Clinical and Genetic Overview of Paroxysmal Movement Disorders and Episodic Ataxias

 , , , , , and
, , , , , and 
Abstract
1. Introduction on Paroxysmal Movement Disorders and Episodic Ataxias
2. Methods
3. Phenotypic Classification and Treatment Options
3.1 Paroxysmal Dyskinesias and Other Paroxysmal Movement Disorders
3.1.1 PKD (Paroxysmal Kinesigenic Dyskinesia)
3.1.2. PNKD (Paroxysmal Nonkinesigenic Dyskinesia)
3.1.3. PED (Paroxysmal Exercise-Induced Dyskinesia)
3.1.4. Paroxysmal Nocturnal Dyskinesia
3.1.5. Developmental Paroxysmal Movement Disorders
3.1.6. Other Paroxysmal Movement Disorders in Pediatric Neurological Diseases
3.2. Episodic Ataxias
3.2.1. EA1
3.2.2. EA2
3.2.3. EA5
3.2.4. EA6
3.2.5. EA8
3.2.6. Other EAs with Associated Disease Loci
4. Genetic Aspects and Pathophysiology
4.1 Genes in Paroxysmal Movement Disorders
4.1.1. PRRT2 (OMIM #614386)
4.1.2. PNKD (Formerly MR-1) (OMIM #609023)
4.1.3. SLC2A1 (OMIM #138140)
4.1.4. Pyruvate Dehydrogenase Complex (PDH) Deficiency (OMIM #300502, #608769, #608770)
4.1.5. ECSH1 and HIBCH (OMIM #602292 and #610690)
4.1.6. ATP1A3 (OMIM #182350)
4.1.7. ADCY5 (OMIM #600293)
4.1.8. TBC1D24 (OMIM #613577)
4.1.9. SLC16A2 (OMIM #300095)
4.1.10. Other Genetic Causes of PMD
4.2. Genes in EAs
4.2.1. KCNA1 (OMIM #176260)
4.2.2. CACNA1A (OMIM #601011)
4.2.3. CACNB4 (OMIM #601949)
4.2.4. SLC1A3 (OMIM #600111)
4.2.5. UBR4 (OMIM #609890)
4.2.6. Metabolic Disorders with EAs, Other Genes in Unclassified EAs, or Neurogenetic Diseases with EAs
Metabolic Disorders with EA
Other Genes in Unclassified EAs or Neurogenetic Diseases with EAs
4.3. Secondary (Acquired) Causes of PMD and EAs
4.3.1. Acquired PMD
4.3.2. Acquired EAs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Erro, R.; Bhatia, K.P. Unravelling of the paroxysmal dyskinesias. J. Neurol. Neurosurg. Psychiatry 2019, 90, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Méneret, A.; Roze, E. Paroxysmal movement disorders: An update. Rev. Neurol. (Paris) 2016, 172, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Thenganatt, M.A.; Jankovic, J. Psychogenic (Functional) Movement Disorders. Continuum (Minneap Minn) 2019, 25, 1121–1140. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.B.P. Paroxysmal non-epileptic motor events in childhood. Dev. Med. Child Neurol. 2012, 54, 299–300. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Alvarez, E. Transient benign paroxysmal movement disorders in infancy. Eur. J. Paediatr. Neurol. 2018, 22, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Koy, A.; Cirak, S.; Gonzalez, V.; Becker, K.; Roujeau, T.; Milesi, C.; Baleine, J.; Cambonie, G.; Boularan, A.; Greco, F.; et al. Deep brain stimulation is effective in pediatric patients with GNAO1 associated severe hyperkinesia. J. Neurol. Sci. 2018, 391, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Shang, L.; Tian, W.T.; Cao, L.; Zhang, X.; Liu, Q. Complicated paroxysmal kinesigenic dyskinesia associated with SACS mutations. Ann. Transl. Med. 2020, 8, 8. [Google Scholar] [CrossRef]
- Hanagasi, H.A.; Bilgiç, B.; Abbink, T.E.M.; Hanagasi, F.; Tüfekçioğlu, Z.; Gürvit, H.; Başak, N.; van der Knaap, M.S.; Emre, M. Secondary paroxysmal kinesigenic dyskinesia associated with CLCN2 gene mutation. Parkinsonism Relat. Disord. 2015, 21, 544–546. [Google Scholar] [CrossRef]
- Van Berge, L.; Hamilton, E.M.; Linnankivi, T.; Uziel, G.; Steenweg, M.E.; Isohanni, P.; Wolf, N.I.; Krägeloh-Mann, I.; Brautaset, N.J.; Andrews, P.I.; et al. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: Clinical and genetic characterization and target for therapy. Brain 2014, 137, 1019–1029. [Google Scholar] [CrossRef]
- Espay, A.J.; Morgante, F.; Merola, A.; Fasano, A.; Marsili, L.; Fox, S.H.; Bezard, E.; Picconi, B.; Calabresi, P.; Lang, A.E. Levodopa-induced dyskinesia in Parkinson disease: Current and evolving concepts. Ann. Neurol. 2018, 84, 797–811. [Google Scholar] [CrossRef]
- Waln, O.; Jankovic, J. Paroxysmal movement disorders. Neurol. Clin. 2015, 33, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Kure, S. An atypical case of Thomsen’s disease. Tokyo Igakukai Zasshi [Journal of the Tokyo Medical Association] 1892, 6, 505–514. [Google Scholar]
- Kato, N.; Sadamatsu, M.; Kikuchi, T.; Niikawa, N.; Fukuyama, Y. Paroxysmal kinesigenic choreoathetosis: From first discovery in 1892 to genetic linkage with benign familial infantile convulsions. Epilepsy Res. 2006, 70, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Mount, L.A.; Reback, S. Familial paroxysmal choreoathetosis: Preliminary Report on a Hitherto Undescribed Clinical Syndrome. Arch. Neurol. Psychiatry 1940, 44, 841–847. [Google Scholar] [CrossRef]
- Kertesz, A. Paroxysmal kinesigenic choreoathetosis. An entity within the paroxysmal choreoathetosis syndrome. Description of 10 cases, including 1 autopsied. Neurology 1967, 17, 680–690. [Google Scholar] [CrossRef]
- Lance, J.W. Familial paroxysmal dystonic choreoathetosis and its differentiation from related syndromes. Ann. Neurol. 1977, 2, 285–293. [Google Scholar] [CrossRef]
- Demirkiran, M.; Jankovic, J. Paroxysmal dyskinesias: Clinical features and classification. Ann. Neurol. 1995, 38, 571–579. [Google Scholar] [CrossRef]
- Lugaresi, E.; Cirignotta, F. Hypnogenic Paroxysmal Dystonia: Epileptic Seizure or a New Syndrome? Sleep 1981, 4, 129–138. [Google Scholar] [CrossRef]
- Tinuper, P.; Cerullo, A.; Cirignotta, F.; Cortelli, P.; Lugaresi, E.; Montagna, P. Nocturnal Paroxysmal Dystonia with Short-Lasting Attacks: Three Cases with Evidence for an Epileptic Frontal Lobe Origin of Seizures. Epilepsia 1990, 31, 549–556. [Google Scholar] [CrossRef]
- Silveira-Moriyama, L.; Kovac, S.; Kurian, M.A.; Houlden, H.; Lees, A.J.; Walker, M.C.; Roze, E.; Paciorkowski, A.R.; Mink, J.W.; Warner, T.T. Phenotypes, genotypes, and the management of paroxysmal movement disorders. Dev. Med. Child Neurol. 2018, 60, 559–565. [Google Scholar] [CrossRef]
