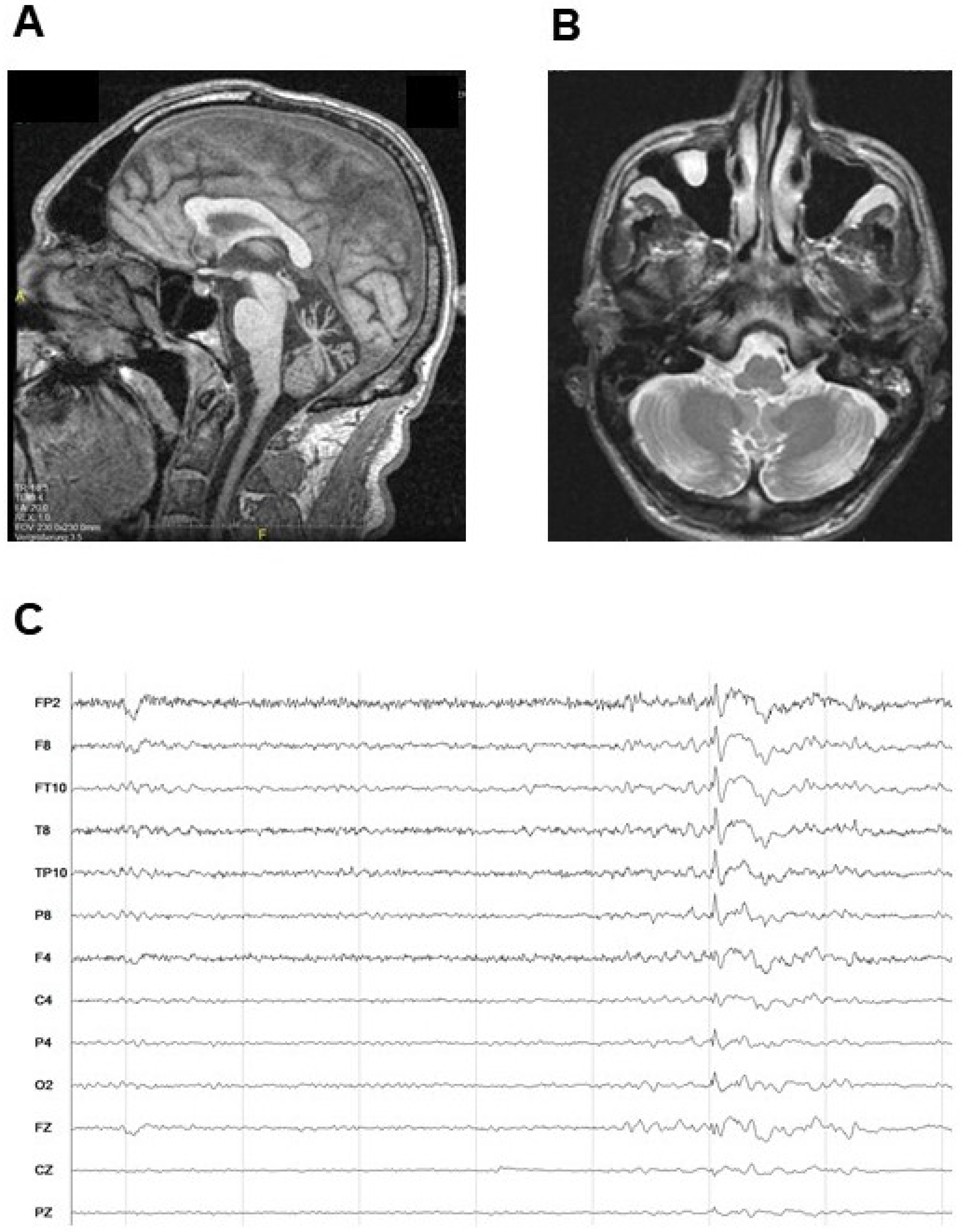
2.1. Case Report
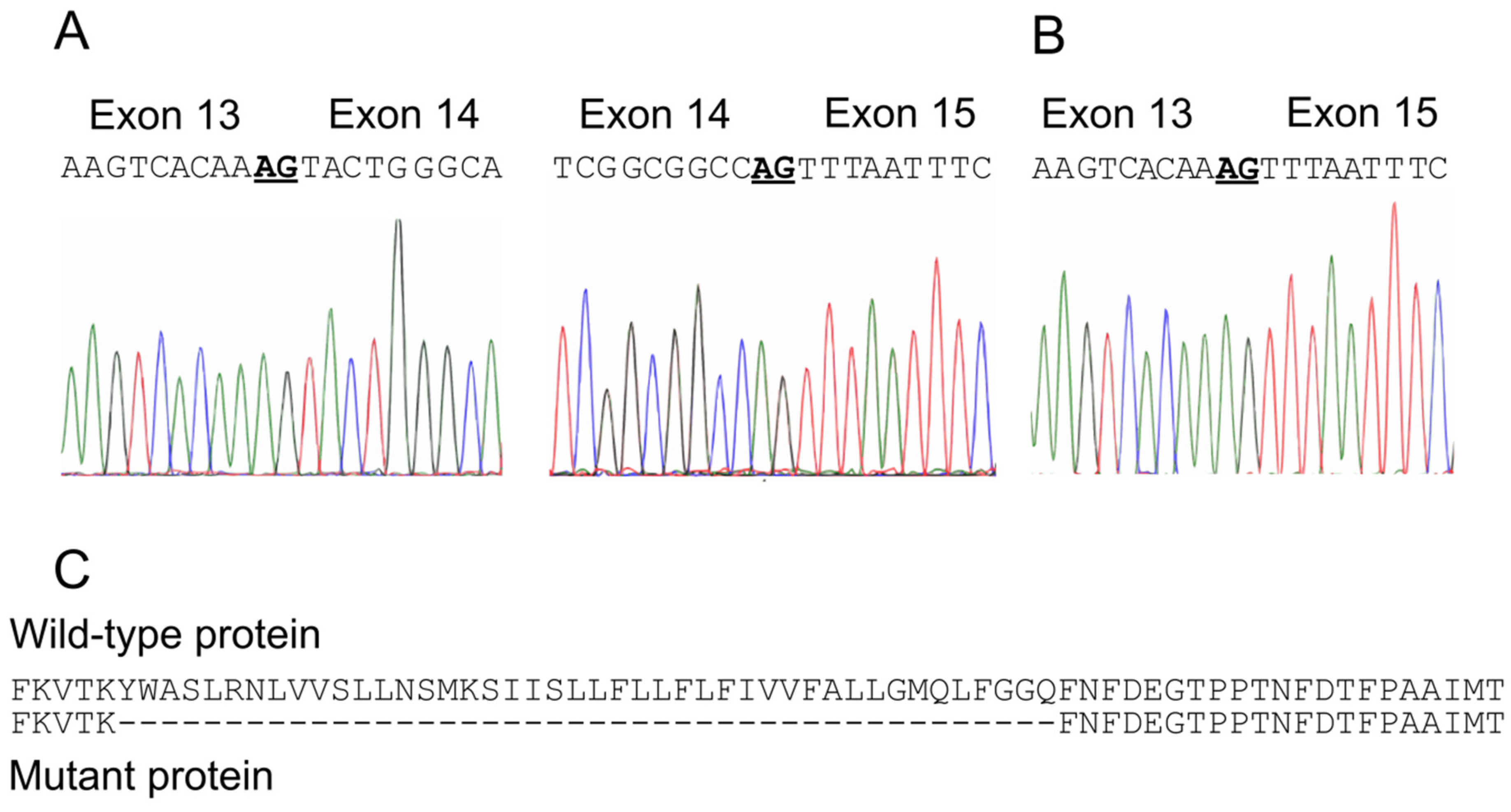
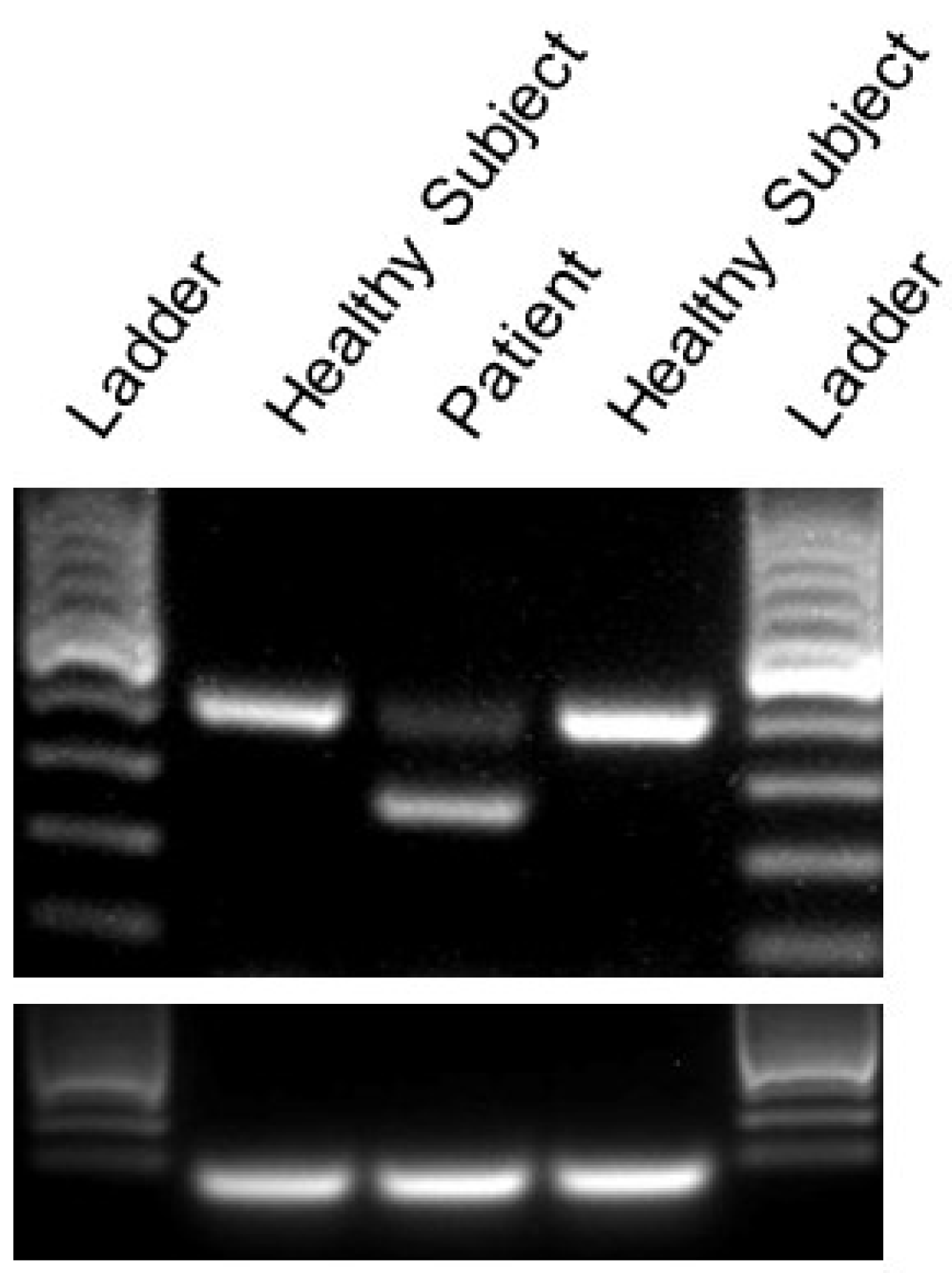
2.2. Identification of A Novel CACNA1A Splice Variant Resulting in Exon 14 Skipping
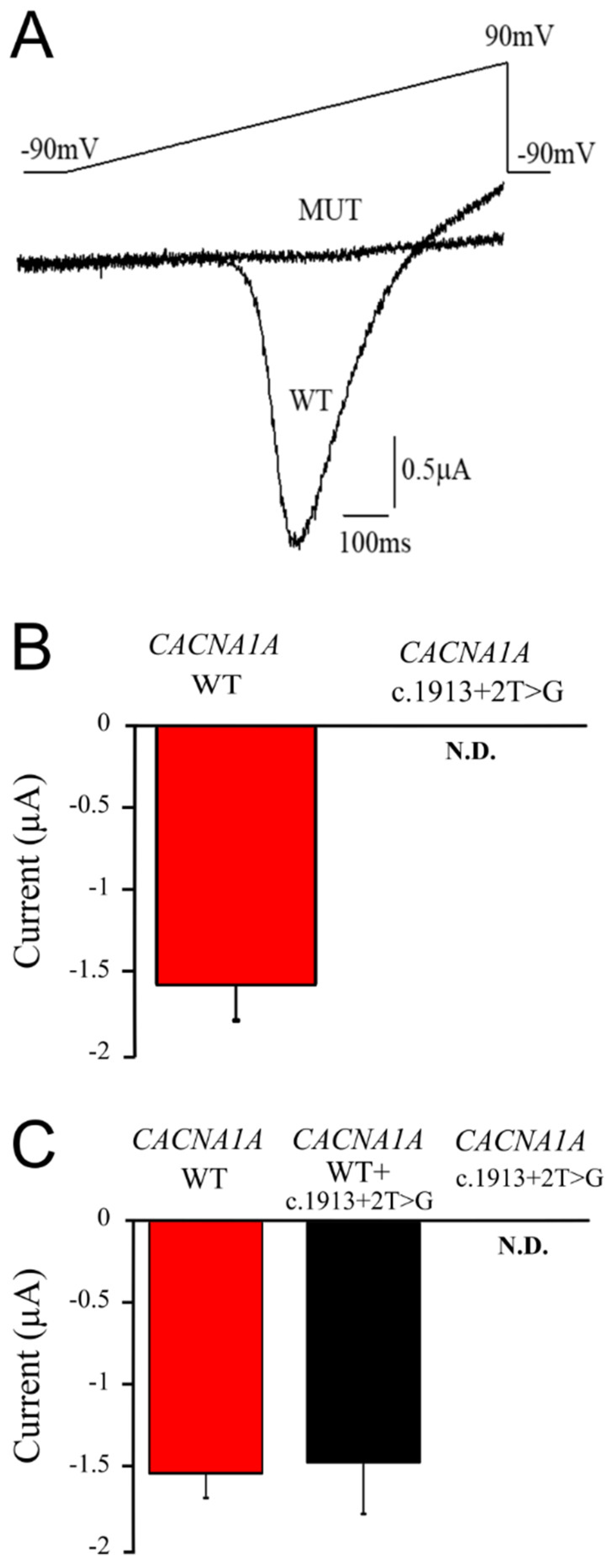
2.3. The CACNA1A Mutation Causes Total Loss of Cav2.1 Channel Function
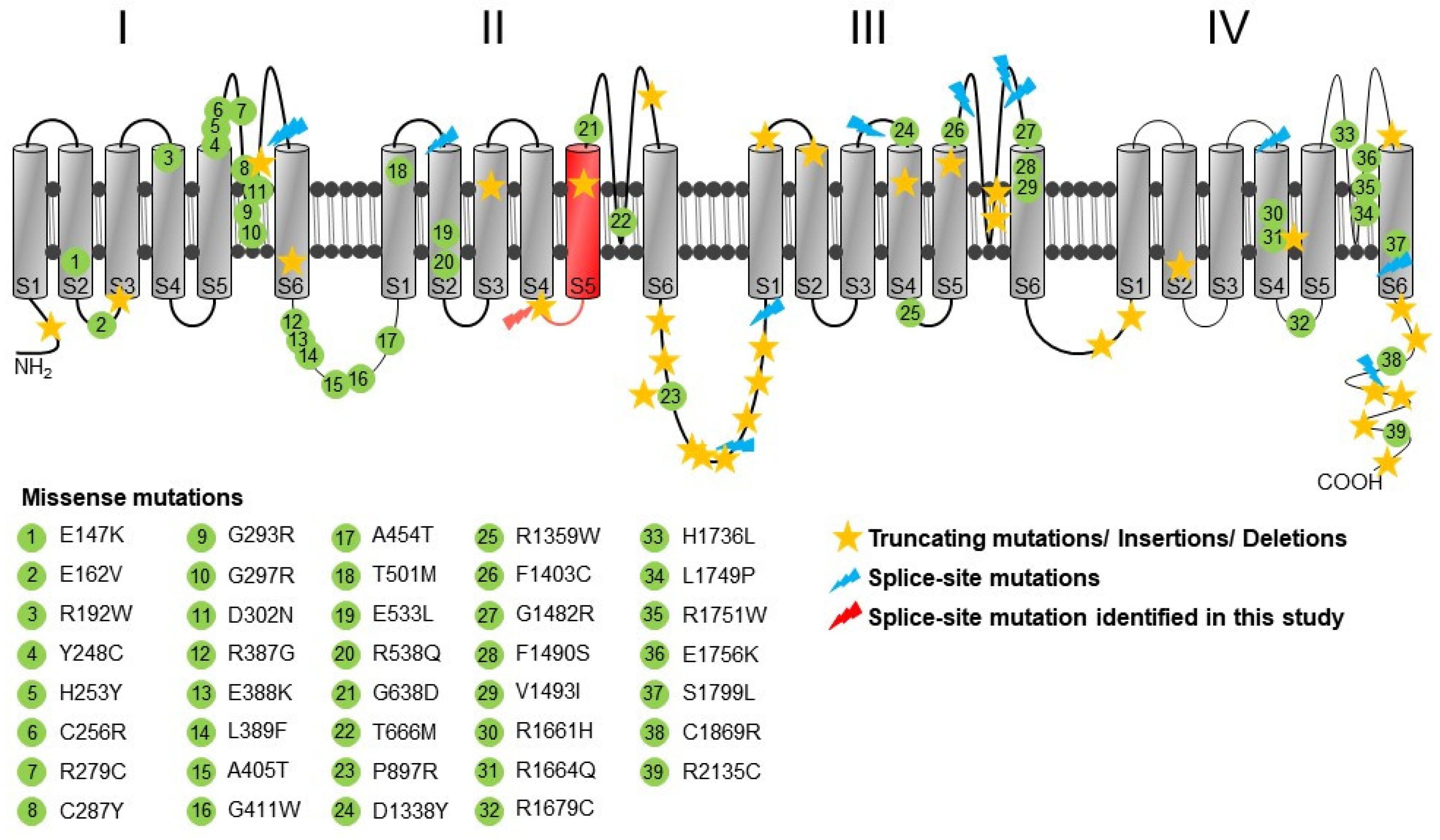
