Overview of Mitochondrial E3 Ubiquitin Ligase MITOL/MARCH5 from Molecular Mechanisms to Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mitochondria and Intracellular Quality Control by MITOL
2.1. Neurodegenerative Diseases
2.2. Innate Immune Response
2.3. Cell Death in Cancer Cells
2.4. Mitophagy
2.5. Aging
3. Regulation of Mitochondrial Dynamics by MITOL
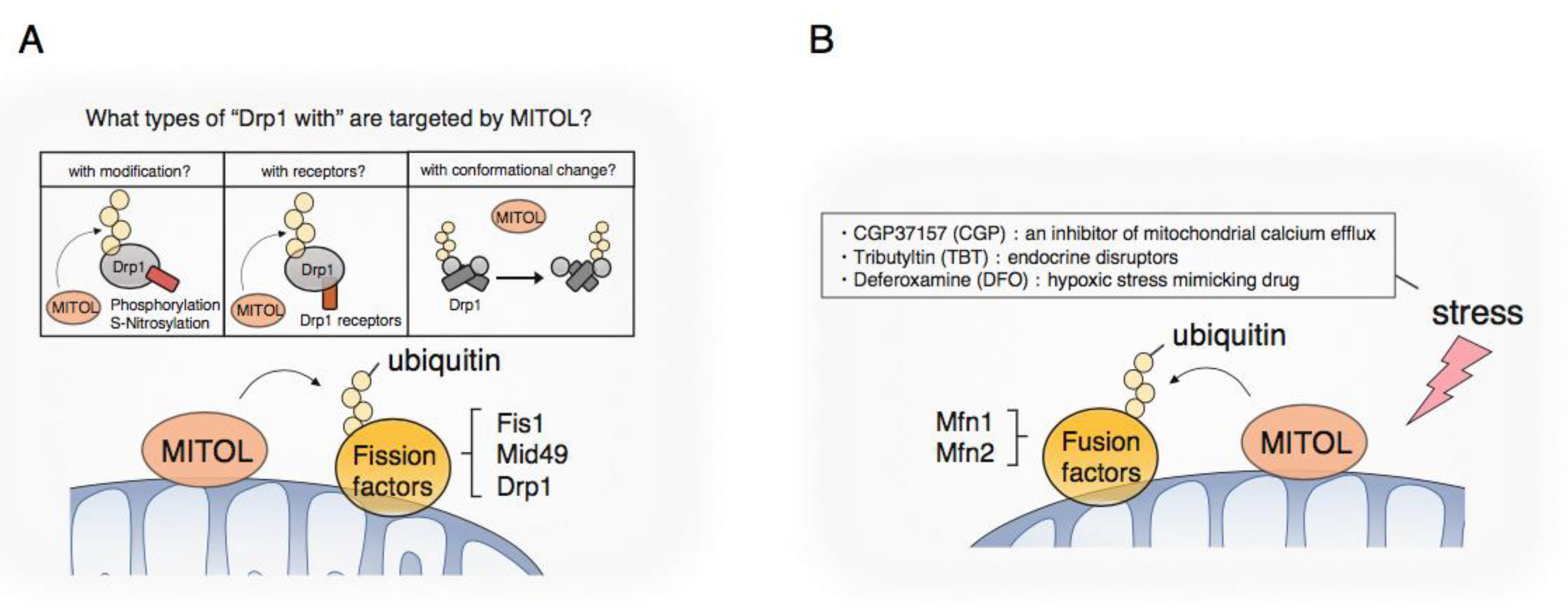
3.1. Mitochondrial Fission
3.2. Mitochondrial Fusion
4. Relation between MITOL and Membrane Bontact Site with the Endoplasmic Reticulum
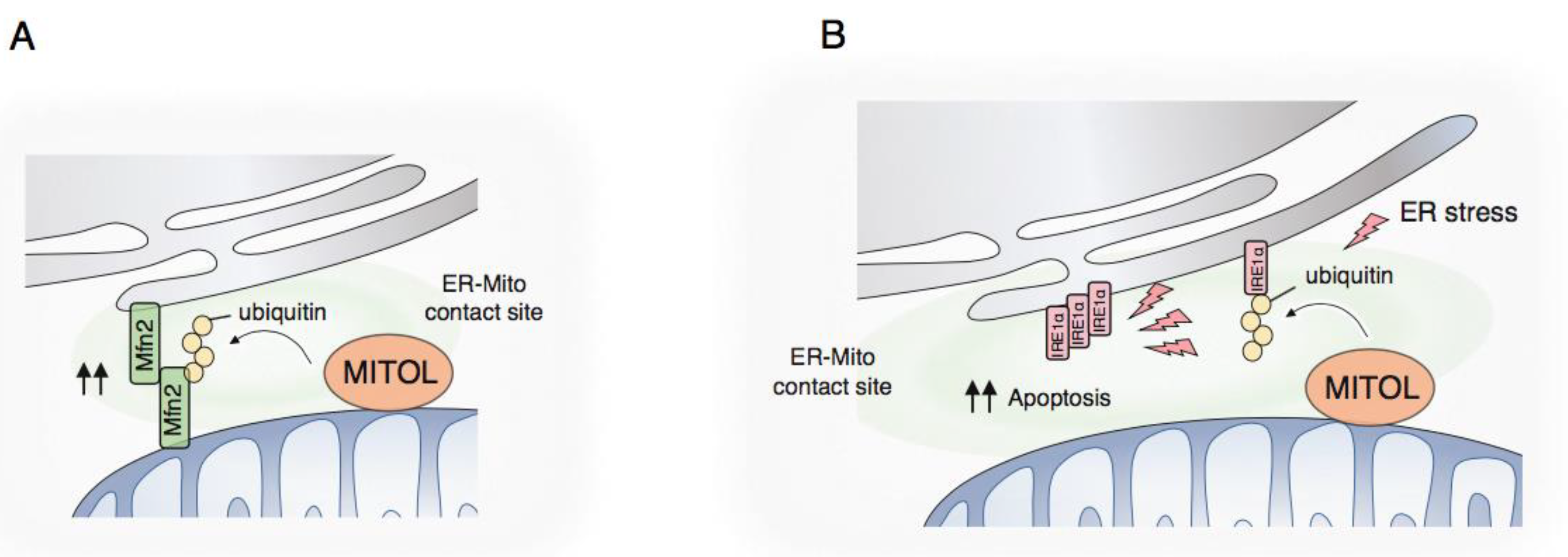
4.1. Membrane Contact Site with the Endoplasmic Reticulum
4.2. MCS Formation by MITOL
4.3. UPR Regulation by MITOL
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Saeki, Y. Ubiquitin recognition by the proteasome. J. Biochem. 2017, 161, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Martensson, C.U.; Priesnitz, C.; Song, J.; Ellenrieder, L.; Doan, K.N.; Boos, F.; Floerchinger, A.; Zufall, N.; Oeljeklaus, S.; Warscheid, B.; et al. Mitochondrial protein translocation-associated degradation. Nature 2019, 569, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Shi, Y.; Furukawa, Y.; Zhai, H.; Fu, R.; Liu, E.; Gorrie, G.H.; Khan, M.S.; Hung, W.Y.; Bigio, E.H.; et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of sod1 in mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7142–7147. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; Yonashiro, R.; Fukuda, T.; Matsushita, N.; Nagashima, S.; Inatome, R.; Yanagi, S. A mitochondrial ubiquitin ligase mitol controls cell toxicity of polyglutamine-expanded protein. Mitochondrion 2011, 11, 139–146. [Google Scholar] [CrossRef]
- Yonashiro, R.; Sugiura, A.; Miyachi, M.; Fukuda, T.; Matsushita, N.; Inatome, R.; Ogata, Y.; Suzuki, T.; Dohmae, N.; Yanagi, S. Mitochondrial ubiquitin ligase mitol ubiquitinates mutant sod1 and attenuates mutant sod1-induced ros generation. Mol. Biol. Cell 2009, 20, 4254–4530. [Google Scholar] [CrossRef]
- Lin, H.; Li, S.; Shu, H.B. The membrane-associated march e3 ligase family: Emerging roles in immune regulation. Front. Immunol. 2019, 10, 1751. [Google Scholar] [CrossRef]
- Park, Y.J.; Oanh, N.T.K.; Heo, J.; Kim, S.G.; Lee, H.S.; Lee, H.; Lee, J.H.; Kang, H.C.; Lim, W.; Yoo, Y.S.; et al. Dual targeting of rig-i and mavs by march5 mitochondria ubiquitin ligase in innate immunity. Cell. Signal. 2020, 67, 109520. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.R.; Zhou, L.; Hu, M.M.; Li, M.; Lin, H.; Yang, Y.; Wang, Y.Y.; Shu, H.B. Pkacs attenuate innate antiviral response by phosphorylating visa and priming it for march5-mediated degradation. PLoS Pathog. 2017, 13, e1006648. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.S.; Park, Y.Y.; Kim, J.H.; Cho, H.; Kim, S.H.; Lee, H.S.; Kim, T.H.; Sun Kim, Y.; Lee, Y.; Kim, C.J.; et al. The mitochondrial ubiquitin ligase march5 resolves mavs aggregates during antiviral signalling. Nat. Commun. 2015, 6, 7910. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.X.; Liu, X.; Wang, Q.; Tang, P.P.; Liu, X.Y.; Shan, Y.F.; Wang, C. Mitochondrial ubiquitin ligase march5 promotes tlr7 signaling by attenuating tank action. PLoS Pathog. 2011, 7, e1002057. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.S.; Park, Y.J.; Lee, H.S.; Oanh, N.T.K.; Cho, M.Y.; Heo, J.; Lee, E.S.; Cho, H.; Park, Y.Y.; Cho, H. Mitochondria ubiquitin ligase, march5 resolves hepatitis b virus x protein aggregates in the liver pathogenesis. Cell Death Dis. 2019, 10, 938. [Google Scholar] [CrossRef] [PubMed]