- Zhang, X.-J.; Xu, Z.-Y.; Wu, Y.-C.; Tan, E.-K. Paroxysmal movement disorders: Recent advances and proposal of a classification system. Parkinsonism Relat. Disord. 2019, 59, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.K.; Hallett, M.; Gwinn-Hardy, K.; Sorensen, B.; Considine, E.; Tucker, S.; Lynch, D.R.; Mathews, K.D.; Swoboda, K.J.; Harris, J.; et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: New diagnostic criteria. Neurology 2004, 63, 2280–2287. [Google Scholar] [CrossRef]
- McGovern, E.M.; Roze, E.; Counihan, T.J. The expanding spectrum of paroxysmal movement disorders: Update from clinical features to therapeutics. Curr. Opin. Neurol. 2018, 31, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-L.; Cao, L.; Li, X.-H.; Hu, Z.-M.; Li, J.-D.; Zhang, J.-G.; Liang, Y.; San-A; Li, N.; Chen, S.-Q.; et al. Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain 2011, 134, 3493–3501. [Google Scholar] [CrossRef] [PubMed]
- Méneret, A.; Grabli, D.; Depienne, C.; Gaudebout, C.; Picard, F.; Dürr, A.; Lagroua, I.; Bouteiller, D.; Mignot, C.; Doummar, D.; et al. PRRT2 mutations: A major cause of paroxysmal kinesigenic dyskinesia in the European population. Neurology 2012, 79, 170–174. [Google Scholar] [CrossRef]
- Groffen, A.J.A.; Klapwijk, T.; van Rootselaar, A.-F.; Groen, J.L.; Tijssen, M.A.J. Genetic and phenotypic heterogeneity in sporadic and familial forms of paroxysmal dyskinesia. J. Neurol. 2013, 260, 93–99. [Google Scholar] [CrossRef]
- Lamperti, C.; Invernizzi, F.; Solazzi, R.; Freri, E.; Carella, F.; Zeviani, M.; Zibordi, F.; Fusco, C.; Zorzi, G.; Granata, T.; et al. Clinical and genetic features of paroxysmal kinesigenic dyskinesia in Italian patients. Eur. J. Paediatr. Neurol. 2016, 20, 152–157. [Google Scholar] [CrossRef]
- Liu, Q.; Qi, Z.; Wan, X.-H.; Li, J.-Y.; Shi, L.; Lu, Q.; Zhou, X.-Q.; Qiao, L.; Wu, L.-W.; Liu, X.-Q.; et al. Mutations in PRRT2 result in paroxysmal dyskinesias with marked variability in clinical expression. J. Med. Genet. 2012, 49, 79–82. [Google Scholar] [CrossRef][Green Version]
- Cao, L.; Huang, X.-J.; Zheng, L.; Xiao, Q.; Wang, X.-J.; Chen, S.-D. Identification of a novel PRRT2 mutation in patients with paroxysmal kinesigenic dyskinesias and c.649dupC as a mutation hot-spot. Parkinsonism Relat. Disord. 2012, 18, 704–706. [Google Scholar] [CrossRef]
- Ono, S.; Yoshiura, K.; Kinoshita, A.; Kikuchi, T.; Nakane, Y.; Kato, N.; Sadamatsu, M.; Konishi, T.; Nagamitsu, S.; Matsuura, M.; et al. Mutations in PRRT2 responsible for paroxysmal kinesigenic dyskinesias also cause benign familial infantile convulsions. J. Hum. Genet. 2012, 57, 338–341. [Google Scholar] [CrossRef]
- Mao, C.-Y.; Shi, C.-H.; Song, B.; Wu, J.; Ji, Y.; Qin, J.; Li, Y.-S.; Wang, J.-J.; Shang, D.-D.; Sun, S.-L.; et al. Genotype-phenotype correlation in a cohort of paroxysmal kinesigenic dyskinesia cases. J. Neurol. Sci. 2014, 340, 91–93. [Google Scholar] [CrossRef]
- Tan, L.C.S.; Methawasin, K.; Teng, E.W.L.; Ng, A.R.J.; Seah, S.H.; Au, W.L.; Liu, J.J.; Foo, J.N.; Zhao, Y.; Tan, E.K. Clinico-genetic comparisons of paroxysmal kinesigenic dyskinesia patients with and without PRRT2 mutations. Eur. J. Neurol. 2014, 21, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Gardella, E.; Becker, F.; Møller, R.S.; Schubert, J.; Lemke, J.R.; Larsen, L.H.G.; Eiberg, H.; Nothnagel, M.; Thiele, H.; Altmüller, J.; et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann. Neurol. 2016, 79, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.-T.; Huang, X.-J.; Mao, X.; Liu, Q.; Liu, X.-L.; Zeng, S.; Guo, X.-N.; Shen, J.-Y.; Xu, Y.-Q.; Tang, H.-D.; et al. Proline-rich transmembrane protein 2–negative paroxysmal kinesigenic dyskinesia: Clinical and genetic analyses of 163 patients. Mov. Disord. 2018, 33, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.-M.; Lin, J.-H.; Cao, L.; Zhang, T.-M.; Zeng, S.; Zhang, K.-L.; Tian, W.-T.; Hu, Z.-M.; Li, N.; Wang, J.-L.; et al. Familial paroxysmal kinesigenic dyskinesia is associated with mutations in the KCNA1 gene. Hum. Mol. Genet. 2018, 27, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Dumitrescu, A.M.; Best, T.T.; Hanefeld, F.; Refetoff, S. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J. Neurol. 2005, 252, 663–666. [Google Scholar] [CrossRef]
- Zhu, M.; Zhu, X.; Wan, H.; Hong, D. Familial IBGC caused by SLC20A2 mutation presenting as paroxysmal kinesigenic dyskinesia. Parkinsonism Relat. Disord. 2014, 20, 353–354. [Google Scholar] [CrossRef]
- Jiang, Y.; Yuan, F.; Yang, Y.; Sun, X.; Song, L.; Jiang, W. CHRNA4 variant causes paroxysmal kinesigenic dyskinesia and genetic epilepsy with febrile seizures plus? Seizure 2018, 56, 88–91. [Google Scholar] [CrossRef]
- Bonakis, A.; Papageorgiou, S.G.; Potagas, C.; Karahalios, G.; Kalfakis, N. A case of refractory secondary paroxysmal kinesigenic dyskinesia with high sensitivity to phenytoin monotherapy. Parkinsonism Relat. Disord. 2009, 15, 68–70. [Google Scholar] [CrossRef]
- Baba, Y.; Wszolek, Z.K.; Normand, M.M. Paroxysmal kinesigenic dyskinesia associated with central pontine myelinolysis. Parkinsonism Relat. Disord. 2003, 10, 113. [Google Scholar] [CrossRef]
- De Seze, J.; Stojkovic, T.; Destée, M.; Destée, A.; Vermersch, P. Paroxysmal kinesigenic choreoathetosis as a presenting symptom of multiple sclerosis. J. Neurol. 2000, 247, 478–480. [Google Scholar] [CrossRef]
- Diaz, G.E.; Wirrell, E.C.; Matsumoto, J.Y.; Krecke, K.N. Bilateral striopallidodentate calcinosis with paroxysmal kinesigenic dyskinesia. Pediatr. Neurol. 2010, 43, 46–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-Y.; Zhang, S.-Q.; Hao, Y.; Zheng, H.-M. Paroxysmal kinesigenic dyskinesia as the initial symptom of Hashimoto encephalopathy. CNS Neurosci. Ther. 2012, 18, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Puri, V.; Chaudhry, N. Paroxysmal kinesigenic dyskinesia manifestation of hyperthyroidism. Neurol. India 2004, 52, 102–103. [Google Scholar]
- Bruno, M.K.; Lee, H.-Y.; Auburger, G.W.J.; Friedman, A.; Nielsen, J.E.; Lang, A.E.; Bertini, E.; Bogaert, P.V.; Averyanov, Y.; Hallett, M.; et al. Genotype–phenotype correlation of paroxysmal nonkinesigenic dyskinesia. Neurology 2007, 68, 1782–1789. [Google Scholar] [CrossRef]