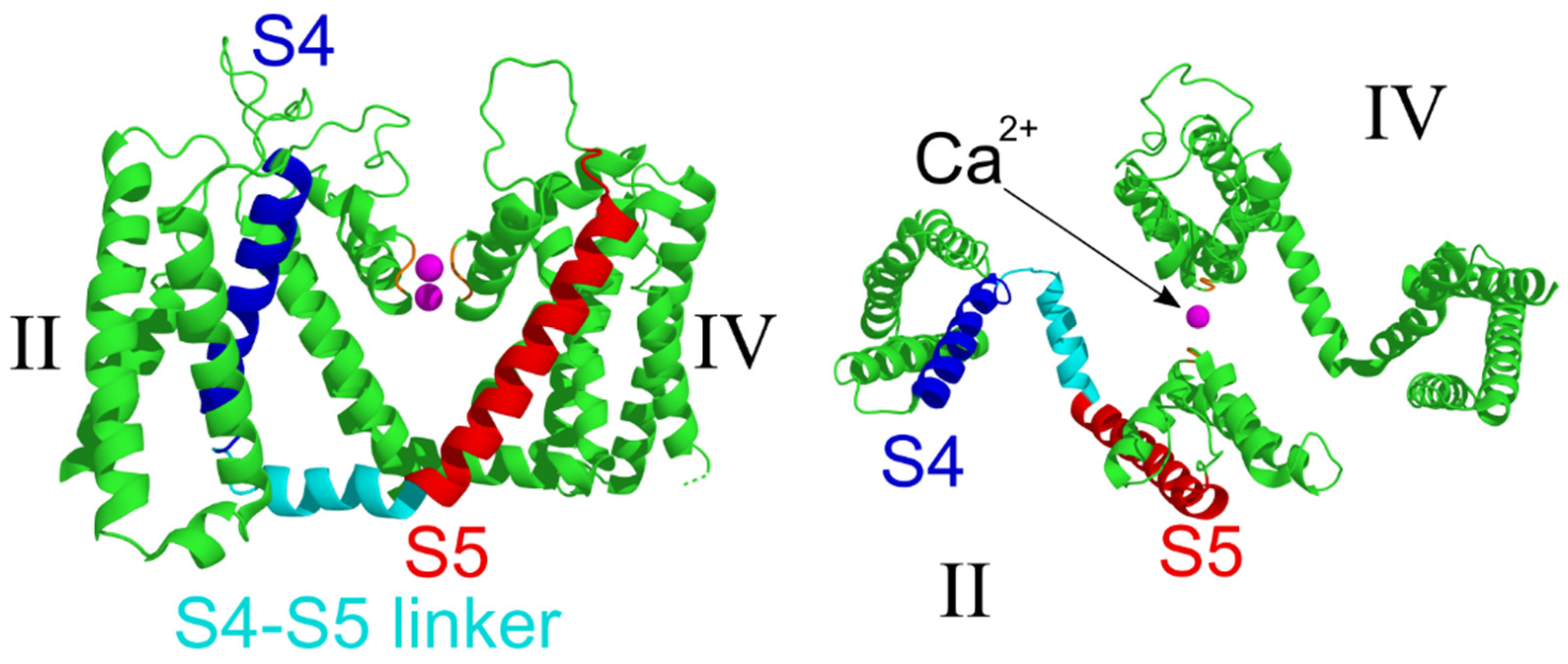
2.4. Structural Analysis
3. Discussion
4. Materials and Methods
4.1. Genetic Analysis
4.2. RT-PCR Analysis
4.3. Cloning of Expression Constructs
4.4. Electrophysiology
4.5. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Haan, J.; Terwindt, G.M.; van den Maagdenberg, A.M.; Stam, A.H.; Ferrari, M.D. A review of the genetic relation between migraine and epilepsy. Cephalalgia 2008, 28, 105–113. [Google Scholar]