- Djajawi, T.M.; Liu, L.; Gong, J.N.; Huang, A.S.; Luo, M.J.; Xu, Z.; Okamoto, T.; Call, M.J.; Huang, D.C.S.; van Delft, M.F. March5 requires mtch2 to coordinate proteasomal turnover of the mcl1:Noxa complex. Cell Death Differ. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Timucin, A.C.; Basaga, H.; Kutuk, O. Selective targeting of antiapoptotic bcl-2 proteins in cancer. Med. Res. Rev. 2019, 39, 146–175. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Andronache, A.; Li, Y.C.; Wade, M. Inhibition of march5 ubiquitin ligase abrogates mcl1-dependent resistance to bh3 mimetics via noxa. Oncotarget 2016, 7, 15986–16002. [Google Scholar] [CrossRef]
- Haschka, M.D.; Karbon, G.; Soratroi, C.; O’Neill, K.L.; Luo, X.; Villunger, A. March5-dependent degradation of mcl1/noxa complexes defines susceptibility to antimitotic drug treatment. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Tang, H.; Peng, S.; Dong, Y.; Yang, X.; Yang, P.; Yang, L.; Yang, B.; Bao, G. March5 overexpression contributes to tumor growth and metastasis and associates with poor survival in breast cancer. Cancer Manag. Res. 2019, 11, 201–215. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Sekine, S.; Youle, R.J. Pink1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-content genome-wide rnai screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Yamano, K.; Kosako, H.; Tanaka, K.; Matsuda, N. Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by mitol. J. Biol. Chem. 2019, 294, 10300–10314. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Yamano, K.; Kosako, H.; Kimura, Y.; Kimura, M.; Fujiki, Y.; Tanaka, K.; Matsuda, N. Parkin-mediated ubiquitylation redistributes mitol/march5 from mitochondria to peroxisomes. EMBO Rep. 2019, 20, e47728. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2784–2790. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, L.; Cheng, Q.; Li, Y.; Wu, H.; Zhang, W.; Wang, Y.; Sehgal, S.A.; Siraj, S.; Wang, X.; et al. Mitochondrial e3 ligase march5 regulates fundc1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017, 18, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.Y.; Lee, S.; Karbowski, M.; Neutzner, A.; Youle, R.J.; Cho, H. Loss of march5 mitochondrial e3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J. Cell Sci. 2010, 123, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.; Ryan, M.T. The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 2017, 130, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [PubMed]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational changes in dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 2011, 18, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Koirala, S.; Guo, Q.; Kalia, R.; Bui, H.T.; Eckert, D.M.; Frost, A.; Shaw, J.M. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc. Natl. Acad. Sci. USA 2013, 110, E1342–E1351. [Google Scholar] [CrossRef] [PubMed]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Prudent, J.; McBride, H.M. The mitochondria-endoplasmic reticulum contact sites: A signalling platform for cell death. Curr. Opin. Cell Biol. 2017, 47, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Braschi, E.; Zunino, R.; McBride, H.M. Mapl is a new mitochondrial sumo e3 ligase that regulates mitochondrial fission. EMBO Rep. 2009, 10, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.R.; Thomenius, M.J.; Johnson, E.S.; Freel, C.D.; Wu, J.Q.; Coloff, J.L.; Yang, C.S.; Tang, W.; An, J.; Ilkayeva, O.R.; et al. Regulation of mitochondrial morphology by apc/ccdh1-mediated control of drp1 stability. Mol. Biol. Cell 2011, 22, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Tang, X.; Christian, W.V.; Yoon, Y.; Tieu, K. Perturbations in mitochondrial dynamics induced by human mutant pink1 can be rescued by the mitochondrial division inhibitor mdivi-1. J. Biol. Chem. 2010, 285, 11740–11752. [Google Scholar] [CrossRef]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin ubiquitinates drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in parkinson disease. J. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef]
- Yonashiro, R.; Ishido, S.; Kyo, S.; Fukuda, T.; Goto, E.; Matsuki, Y.; Ohmura-Hoshino, M.; Sada, K.; Hotta, H.; Yamamura, H.; et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006, 25, 3618–3626. [Google Scholar] [CrossRef]
- Nakamura, N.; Kimura, Y.; Tokuda, M.; Honda, S.; Hirose, S. March-v is a novel mitofusin 2- and drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006, 7, 1019–1022. [Google Scholar] [CrossRef]
- Karbowski, M.; Neutzner, A.; Youle, R.J. The mitochondrial e3 ubiquitin ligase march5 is required for drp1 dependent mitochondrial division. J. Cell Biol. 2007, 178, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Cherok, E.; Das, S.; Li, S.; Roelofs, B.A.; Ge, S.X.; Polster, B.M.; Boyman, L.; Lederer, W.J.; Wang, C.; et al. Mitochondrial e3 ubiquitin ligase march5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of mid49 protein. Mol. Biol. Cell 2016, 27, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Rizza, S.; Cardaci, S.; Montagna, C.; Di Giacomo, G.; De Zio, D.; Bordi, M.; Maiani, E.; Campello, S.; Borreca, A.; Puca, A.A.; et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E3388–E3397. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Stroissnigg, H.; Trancikova, A.; Descovich, L.; Fuhrmann, J.; Kutschera, W.; Kostan, J.; Meixner, A.; Nothias, F.; Propst, F. S-nitrosylation of microtubule-associated protein 1b mediates nitric-oxide-induced axon retraction. Nat. Cell Biol. 2007, 9, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Vo, A.; Liu, G.; McKeehan, W.L. Distinct structural domains within c19orf5 support association with stabilized microtubules and mitochondrial aggregation and genome destruction. Cancer Res. 2005, 65, 4191–4201. [Google Scholar] [CrossRef][Green Version]