- Yeh, T.-H.; Lin, J.-J.; Lai, S.-C.; Wu-Chou, Y.-H.; Chen, A.-C.; Yueh, K.-C.; Chen, R.-S.; Lu, C.-S. Familial paroxysmal nonkinesigenic dyskinesia: Clinical and genetic analysis of a Taiwanese family. J. Neurol. Sci. 2012, 323, 80–84. [Google Scholar] [CrossRef]
- Stefanova, E.; Djarmati, A.; Momcilović, D.; Dragasević, N.; Svetel, M.; Klein, C.; Kostić, V.S. Clinical characteristics of paroxysmal nonkinesigenic dyskinesia in Serbian family with Myofibrillogenesis regulator 1 gene mutation. Mov. Disord. 2006, 21, 2010–2015. [Google Scholar] [CrossRef]
- Erro, R.; Sheerin, U.-M.; Bhatia, K.P. Paroxysmal dyskinesias revisited: A review of 500 genetically proven cases and a new classification. Mov. Disord. 2014, 29, 1108–1116. [Google Scholar] [CrossRef]
- Zittel, S.; Ganos, C.; Münchau, A. Fatal paroxysmal non-kinesigenic dyskinesia. Eur. J. Neurol. 2015, 22, e30–e31. [Google Scholar] [CrossRef]
- Pons, R.; Cuenca-León, E.; Miravet, E.; Pons, M.; Xaidara, A.; Youroukos, S.; Macaya, A. Paroxysmal non-kinesigenic dyskinesia due to a PNKD recurrent mutation: Report of two Southern European families. Eur. J. Paediatr. Neurol. 2012, 16, 86–89. [Google Scholar] [CrossRef]
- Kumar, A.; Szekely, A.; Jabbari, B. Effective Treatment of Paroxysmal Nonkinesigenic Dyskinesia With Oxcarbazepine. Clin. Neuropharmacol. 2016, 39, 201–205. [Google Scholar] [CrossRef]
- Chudnow, R.S.; Mimbela, R.A.; Owen, D.B.; Roach, E.S. Gabapentin for familial paroxysmal dystonic choreoathetosis. Neurology 1997, 49, 1441–1442. [Google Scholar] [CrossRef] [PubMed]
- Van Coller, R.; Slabbert, P.; Vaidyanathan, J.; Schutte, C. Successful treatment of disabling paroxysmal nonkinesigenic dyskinesia with deep brain stimulation of the globus pallidus internus. Stereotact. Funct. Neurosurg. 2014, 92, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Zimmern, V.; Riant, F.; Roze, E.; Ranza, E.; Lehmann-Horn, F.; de Bellescize, J.; Ville, D.; Lesca, G.; Korff, C.M. Infantile-Onset Paroxysmal Movement Disorder and Episodic Ataxia Associated with a TBC1D24 Mutation. Neuropediatrics 2019, 50, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Set, K.K.; Ghosh, D.; Huq, A.H.M.; Luat, A.F. Episodic Ataxia Type 1 (K-channelopathy) Manifesting as Paroxysmal Nonkinesogenic Dyskinesia: Expanding the Phenotype. Mov. Disord. Clin. Pract. 2017, 4, 784–786. [Google Scholar] [CrossRef]
- Narayanan, D.L.; Deshpande, D.; Das Bhowmik, A.; Varma, D.R.; Dalal, A. Familial choreoathetosis due to novel heterozygous mutation in PDE10A. Am. J. Med. Genet. A 2018, 176, 146–150. [Google Scholar] [CrossRef]
- Weber, Y.G.; Storch, A.; Wuttke, T.V.; Brockmann, K.; Kempfle, J.; Maljevic, S.; Margari, L.; Kamm, C.; Schneider, S.A.; Huber, S.M.; et al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J. Clin. Investig. 2008, 118, 2157–2168. [Google Scholar] [CrossRef]
- Gardiner, A.R.; Jaffer, F.; Dale, R.C.; Labrum, R.; Erro, R.; Meyer, E.; Xiromerisiou, G.; Stamelou, M.; Walker, M.; Kullmann, D.; et al. The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain 2015, 138, 3567–3580. [Google Scholar] [CrossRef]
- Castiglioni, C.; Verrigni, D.; Okuma, C.; Diaz, A.; Alvarez, K.; Rizza, T.; Carrozzo, R.; Bertini, E.; Miranda, M. Pyruvate dehydrogenase deficiency presenting as isolated paroxysmal exercise induced dystonia successfully reversed with thiamine supplementation. Case report and mini-review. Eur. J. Paediatr. Neurol. 2015, 19, 497–503. [Google Scholar] [CrossRef]
- Friedman, J.; Feigenbaum, A.; Chuang, N.; Silhavy, J.; Gleeson, J.G. Pyruvate dehydrogenase complex-E2 deficiency causes paroxysmal exercise-induced dyskinesia. Neurology 2017, 89, 2297–2298. [Google Scholar] [CrossRef]
- Chandra, S.R.; Issac, T.G. A case of mitochondrial cytopathy with exertion induced dystonia. J. Pediatr. Neurosci. 2015, 10, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Constantinou, J.; Sidiropoulos, C. ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: Case presentation and initial benefit of intervention. J. Neurol. 2017, 264, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Olgiati, S.; Skorvanek, M.; Quadri, M.; Minneboo, M.; Graafland, J.; Breedveld, G.J.; Bonte, R.; Ozgur, Z.; van den Hout, M.C.G.N.; Schoonderwoerd, K.; et al. Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov. Disord. 2016, 31, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, J.; Yu, K.; Feng, F.; Sun, X.; Li, C.; Li, H.; Cui, L. A therapeutic regimen for 3-hydroxyisobutyryl-CoA hydrolase deficiency with exercise-induced dystonia. Eur. J. Paediatr. Neurol. 2019, 23, 755–759. [Google Scholar] [CrossRef]
- Bruno, M.K.; Ravina, B.; Garraux, G.; Hallett, M.; Ptacek, L.; Singleton, A.; Johnson, J.; Singleton, A.; Hanson, M.; Considine, E.; et al. Exercise-induced dystonia as a preceding symptom of familial Parkinson’s disease. Mov. Disord. 2004, 19, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Dale, R.C.; Melchers, A.; Fung, V.S.C.; Grattan-Smith, P.; Houlden, H.; Earl, J. Familial paroxysmal exercise-induced dystonia: Atypical presentation of autosomal dominant GTP-cyclohydrolase 1 deficiency. Dev. Med. Child Neurol. 2010, 52, 583–586. [Google Scholar] [CrossRef]
- Yoshimura, K.; Kanki, R. Child-onset paroxysmal exercise-induced dystonia as the initial manifestation of hereditary Parkinson’s disease. Parkinsonism Relat. Disord. 2018, 49, 108–109. [Google Scholar] [CrossRef]
- Méneret, A.; Roze, E.; Maranci, J.-B.; Dodet, P.; Doummar, D.; Riant, F.; Tranchant, C.; Fraix, V.; Anheim, M.; Ekmen, A.; et al. Sleep in ADCY5-Related Dyskinesia: Prolonged Awakenings Caused by Abnormal Movements. J. Clin. Sleep Med. 2019, 15, 1021–1029. [Google Scholar] [CrossRef]
- Friedman, J.R.; Méneret, A.; Chen, D.-H.; Trouillard, O.; Vidailhet, M.; Raskind, W.H.; Roze, E. ADCY5 Mutation Carriers Display Pleiotropic Paroxysmal Day and Nighttime Dyskinesias. Mov. Disord. 2016, 31, 147–148. [Google Scholar] [CrossRef]
- Liu, X.-R.; Huang, D.; Wang, J.; Wang, Y.-F.; Sun, H.; Tang, B.; Li, W.; Lai, J.-X.; He, N.; Wu, M.; et al. Paroxysmal hypnogenic dyskinesia is associated with mutations in the PRRT2 gene. Neurol. Genet. 2016, 2, e66. [Google Scholar] [CrossRef]