- Holtmann, M.; Opp, J.; Tokarzewski, M.; Korn-Merker, E. Human epilepsy, episodic ataxia type 2, and migraine. Lancet 2002, 359, 170–171. [Google Scholar] [CrossRef]
- Damaj, L.; Lupien-Meilleur, A.; Lortie, A.; Riou, E.; Ospina, L.H.; Gagnon, L.; Vanasse, C.; Rossignol, E. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet 2015, 23, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, M.C.; Catacuzzeno, L.; Di Giovanni, G.; Franciolini, F.; Pessia, M. K(+) channelepsy: Progress in the neurobiology of potassium channels and epilepsy. Front Cell Neurosci. 2013, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, M.C.; Hasan, S.; Guglielmi, L.; Servettini, I.; Cenciarini, M.; Catacuzzeno, L.; Franciolini, F. New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front Cell Neurosci. 2015, 9, 317. [Google Scholar] [CrossRef]
- Bris, C.; Goudenege, D.; Desquiret-Dumas, V.; Charif, M.; Colin, E.; Bonneau, D.; Amati-Bonneau, P.; Lenaers, G.; Reynier, P.; Procaccio, V. Bioinformatics Tools and Databases to Assess the Pathogenicity of Mitochondrial DNA Variants in the Field of Next Generation Sequencing. Front Genet 2018, 9, 632. [Google Scholar] [CrossRef]
- Spacey, S. Episodic Ataxia Type 2. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1116/ (accessed on 22 May 2020).
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet 1997, 15, 62–69. [Google Scholar] [CrossRef]
- Casey, H.L.; Gomez, C.M. Spinocerebellar Ataxia Type 6. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1116/ (accessed on 22 May 2020).