- Yonashiro, R.; Kimijima, Y.; Shimura, T.; Kawaguchi, K.; Fukuda, T.; Inatome, R.; Yanagi, S. Mitochondrial ubiquitin ligase mitol blocks s-nitrosylated map1b-light chain 1-mediated mitochondrial dysfunction and neuronal cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 2382–2387. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. Er tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Murley, A.; Nunnari, J. The emerging network of mitochondria-organelle contacts. Mol. Cell 2016, 61, 648–653. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via rab7 gtp hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Nagashima, S.; Tabara, L.C.; Tilokani, L.; Paupe, V.; Anand, H.; Pogson, J.H.; Zunino, R.; McBride, H.M.; Prudent, J. Golgi-derived pi(4)p-containing vesicles drive late steps of mitochondrial division. Science 2020, 367, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Onoue, K.; Jofuku, A.; Ban-Ishihara, R.; Ishihara, T.; Maeda, M.; Koshiba, T.; Itoh, T.; Fukuda, M.; Otera, H.; Oka, T.; et al. Fis1 acts as a mitochondrial recruitment factor for tbc1d15 that is involved in regulation of mitochondrial morphology. J. Cell Sci. 2013, 126, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial rab gaps govern autophagosome biogenesis during mitophagy. eLife 2014, 3, e01612. [Google Scholar] [CrossRef] [PubMed]
- Legros, F.; Lombes, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 2002, 13, 4343–4354. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins mfn1 and mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via gtpase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; St-Pierre, P.; Shankar, J.; Wang, P.T.; Joshi, B.; Nabi, I.R. Regulation of mitophagy by the gp78 e3 ubiquitin ligase. Mol. Biol. Cell 2013, 24, 1153–1162. [Google Scholar] [CrossRef]
- Shankar, J.; Kojic, L.D.; St-Pierre, P.; Wang, P.T.; Fu, M.; Joshi, B.; Nabi, I.R. Raft endocytosis of amf regulates mitochondrial dynamics through rac1 signaling and the gp78 ubiquitin ligase. J. Cell Sci. 2013, 126, 3295–3304. [Google Scholar] [CrossRef]
- Bagher, P.; Jiao, J.; Owen Smith, C.; Cota, C.D.; Gunn, T.M. Characterization of mahogunin ring finger-1 expression in mice. Pigment Cell Res. 2006, 19, 635–643. [Google Scholar] [CrossRef]
- Yue, W.; Chen, Z.; Liu, H.; Yan, C.; Chen, M.; Feng, D.; Yan, C.; Wu, H.; Du, L.; Wang, Y.; et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase usp30. Cell Res. 2014, 24, 482–496. [Google Scholar] [CrossRef]
- Tang, F.L.; Liu, W.; Hu, J.X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.C. Vps35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, V.; Kaddour-Djebbar, I.; Alaisami, R.; Kumar, M.V.; Bollag, W.B. Mitofusin 1 degradation is induced by a disruptor of mitochondrial calcium homeostasis, cgp37157: A role in apoptosis in prostate cancer cells. Int. J. Oncol. 