- Ahn, B.J.; Kwon, K.-Y. Recurrent episodes of nocturnal hemiballism: A post-stroke movement disorder. Parkinsonism Relat. Disord. 2017, 42, 102–104. [Google Scholar] [CrossRef]
- Morales-Briceño, H.; Fung, V.S.C. Isolated Nocturnal Occurrence of Orofacial Dyskinesias in N-methyl-D-aspartate Receptor Encephalitis-A New Diagnostic Clue. Mov. Disord. Clin. Pract. 2017, 4, 884–886. [Google Scholar] [CrossRef]
- Bonnet, C.; Roubertie, A.; Doummar, D.; Bahi-Buisson, N.; de Cock, V.C.; Roze, E. Developmental and benign movement disorders in childhood. Mov. Disord. 2010, 25, 1317–1334. [Google Scholar] [CrossRef] [PubMed]
- Blumkin, L.; Leshinsky-Silver, E.; Michelson, M.; Zerem, A.; Kivity, S.; Lev, D.; Lerman-Sagie, T. Paroxysmal tonic upward gaze as a presentation of de-novo mutations in CACNA1A. Eur. J. Paediatr. Neurol. 2015, 19, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.; Douglass, L.M.; Milunsky, J.M.; Rosman, N.P. The Genetics of Benign Paroxysmal Torticollis of Infancy: Is There an Association With Mutations in the CACNA1A Gene? J. Child Neurol. 2016, 31, 1057–1061. [Google Scholar] [CrossRef]
- Maini, I.; Iodice, A.; Spagnoli, C.; Salerno, G.G.; Bertani, G.; Frattini, D.; Fusco, C. Expanding phenotype of PRRT2 gene mutations: A new case with epilepsy and benign myoclonus of early infancy. Eur. J. Paediatr. Neurol. 2016, 20, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Maurer, V.O.; Rizzi, M.; Bianchetti, M.G.; Ramelli, G.P. Benign Neonatal Sleep Myoclonus: A Review of the Literature. Pediatrics 2010, 125, e919–e924. [Google Scholar] [CrossRef] [PubMed]
- Fokke, C.; Fock, J.M.; Brouwer, O.F.; Elting, J.W.J. Benign neonatal sleep myoclonus: A case with a spinal generator? Neurology 2011, 77, 1308–1309. [Google Scholar] [CrossRef]
- Ouvrier, R.; Billson, F. Paroxysmal tonic upgaze of childhood--a review. Brain Dev. 2005, 27, 185–188. [Google Scholar] [CrossRef]
- Salmina, C.; Taddeo, I.; Falesi, M.; Weber, P.; Bianchetti, M.G.; Ramelli, G.P. Paroxysmal tonic upgaze in normal children: A case series and a review of the literature. Eur. J. Paediatr. Neurol. 2012, 16, 683–687. [Google Scholar] [CrossRef]
- Quade, A.; Thiel, A.; Kurth, I.; Holtgrewe, M.; Elbracht, M.; Beule, D.; Eggermann, K.; Scholl, U.I.; Häusler, M. Paroxysmal tonic upgaze: A heterogeneous clinical condition responsive to carbonic anhydrase inhibition. Eur. J. Paediatr. Neurol. 2019. [Google Scholar] [CrossRef]
- Roubertie, A.; Echenne, B.; Leydet, J.; Soete, S.; Krams, B.; Rivier, F.; Riant, F.; Tournier-Lasserve, E. Benign paroxysmal tonicupgaze, benign paroxysmal torticollis, episodic ataxia and CACNA1A mutation in a family. J. Neurol. 2008, 255, 1600–1602. [Google Scholar] [CrossRef] [PubMed]
- Tantsis, E.M.; Gill, D.; Griffiths, L.; Gupta, S.; Lawson, J.; Maksemous, N.; Ouvrier, R.; Riant, F.; Smith, R.; Troedson, C.; et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev. Med. Child Neurol. 2016, 58, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Hills, L.B.; Masri, A.; Konno, K.; Kakegawa, W.; Lam, A.-T.N.; Lim-Melia, E.; Chandy, N.; Hill, R.S.; Partlow, J.N.; Al-Saffar, M.; et al. Deletions in GRID2 lead to a recessive syndrome of cerebellar ataxia and tonic upgaze in humans. Neurology 2013, 81, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Campistol, J.; Prats, J.M.; Garaizar, C. Benign Paroxysmal Tonic Upgaze of Childhood With Ataxia, A Neuroophthalmological Syndrome of Familial Origin? Dev. Med. Child Neurol. 1993, 35, 436–439. [Google Scholar] [CrossRef]
- Deonna, T.; Martin, D. Benign paroxysmal torticollis in infancy. Arch. Dis. Child. 1981, 56, 956–959. [Google Scholar] [CrossRef]
- Drigo, P.; Carli, G.; Laverda, A.M. Benign paroxysmal torticollis of infancy. Brain Dev. 2000, 22, 169–172. [Google Scholar] [CrossRef]
- Moavero, R.; Papetti, L.; Bernucci, M.C.; Cenci, C.; Ferilli, M.A.N.; Sforza, G.; Vigevano, F.; Valeriani, M. Cyclic vomiting syndrome and benign paroxysmal torticollis are associated with a high risk of developing primary headache: A longitudinal study. Cephalalgia 2019, 39, 1236–1240. [Google Scholar] [CrossRef]
- Humbertclaude, V.; Krams, B.; Nogue, E.; Nagot, N.; Annequin, D.; Tourniaire, B.; Tournier-Lasserve, E.; Riant, F.; Roubertie, A. Episodic Syndromes Consortium Benign paroxysmal torticollis, benign paroxysmal vertigo, and benign tonic upward gaze are not benign disorders. Dev. Med. Child Neurol. 2018, 60, 1256–1263. [Google Scholar] [CrossRef]
- Giffin, N.J.; Benton, S.; Goadsby, P.J. Benign paroxysmal torticollis of infancy: Four new cases and linkage to CACNA1A mutation. Dev. Med. Child Neurol. 2002, 44, 490–493. [Google Scholar] [CrossRef]
- Danielsson, A.; Anderlid, B.-M.; Stödberg, T.; Lagerstedt-Robinson, K.; Klackenberg Arrhenius, E.; Tedroff, K. Benign paroxysmal torticollis of infancy does not lead to neurological sequelae. Dev. Med. Child Neurol. 2018, 60, 1251–1255. [Google Scholar] [CrossRef]
- Blumkin, L. Paroxysmal torticollis of infancy: A benign phenomenon? Dev. Med. Child Neurol. 2018, 60, 1196–1197. [Google Scholar] [CrossRef] [PubMed]
- Angelini, L.; Rumi, V.; Lamperti, E.; Nardocci, N. Transient paroxysmal dystonia in infancy. Neuropediatrics 1988, 19, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.; Monteiro, J.P.; Fonseca, M.J. Transient idiopathic dystonia in infancy. Acta Paediatr. 2011, 100, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Deonna, T.W.; Ziegler, A.L.; Nielsen, J. Transient idiopathic dystonia in infancy. Neuropediatrics 1991, 22, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Lombroso, C.T.; Fejerman, N. Benign myoclonus of early infancy. Ann. Neurol. 1977, 1, 138–143. [Google Scholar] [CrossRef]
- Vanasse, M.; Bedard, P.; Andermann, F. Shuddering attacks in children: An early clinical manifestation of essential tremor. Neurology 1976, 26, 1027–1030. [Google Scholar] [CrossRef]
- Caraballo, R.H.; Capovilla, G.; Vigevano, F.; Beccaria, F.; Specchio, N.; Fejerman, N. The spectrum of benign myoclonus of early infancy: Clinical and neurophysiologic features in 102 patients. Epilepsia 2009, 50, 1176–1183. [Google Scholar] [CrossRef]