- Schols, L.; Kruger, R.; Amoiridis, G.; Przuntek, H.; Epplen, J.T.; Riess, O. Spinocerebellar ataxia type 6: Genotype and phenotype in German kindreds. J. Neurol. Neurosurg. Psychiatry 1998, 64, 67–73. [Google Scholar] [CrossRef]
- Jodice, C.; Mantuano, E.; Veneziano, L.; Trettel, F.; Sabbadini, G.; Calandriello, L.; Francia, A.; Spadaro, M.; Pierelli, F.; Salvi, F.; et al. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum. Mol Genet 1997, 6, 1973–1978. [Google Scholar] [CrossRef]
- Romaniello, R.; Zucca, C.; Tonelli, A.; Bonato, S.; Baschirotto, C.; Zanotta, N.; Epifanio, R.; Righini, A.; Bresolin, N.; Bassi, M.T.; et al. A wide spectrum of clinical, neurophysiological and neuroradiological abnormalities in a family with a novel CACNA1A mutation. J. Neurol. Neurosurg. Psychiatry 2010, 81, 840–843. [Google Scholar] [CrossRef]
- Tonelli, A.; D’Angelo, M.G.; Salati, R.; Villa, L.; Germinasi, C.; Frattini, T.; Meola, G.; Turconi, A.C.; Bresolin, N.; Bassi, M.T. Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J. Neurol. Sci. 2006, 241, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Yabe, I.; Kitagawa, M.; Suzuki, Y.; Fujiwara, K.; Wada, T.; Tsubuku, T.; Takeichi, N.; Sakushima, K.; Soma, H.; Tsuji, S.; et al. Downbeat positioning nystagmus is a common clinical feature despite variable phenotypes in an FHM1 family. J. Neurol. 2008, 255, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Sintas, C.; Carreno, O.; Fernandez-Castillo, N.; Corominas, R.; Vila-Pueyo, M.; Toma, C.; Cuenca-Leon, E.; Barroeta, I.; Roig, C.; Volpini, V.; et al. Mutation Spectrum in the CACNA1A Gene in 49 Patients with Episodic Ataxia. Sci. Rep. 2017, 7, 2514. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, D. Calcium channels and migraine. Biochim. Biophys Acta 2013, 1828, 1655–1665. [Google Scholar] [CrossRef]
- Shapiro, M.S.; Gomeza, J.; Hamilton, S.E.; Hille, B.; Loose, M.D.; Nathanson, N.M.; Roche, J.P.; Wess, J. Identification of subtypes of muscarinic receptors that regulate Ca2+ and K+ channel activity in sympathetic neurons. Life Sci. 2001, 68, 2481–2487. [Google Scholar] [CrossRef]
- Hans, M.; Luvisetto, S.; Williams, M.E.; Spagnolo, M.; Urrutia, A.; Tottene, A.; Brust, P.F.; Johnson, E.C.; Harpold, M.M.; Stauderman, K.A.; et al. Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine. J. Neurosci. 1999, 19, 1610–1619. [Google Scholar] [CrossRef]
- Guida, S.; Trettel, F.; Pagnutti, S.; Mantuano, E.; Tottene, A.; Veneziano, L.; Fellin, T.; Spadaro, M.; Stauderman, K.; Williams, M.; et al. Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am J. Hum. Genet 2001, 68, 759–764. [Google Scholar] [CrossRef]
- Sinke, R.J.; Ippel, E.F.; Diepstraten, C.M.; Beemer, F.A.; Wokke, J.H.; van Hilten, B.J.; Knoers, N.V.; van Amstel, H.K.; Kremer, H.P. Clinical and molecular correlations in spinocerebellar ataxia type 6: A study of 24 Dutch families. Arch Neurol. 2001, 58, 1839–1844. [Google Scholar] [CrossRef][Green Version]
- Wu, J.; Yan, Z.; Li, Z.; Qian, X.; Lu, S.; Dong, M.; Zhou, Q.; Yan, N. Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 A resolution. Nature 2016, 537, 191–196. [Google Scholar] [CrossRef]
- Garcia-Baro-Huarte, M.; Iglesias-Mohedano, A.M.; Slocker-Barrio, M.; Vazquez-Lopez, M.; Garcia-Morin, M.; Miranda-Herrero, M.C.; Castro-Castro, P. Phenotypic variability in a four generation family with a p.Thr666Met CACNA1A gene mutation. Pediatr. Neurol. 2014, 51, 557–559. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, M.C.; Liantonio, A.; Conte, E.; Pessia, M.; Imbrici, P. Ion channels involvement in neurodevelopmental disorders. Neuroscience 2020, in press. [Google Scholar]