2014, 44, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.Y.; Cho, H. Mitofusin 1 is degraded at g2/m phase through ubiquitylation by march5. Cell Div. 2012, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Nagano, Y.; Choi, S.J.; Park, S.Y.; Kim, H.; Yao, T.P.; Lee, J.Y. Hdac6 maintains mitochondrial connectivity under hypoxic stress by suppressing march5/mitol dependent mfn2 degradation. Biochem. Biophys. Res. Commun. 2015, 464, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Weaver, D.; Hajnoczky, G. Endoplasmic reticulum-mitochondrial contactology: Structure and signaling functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Kwon, S.K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. Er-mitochondria tethering by pdzd8 regulates ca(2+) dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef]
- Tamura, Y.; Kawano, S.; Endo, T. Organelle contact zones as sites for lipid transfer. J. Biochem. 2019, 165, 115–123. [Google Scholar] [CrossRef]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the center of cell signaling: Interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnoczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1287. [Google Scholar] [CrossRef]
- Sugiura, A.; Nagashima, S.; Tokuyama, T.; Amo, T.; Matsuki, Y.; Ishido, S.; Kudo, Y.; McBride, H.M.; Fukuda, T.; Matsushita, N.; et al. Mitol regulates endoplasmic reticulum-mitochondria contacts via mitofusin2. Mol. Cell 2013, 51, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial gtpase mitofusin 2 cause charcot-marie-tooth neuropathy type 2a. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in sigmar1- and sod1-linked als. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Takeda, K.; Ohno, N.; Ishido, S.; Aoki, M.; Saitoh, Y.; Takada, T.; Tokuyama, T.; Sugiura, A.; Fukuda, T.; et al. Mitol deletion in the brain impairs mitochondrial structure and er tethering leading to oxidative stress. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Nagashima, S.; Shiiba, I.; Uda, A.; Tokuyama, T.; Ito, N.; Fukuda, T.; Matsushita, N.; Ishido, S.; Iwawaki, T.; et al. Mitol prevents er stress-induced apoptosis by ire1alpha ubiquitylation at er-mitochondria contact sites. EMBO J. 2019, 38, e100999. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiiba, I.; Takeda, K.; Nagashima, S.; Yanagi, S. Overview of Mitochondrial E3 Ubiquitin Ligase MITOL/MARCH5 from Molecular Mechanisms to Diseases. Int. J. Mol. Sci. 2020, 21, 3781. https://doi.org/10.3390/ijms21113781
Shiiba I, Takeda K, Nagashima S, Yanagi S. Overview of Mitochondrial E3 Ubiquitin Ligase MITOL/MARCH5 from Molecular Mechanisms to Diseases. International Journal of Molecular Sciences. 2020; 21(11):3781. https://doi.org/10.3390/ijms21113781
Chicago/Turabian StyleShiiba, Isshin, Keisuke Takeda, Shun Nagashima, and Shigeru Yanagi. 2020. "Overview of Mitochondrial E3 Ubiquitin Ligase MITOL/MARCH5 from Molecular Mechanisms to Diseases" International Journal of Molecular Sciences 21, no. 11: 3781. https://doi.org/10.3390/ijms21113781
APA StyleShiiba, I., Takeda, K., Nagashima, S., & Yanagi, S. (2020). Overview of Mitochondrial E3 Ubiquitin Ligase MITOL/MARCH5 from Molecular Mechanisms to Diseases. International Journal of Molecular Sciences, 21(11), 3781. https://doi.org/10.3390/ijms21113781