- Capovilla, G.; Montagnini, A.; Peruzzi, C.; Beccaria, F. Head atonic attacks: A new type of benign non-epileptic attack in infancy strongly mimicking epilepsy. Epileptic Disord. 2013, 15, 44–48. [Google Scholar] [CrossRef]
- Capovilla, G. Shaking body attacks: A new type of benign non-epileptic attack in infancy. Epileptic Disord. 2011, 13, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Meinck, H.-M. Startle and its disorders. Neurophysiol. Clin./Clin. Neurophysiol. 2006, 36, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Bakker, M.J.; van Dijk, J.G.; van den Maagdenberg, A.M.; Tijssen, M.A. Startle syndromes. Lancet Neurol. 2006, 5, 513–524. [Google Scholar] [CrossRef]
- Orivoli, S.; Facini, C.; Pisani, F. Paroxysmal nonepileptic motor phenomena in newborn. Brain Dev. 2015, 37, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Sprovieri, T.; Ungaro, C.; Sivo, S.; Quintiliani, M.; Contaldo, I.; Veredice, C.; Citrigno, L.; Muglia, M.; Cavalcanti, F.; Cavallaro, S.; et al. Clinical features and genetic analysis of two siblings with startle disease in an Italian family: A case report. BMC Med. Genet. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, I.; Marsden, C.D.; Schneider, S.A.; Bhatia, K.P. Brainstem myoclonus and startle syndromes. In Marsden’s Book of Movement Disorders; Oxford University Press: Oxford, UK, 2012; pp. 993–1006. [Google Scholar] [CrossRef]
- Pons, L.; Lesca, G.; Sanlaville, D.; Chatron, N.; Labalme, A.; Manel, V.; Arzimanoglou, A.; de Bellescize, J.; Lion-François, L. Neonatal tremor episodes and hyperekplexia-like presentation at onset in a child with SCN8A developmental and epileptic encephalopathy. Epileptic Disord. 2018, 20, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Carecchio, M.; Zorzi, G.; Ragona, F.; Zibordi, F.; Nardocci, N. ATP1A3-related disorders: An update. Eur. J. Paediatr. Neurol. 2018, 22, 257–263. [Google Scholar] [CrossRef]
- Ragona, F.; Castellotti, B.; Salis, B.; Magri, S.; DiFrancesco, J.C.; Nardocci, N.; Franceschetti, S.; Gellera, C.; Granata, T. Alternating Hemiplegia and Epilepsia Partialis Continua: A new phenotype for a novel compound TBC1D24 mutation. Seizure 2017, 47, 71–73. [Google Scholar] [CrossRef]
- Sen, K.; Hicks, M.A.; Huq, A.H.M.; Agarwal, R. Homozygous TANGO2 Single Nucleotide Variants Presenting with Additional Manifestations Resembling Alternating Hemiplegia of Childhood-Expanding the Phenotype of a Recently Reported Condition. Neuropediatrics 2019, 50, 122–125. [Google Scholar] [CrossRef]
- Westenberger, A.; Max, C.; Brüggemann, N.; Domingo, A.; Grütz, K.; Pawlack, H.; Weissbach, A.; Kühn, A.A.; Spiegler, J.; Lang, A.E.; et al. Alternating Hemiplegia of Childhood as a New Presentation of Adenylate Cyclase 5-Mutation-Associated Disease: A Report of Two Cases. J. Pediatr. 2017, 181, 306–308.e1. [Google Scholar] [CrossRef]
- Duan, B.C.; Wong, L.-C.; Lee, W.-T. Alternating hemiplegia and paroxysmal torticollis caused by SCN4A mutation: A new phenotype? Neurology 2019, 93, 673–674. [Google Scholar] [CrossRef]
- Graziola, F.; Garone, G.; Stregapede, F.; Bosco, L.; Vigevano, F.; Curatolo, P.; Bertini, E.; Travaglini, L.; Capuano, A. Diagnostic Yield of a Targeted Next-Generation Sequencing Gene Panel for Pediatric-Onset Movement Disorders: A 3-Year Cohort Study. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Klepper, J.; Leiendecker, B.; Eltze, C.; Heussinger, N. Paroxysmal Nonepileptic Events in Glut1 Deficiency. Mov. Disord. Clin. Pract. 2016, 3, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Capuano, A.; Garone, G.; Tiralongo, G.; Graziola, F. Alternating Hemiplegia of Childhood: Understanding the Genotype-Phenotype Relationship of ATP1A3 Variations. TACG 2020, 13, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Slow, E.J.; Lang, A.E. Oculogyric crises: A review of phenomenology, etiology, pathogenesis, and treatment. Mov. Disord. 2017, 32, 193–202. [Google Scholar] [CrossRef]
- Pearson, T.S.; Pons, R.; Engelstad, K.; Kane, S.A.; Goldberg, M.E.; De Vivo, D.C. Paroxysmal eye-head movements in Glut1 deficiency syndrome. Neurology 2017, 88, 1666–1673. [Google Scholar] [CrossRef]
- Reis, S.; Matias, J.; Machado, R.; Monteiro, J.P. Paroxysmal ocular movements - an early sign in Glut1 deficiency Syndrome. Metab. Brain Dis. 2018, 33, 1381–1383. [Google Scholar] [CrossRef]
- Sweney, M.T.; Silver, K.; Gerard-Blanluet, M.; Pedespan, J.-M.; Renault, F.; Arzimanoglou, A.; Schlesinger-Massart, M.; Lewelt, A.J.; Reyna, S.P.; Swoboda, K.J. Alternating hemiplegia of childhood: Early characteristics and evolution of a neurodevelopmental syndrome. Pediatrics 2009, 123, e534–e541. [Google Scholar] [CrossRef]
- Jen, J.C.; Graves, T.D.; Hess, E.J.; Hanna, M.G.; Griggs, R.C.; Baloh, R.W. CINCH investigators Primary episodic ataxias: Diagnosis, pathogenesis and treatment. Brain 2007, 130, 2484–2493. [Google Scholar] [CrossRef]
- Tomlinson, S.E.; Hanna, M.G.; Kullmann, D.M.; Tan, S.V.; Burke, D. Clinical neurophysiology of the episodic ataxias: Insights into ion channel dysfunction in vivo. Clin. Neurophysiol. 2009, 120, 1768–1776. [Google Scholar] [CrossRef]
- Choi, K.-D.; Choi, J.-H. Episodic Ataxias: Clinical and Genetic Features. J. Mov. Disord. 2016, 9, 129–135. [Google Scholar] [CrossRef]
- Orsucci, D.; Raglione, L.M.; Mazzoni, M.; Vista, M. Therapy of episodic ataxias: Case report and review of the literature. Drugs Context 2019, 8, 212576. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, M.C.; Hasan, S.; Guglielmi, L.; Servettini, I.; Cenciarini, M.; Catacuzzeno, L.; Franciolini, F. New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front. Cell. Neurosci. 2015, 9, 317. [Google Scholar] [CrossRef]
- De Marcos, F.A.; Ghizoni, E.; Kobayashi, E.; Li, L.M.; Cendes, F. Cerebellar volume and long-term use of phenytoin. Seizure 2003, 12, 312–315. [Google Scholar] [CrossRef]
- VanDyke, D.H.; Griggs, R.C.; Murphy, M.J.; Goldstein, M.N. Hereditary myokymia and periodic ataxia. J. Neurol. Sci. 1975, 25, 109–118. [Google Scholar] [CrossRef]