- Graves, T.D.; Imbrici, P.; Kors, E.E.; Terwindt, G.M.; Eunson, L.H.; Frants, R.R.; Haan, J.; Ferrari, M.D.; Goadsby, P.J.; Hanna, M.G.; et al. Premature stop codons in a facilitating EF-hand splice variant of CaV2.1 cause episodic ataxia type 2. Neurobiol. Dis. 2008, 32, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Salvi, J.; Bertaso, F.; Mausset-Bonnefont, A.L.; Metz, A.; Lemmers, C.; Ango, F.; Fagni, L.; Lory, P.; Mezghrani, A. RNAi silencing of P/Q-type calcium channels in Purkinje neurons of adult mouse leads to episodic ataxia type 2. Neurobiol. Dis. 2014, 68, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Eunson, L.H.; Graves, T.D.; Hanna, M.G. New calcium channel mutations predict aberrant RNA splicing in episodic ataxia. Neurology 2005, 65, 308–310. [Google Scholar] [CrossRef]
- Wan, J.; Khanna, R.; Sandusky, M.; Papazian, D.M.; Jen, J.C.; Baloh, R.W. CACNA1A mutations causing episodic and progressive ataxia alter channel trafficking and kinetics. Neurology 2005, 64, 2090–2097. [Google Scholar] [CrossRef] [PubMed]
- Nachbauer, W.; Nocker, M.; Karner, E.; Stankovic, I.; Unterberger, I.; Eigentler, A.; Schneider, R.; Poewe, W.; Delazer, M.; Boesch, S. Episodic ataxia type 2: Phenotype characteristics of a novel CACNA1A mutation and review of the literature. J. Neurol. 2014, 261, 983–991. [Google Scholar] [CrossRef]
- Imbrici, P.; Jaffe, S.L.; Eunson, L.H.; Davies, N.P.; Herd, C.; Robertson, R.; Kullmann, D.M.; Hanna, M.G. Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain 2004, 127, 2682–2692. [Google Scholar] [CrossRef]
- Imbrici, P.; D’Adamo, M.C.; Cusimano, A.; Pessia, M. Episodic Ataxia Type 1 Mutation F184C Alters Zn2+-Induced Modulation of the Human Potassium Channel Kv1.4-Kv1.1/Kvβ1.1. Am. J. Physiol.-Cell Physiol. 2007, 292, C778–C787. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stendel, C.; D’Adamo, M.C.; Wiessner, M.; Dusl, M.; Cenciarini, M.; Belia, S.; Nematian-Ardestani, E.; Bauer, P.; Senderek, J.; Klopstock, T.; et al. Association of A Novel Splice Site Mutation in P/Q-Type Calcium Channels with Childhood Epilepsy and Late-Onset Slowly Progressive Non-Episodic Cerebellar Ataxia. Int. J. Mol. Sci. 2020, 21, 3810. https://doi.org/10.3390/ijms21113810
Stendel C, D’Adamo MC, Wiessner M, Dusl M, Cenciarini M, Belia S, Nematian-Ardestani E, Bauer P, Senderek J, Klopstock T, et al. Association of A Novel Splice Site Mutation in P/Q-Type Calcium Channels with Childhood Epilepsy and Late-Onset Slowly Progressive Non-Episodic Cerebellar Ataxia. International Journal of Molecular Sciences. 2020; 21(11):3810. https://doi.org/10.3390/ijms21113810
Chicago/Turabian StyleStendel, Claudia, Maria Cristina D’Adamo, Manuela Wiessner, Marina Dusl, Marta Cenciarini, Silvia Belia, Ehsan Nematian-Ardestani, Peter Bauer, Jan Senderek, Thomas Klopstock, and et al. 2020. "Association of A Novel Splice Site Mutation in P/Q-Type Calcium Channels with Childhood Epilepsy and Late-Onset Slowly Progressive Non-Episodic Cerebellar Ataxia" International Journal of Molecular Sciences 21, no. 11: 3810. https://doi.org/10.3390/ijms21113810
APA StyleStendel, C., D’Adamo, M. C., Wiessner, M., Dusl, M., Cenciarini, M., Belia, S., Nematian-Ardestani, E., Bauer, P., Senderek, J., Klopstock, T., & Pessia, M. (2020). Association of A Novel Splice Site Mutation in P/Q-Type Calcium Channels with Childhood Epilepsy and Late-Onset Slowly Progressive Non-Episodic Cerebellar Ataxia. International Journal of Molecular Sciences, 21(11), 3810. https://doi.org/10.3390/ijms21113810