- Graves, T.D.; Cha, Y.-H.; Hahn, A.F.; Barohn, R.; Salajegheh, M.K.; Griggs, R.C.; Bundy, B.N.; Jen, J.C.; Baloh, R.W.; Hanna, M.G.; et al. Episodic ataxia type 1: Clinical characterization, quality of life and genotype-phenotype correlation. Brain 2014, 137, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, M.C.; Gallenmüller, C.; Servettini, I.; Hartl, E.; Tucker, S.J.; Arning, L.; Biskup, S.; Grottesi, A.; Guglielmi, L.; Imbrici, P.; et al. Novel phenotype associated with a mutation in the KCNA1(Kv1.1) gene. Front. Physiol. 2014, 5, 525. [Google Scholar] [CrossRef]
- Rajakulendran, S.; Schorge, S.; Kullmann, D.M.; Hanna, M.G. Episodic ataxia type 1: A neuronal potassium channelopathy. Neurotherapeutics 2007, 4, 258–266. [Google Scholar] [CrossRef]
- Lee, H.; Wang, H.; Jen, J.C.; Sabatti, C.; Baloh, R.W.; Nelson, S.F. A novel mutation in KCNA1 causes episodic ataxia without myokymia. Hum. Mutat. 2004, 24, 536. [Google Scholar] [CrossRef]
- Shook, S.J.; Mamsa, H.; Jen, J.C.; Baloh, R.W.; Zhou, L. Novel mutation in KCNA1 causes episodic ataxia with paroxysmal dyspnea. Muscle Nerve 2008, 37, 399–402. [Google Scholar] [CrossRef]
- Poujois, A.; Antoine, J.-C.; Combes, A.; Touraine, R.L. Chronic neuromyotonia as a phenotypic variation associated with a new mutation in the KCNA1 gene. J. Neurol. 2006, 253, 957–959. [Google Scholar] [CrossRef]
- Mestre, T.A.; Manole, A.; MacDonald, H.; Riazi, S.; Kraeva, N.; Hanna, M.G.; Lang, A.E.; Männikkö, R.; Yoon, G. A novel KCNA1 mutation in a family with episodic ataxia and malignant hyperthermia. Neurogenetics 2016, 17, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, C.A.; Beggs, A.H.; Rodan, L.; Shi, J.; Towne, M.C.; Pelletier, R.; Cao, S.; Rosenberg, P.A.; Urion, D.K.; Picker, J.; et al. Clinical heterogeneity associated with KCNA1 mutations include cataplexy and nonataxic presentations. Neurogenetics 2016, 17, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.V.; Wraige, E.; Lascelles, K.; Bostock, H. Episodic ataxia type 1 without episodic ataxia: The diagnostic utility of nerve excitability studies in individuals with KCNA1 mutations. Dev. Med. Child Neurol. 2013, 55, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Rajakulendran, S.; Graves, T.D.; Labrum, R.W.; Kotzadimitriou, D.; Eunson, L.; Davis, M.B.; Davies, R.; Wood, N.W.; Kullmann, D.M.; Hanna, M.G.; et al. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J. Physiol. (Lond.) 2010, 588, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Jen, J.; Kim, G.W.; Baloh, R.W. Clinical spectrum of episodic ataxia type 2. Neurology 2004, 62, 17–22. [Google Scholar] [CrossRef]
- Imbrici, P.; Eunson, L.H.; Graves, T.D.; Bhatia, K.P.; Wadia, N.H.; Kullmann, D.M.; Hanna, M.G. Late-onset episodic ataxia type 2 due to an in-frame insertion in CACNA1A. Neurology 2005, 65, 944–946. [Google Scholar] [CrossRef]
- Wan, J.; Khanna, R.; Sandusky, M.; Papazian, D.M.; Jen, J.C.; Baloh, R.W. CACNA1A mutations causing episodic and progressive ataxia alter channel trafficking and kinetics. Neurology 2005, 64, 2090–2097. [Google Scholar] [CrossRef]
- Yue, Q.; Jen, J.C.; Nelson, S.F.; Baloh, R.W. Progressive ataxia due to a missense mutation in a calcium-channel gene. Am. J. Hum. Genet. 1997, 61, 1078–1087. [Google Scholar] [CrossRef]
- Denier, C.; Ducros, A.; Vahedi, K.; Joutel, A.; Thierry, P.; Ritz, A.; Castelnovo, G.; Deonna, T.; Gérard, P.; Devoize, J.L.; et al. High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology 1999, 52, 1816–1821. [Google Scholar] [CrossRef]
- Romaniello, R.; Zucca, C.; Tonelli, A.; Bonato, S.; Baschirotto, C.; Zanotta, N.; Epifanio, R.; Righini, A.; Bresolin, N.; Bassi, M.T.; et al. A wide spectrum of clinical, neurophysiological and neuroradiological abnormalities in a family with a novel CACNA1A mutation. J. Neurol. Neurosurg. Psychiatry 2010, 81, 840–843. [Google Scholar] [CrossRef]
- Strupp, M.; Kalla, R.; Claassen, J.; Adrion, C.; Mansmann, U.; Klopstock, T.; Freilinger, T.; Neugebauer, H.; Spiegel, R.; Dichgans, M.; et al. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology 2011, 77, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jang, D.-H.; Jang, J.-H.; Kim, T. Effectiveness of levetiracetam in an acetazolamide-unresponsive patient with episodic ataxia type 2 by a novel CACNA1A nonsense mutation. Eur. J. Neurol. 2017, 24, e43–e44. [Google Scholar] [CrossRef] [PubMed]
- Escayg, A.; De Waard, M.; Lee, D.D.; Bichet, D.; Wolf, P.; Mayer, T.; Johnston, J.; Baloh, R.; Sander, T.; Meisler, M.H. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am. J. Hum. Genet. 2000, 66, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- González Sánchez, M.; Izquierdo, S.; Álvarez, S.; Bautista Alonso, R.E.; Berciano, J.; Gazulla, J. Clinical manifestations of episodic ataxia type 5. Neurol. Clin. Pract. 2019, 9, 503–504. [Google Scholar] [CrossRef]
- Jen, J.C.; Wan, J.; Palos, T.P.; Howard, B.D.; Baloh, R.W. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 2005, 65, 529–534. [Google Scholar] [CrossRef]
- De Vries, B.; Mamsa, H.; Stam, A.H.; Wan, J.; Bakker, S.L.M.; Vanmolkot, K.R.J.; Haan, J.; Terwindt, G.M.; Boon, E.M.J.; Howard, B.D.; et al. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch. Neurol. 2009, 66, 97–101. [Google Scholar] [CrossRef]
- Iwama, K.; Iwata, A.; Shiina, M.; Mitsuhashi, S.; Miyatake, S.; Takata, A.; Miyake, N.; Ogata, K.; Ito, S.; Mizuguchi, T.; et al. A novel mutation in SLC1A3 causes episodic ataxia. J. Hum. Genet. 2018, 63, 207–211. [Google Scholar] [CrossRef]
- Pyle, A.; Smertenko, T.; Bargiela, D.; Griffin, H.; Duff, J.; Appleton, M.; Douroudis, K.; Pfeffer, G.; Santibanez-Koref, M.; Eglon, G.; et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 2015, 138, 276–283. [Google Scholar] [CrossRef]
- Choi, K.-D.; Jen, J.C.; Choi, S.Y.; Shin, J.-H.; Kim, H.-S.; Kim, H.-J.; Kim, J.-S.; Choi, J.-H. Late-onset episodic ataxia associated with SLC1A3 mutation. J. Hum. Genet. 2017, 62, 443–446. [Google Scholar] [CrossRef]
- Choi, K.-D.; Kim, J.-S.; Kim, H.-J.; Jung, I.; Jeong, S.-H.; Lee, S.-H.; Kim, D.U.; Kim, S.-H.; Choi, S.Y.; Shin, J.-H.; et al. Genetic Variants Associated with Episodic Ataxia in Korea. Sci. Rep. 2017, 7, 13855. [Google Scholar] [CrossRef]
- Conroy, J.; McGettigan, P.; Murphy, R.; Webb, D.; Murphy, S.M.; McCoy, B.; Albertyn, C.; McCreary, D.; McDonagh, C.; Walsh, O.; et al. A novel locus for episodic ataxia:UBR4 the likely candidate. Eur. J. Hum. Genet. 2014, 22, 505–510. [Google Scholar] [CrossRef]
- Steckley, J.L.; Ebers, G.C.; Cader, M.Z.; McLachlan, R.S. An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology 2001, 57, 1499–1502. [Google Scholar] [CrossRef]
- Cader, M.Z.; Steckley, J.L.; Dyment, D.A.; McLachlan, R.S.; Ebers, G.C. A genome-wide screen and linkage mapping for a large pedigree with episodic ataxia. Neurology 2005, 65, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Farmer, T.W.; Mustian, V.M. Vestibulocerebellar ataxia. A newly defined hereditary syndrome with periodic manifestations. Arch. Neurol. 1963, 8, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.M.; Pericak-Vance, M.A.; Payne, C.S.; Coin, J.T.; Olanow, C.W. Linkage and genetic analysis in adult onset periodic vestibulo-cerebellar ataxia: Report of a new family (Abstract). Am. J. Hum. Genet. 1984, 36. [Google Scholar]
- Damji, K.F.; Allingham, R.R.; Pollock, S.C.; Small, K.; Lewis, K.E.; Stajich, J.M.; Yamaoka, L.H.; Vance, J.M.; Pericak-Vance, M.A. Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias. Arch. Neurol. 1996, 53, 338–344. [Google Scholar] [CrossRef]
- Merrill, M.J.; Nai, D.; Ghosh, P.; Edwards, N.A.; Hallett, M.; Ray-Chaudhury, A. Neuropathology in a case of episodic ataxia type 4. Neuropathol. Appl. Neurobiol. 2016, 42, 296–300. [Google Scholar] [CrossRef]
- Kerber, K.A.; Jen, J.C.; Lee, H.; Nelson, S.F.; Baloh, R.W. A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch. Neurol. 2007, 64, 749–752. [Google Scholar] [CrossRef]
- Erro, R.; Bhatia, K.P.; Espay, A.J.; Striano, P. The epileptic and nonepileptic spectrum of paroxysmal dyskinesias: Channelopathies, synaptopathies, and transportopathies. Mov. Disord. 2017, 32, 310–318. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Saffari, A.; Westenberger, A.; Klein, C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain 2015, 138, 3476–3495. [Google Scholar] [CrossRef]
- Dale, R.C.; Grattan-Smith, P.; Nicholson, M.; Peters, G.B. Microdeletions detected using chromosome microarray in children with suspected genetic movement disorders: A single-centre study. Dev. Med. Child Neurol. 2012, 54, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Köhler, A.; Hahn, A.; Neubauer, B.; Müller, U. Benign infantile convulsions (IC) and subsequent paroxysmal kinesigenic dyskinesia (PKD) in a patient with 16p11.2 microdeletion syndrome. Neurogenetics 2013, 14, 251–253. [Google Scholar] [CrossRef]
- Termsarasab, P.; Yang, A.C.; Reiner, J.; Mei, H.; Scott, S.A.; Frucht, S.J. Paroxysmal kinesigenic dyskinesia caused by 16p11.2 microdeletion. Tremor Other Hyperkinet. Mov. (N Y) 2014, 4, 274. [Google Scholar] [CrossRef]
- Delcourt, M.; Riant, F.; Mancini, J.; Milh, M.; Navarro, V.; Roze, E.; Humbertclaude, V.; Korff, C.; Des Portes, V.; Szepetowski, P.; et al. Severe phenotypic spectrum of biallelic mutations in PRRT2 gene. J. Neurol. Neurosurg. Psychiatry 2015, 86, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Labate, A.; Tarantino, P.; Viri, M.; Mumoli, L.; Gagliardi, M.; Romeo, A.; Zara, F.; Annesi, G.; Gambardella, A. Homozygous c.649dupC mutation in PRRT2 worsens the BFIS/PKD phenotype with mental retardation, episodic ataxia, and absences. Epilepsia 2012, 53, e196–e199. [Google Scholar] [CrossRef]
- Huang, X.-J.; Wang, T.; Wang, J.-L.; Liu, X.-L.; Che, X.-Q.; Li, J.; Mao, X.; Zhang, M.; Bi, G.-H.; Wu, L.; et al. Paroxysmal kinesigenic dyskinesia: Clinical and genetic analyses of 110 patients. Neurology 2015, 85, 1546–1553. [Google Scholar] [CrossRef]
- Li, H.-F.; Chen, W.-J.; Ni, W.; Wang, K.-Y.; Liu, G.-L.; Wang, N.; Xiong, Z.-Q.; Xu, J.; Wu, Z.-Y. PRRT2 mutation correlated with phenotype of paroxysmal kinesigenic dyskinesia and drug response. Neurology 2013, 80, 1534–1535. [Google Scholar] [CrossRef]
- Van Vliet, R.; Breedveld, G.; de Rijk-van Andel, J.; Brilstra, E.; Verbeek, N.; Verschuuren-Bemelmans, C.; Boon, M.; Samijn, J.; Diderich, K.; van de Laar, I.; et al. PRRT2 phenotypes and penetrance of paroxysmal kinesigenic dyskinesia and infantile convulsions. Neurology 2012, 79, 777–784. [Google Scholar] [CrossRef]
- Heron, S.E.; Grinton, B.E.; Kivity, S.; Afawi, Z.; Zuberi, S.M.; Hughes, J.N.; Pridmore, C.; Hodgson, B.L.; Iona, X.; Sadleir, L.G.; et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am. J. Hum. Genet. 2012, 90, 152–160. [Google Scholar] [CrossRef]
- Chen, W.-J.; Lin, Y.; Xiong, Z.-Q.; Wei, W.; Ni, W.; Tan, G.-H.; Guo, S.-L.; He, J.; Chen, Y.-F.; Zhang, Q.-J.; et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat. Genet. 2011, 43, 1252–1255. [Google Scholar] [CrossRef]
- Valtorta, F.; Benfenati, F.; Zara, F.; Meldolesi, J. PRRT2: From Paroxysmal Disorders to Regulation of Synaptic Function. Trends Neurosci. 2016, 39, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.; Jouannot, O.; Ramakrishnan, S.K.; Zanetti, M.N.; Wang, J.; Salpietro, V.; Houlden, H.; Rothman, J.E.; Krishnakumar, S.S. PRRT2 Regulates Synaptic Fusion by Directly Modulating SNARE Complex Assembly. Cell Rep. 2018, 22, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Rossi, P.; Sterlini, B.; Castroflorio, E.; Marte, A.; Onofri, F.; Valtorta, F.; Maragliano, L.; Corradi, A.; Benfenati, F. A Novel Topology of Proline-rich Transmembrane Protein 2 (PRRT2) HINTS FOR AN INTRACELLULAR FUNCTION AT THE SYNAPSE. J. Biol. Chem. 2016, 291, 6111–6123. [Google Scholar] [CrossRef] [PubMed]
- Michetti, C.; Castroflorio, E.; Marchionni, I.; Forte, N.; Sterlini, B.; Binda, F.; Fruscione, F.; Baldelli, P.; Valtorta, F.; Zara, F.; et al. The PRRT2 knockout mouse recapitulates the neurological diseases associated with PRRT2 mutations. Neurobiol. Dis. 2017, 99, 66–83. [Google Scholar] [CrossRef]
- Rainier, S.; Thomas, D.; Tokarz, D.; Ming, L.; Bui, M.; Plein, E.; Zhao, X.; Lemons, R.; Albin, R.; Delaney, C.; et al. Myofibrillogenesis regulator 1 gene mutations cause paroxysmal dystonic choreoathetosis. Arch. Neurol. 2004, 61, 1025–1029. [Google Scholar] [CrossRef]
- Lee, H.; Nakayama, J.; Xu, Y.; Fan, X.; Karouani, M.; Shen, Y.; Pothos, E.N.; Hess, E.J.; Fu, Y.-H.; Edwards, R.H.; et al. Dopamine dysregulation in a mouse model of paroxysmal nonkinesigenic dyskinesia. J. Clin. Investig. 2012, 122, 507–518. [Google Scholar] [CrossRef]
- Shen, Y.; Ge, W.-P.; Li, Y.; Hirano, A.; Lee, H.-Y.; Rohlmann, A.; Missler, M.; Tsien, R.W.; Jan, L.Y.; Fu, Y.-H.; et al. Protein mutated in paroxysmal dyskinesia interacts with the active zone protein RIM and suppresses synaptic vesicle exocytosis. Proc. Natl. Acad. Sci. USA 2015, 112, 2935–2941. [Google Scholar] [CrossRef]
- Wang, D.; Pascual, J.M.; De Vivo, D. Glucose Transporter Type 1 Deficiency Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Scheepers, A.; Joost, H.G.; Schurmann, A. The glucose transporter families SGLT and GLUT: Molecular basis of normal and aberrant function. J. Parenter. Enter. Nutr. 2004, 28, 364–371. [Google Scholar] [CrossRef]
- Suls, A.; Dedeken, P.; Goffin, K.; Van Esch, H.; Dupont, P.; Cassiman, D.; Kempfle, J.; Wuttke, T.V.; Weber, Y.; Lerche, H.; et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain 2008, 131, 1831–1844. [Google Scholar] [CrossRef]
- Urbizu, A.; Cuenca-León, E.; Raspall-Chaure, M.; Gratacòs, M.; Conill, J.; Redecillas, S.; Roig-Quilis, M.; Macaya, A. Paroxysmal exercise-induced dyskinesia, writer’s cramp, migraine with aura and absence epilepsy in twin brothers with a novel SLC2A1 missense mutation. J. Neurol. Sci. 2010, 295, 110–113. [Google Scholar] [CrossRef]
- Zorzi, G.; Castellotti, B.; Zibordi, F.; Gellera, C.; Nardocci, N. Paroxysmal movement disorders in GLUT1 deficiency syndrome. Neurology 2008, 71, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Bovi, T.; Fasano, A.; Juergenson, I.; Gellera, C.; Castellotti, B.; Fontana, E.; Tinazzi, M. Paroxysmal exercise-induced dyskinesia with self-limiting partial epilepsy: A novel GLUT-1 mutation with benign phenotype. Parkinsonism Relat. Disord. 2011, 17, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Almuqbil, M.; Rivkin, M.J.; Takeoka, M.; Yang, E.; Rodan, L.H. Transient regional cerebral hypoperfusion during a paroxysmal hemiplegic event in GLUT1 deficiency syndrome. Eur. J. Paediatr. Neurol. 2018, 22, 544–547. [Google Scholar] [CrossRef]
- Pellegrin, S.; Cantalupo, G.; Opri, R.; Dalla Bernardina, B.; Darra, F. EEG findings during “paroxysmal hemiplegia” in a patient with GLUT1-deficiency. Eur. J. Paediatr. Neurol. 2017, 21, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Roubergue, A.; Apartis, E.; Mesnage, V.; Doummar, D.; Trocello, J.-M.; Roze, E.; Taieb, G.; De Villemeur, T.B.; Vuillaumier-Barrot, S.; Vidailhet, M.; et al. Dystonic tremor caused by mutation of the glucose transporter gene GLUT1. J. Inherit. Metab. Dis. 2011, 34, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Tchapyjnikov, D.; Mikati, M.A. Acetazolamide-responsive Episodic Ataxia Without Baseline Deficits or Seizures Secondary to GLUT1 Deficiency: A Case Report and Review of the Literature. Neurologist 2018, 23, 17–18. [Google Scholar] [CrossRef]
- Klepper, J.; Scheffer, H.; Elsaid, M.F.; Kamsteeg, E.-J.; Leferink, M.; Ben-Omran, T. Autosomal recessive inheritance of GLUT1 deficiency syndrome. Neuropediatrics 2009, 40, 207–210. [Google Scholar] [CrossRef]
- Rotstein, M.; Engelstad, K.; Yang, H.; Wang, D.; Levy, B.; Chung, W.K.; De Vivo, D.C. Glut1 Deficiency: Inheritance Pattern Determined by Haploinsufficiency. Ann. Neurol. 2010, 68, 955–958. [Google Scholar] [CrossRef]
- Leen, W.G.; Klepper, J.; Verbeek, M.M.; Leferink, M.; Hofste, T.; van Engelen, B.G.; Wevers, R.A.; Arthur, T.; Bahi-Buisson, N.; Ballhausen, D.; et al. Glucose transporter-1 deficiency syndrome: The expanding clinical and genetic spectrum of a treatable disorder. Brain 2010, 133, 655–670. [Google Scholar] [CrossRef]
- Yang, H.; Wang, D.; Engelstad, K.; Bagay, L.; Wei, Y.; Rotstein, M.; Aggarwal, V.; Levy, B.; Ma, L.; Chung, W.K.; et al. Glut1 deficiency syndrome and erythrocyte glucose uptake assay. Ann. Neurol. 2011, 70, 996–1005. [Google Scholar] [CrossRef]
- Gras, D.; Cousin, C.; Kappeler, C.; Fung, C.-W.; Auvin, S.; Essid, N.; Chung, B.H.; Da Costa, L.; Hainque, E.; Luton, M.-P.; et al. A simple blood test expedites the diagnosis of glucose transporter type 1 deficiency syndrome. Ann. Neurol. 2017, 82, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Leen, W.G.; Mewasingh, L.; Verbeek, M.M.; Kamsteeg, E.-J.; van de Warrenburg, B.P.; Willemsen, M.A. Movement disorders in GLUT1 deficiency syndrome respond to the modified Atkins diet. Mov. Disord. 2013, 28, 1439–1442. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; Hainque, E.; Gras, D.; Adanyeguh, I.M.; Caillet, S.; Héron, B.; Roubertie, A.; Kaphan, E.; Valabregue, R.; Rinaldi, D.; et al. Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J. Neurol. Neurosurg. Psychiatry 2016, 87, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Hainque, E.; Gras, D.; Meneret, A.; Atencio, M.; Luton, M.-P.; Barbier, M.; Doulazmi, M.; Habarou, F.; Ottolenghi, C.; Roze, E.; et al. Long-term follow-up in an open-label trial of triheptanoin in GLUT1 deficiency syndrome: A sustained dramatic effect. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Hainque, E.; Meneret, A.; Gras, D.; Atencio, M.; Luton, M.-P.; Barbier, M.; De Saint Martin, A.; Billette de Villemeur, T.; Ottolenghi, C.; Roze, E.; et al. Transition from ketogenic diet to triheptanoin in patients with GLUT1 deficiency syndrome. J. Neurol. Neurosurg. Psychiatry 2020, 91, 444–445. [Google Scholar] [CrossRef] [PubMed]
- Barnerias, C.; Saudubray, J.-M.; Touati, G.; De Lonlay, P.; Dulac, O.; Ponsot, G.; Marsac, C.; Brivet, M.; Desguerre, I. Pyruvate dehydrogenase complex deficiency: Four neurological phenotypes with differing pathogenesis. Dev. Med. Child Neurol. 2010, 52, e1–e9. [Google Scholar] [CrossRef]
- Patel, K.P.; O’Brien, T.W.; Subramony, S.H.; Shuster, J.; Stacpoole, P.W. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab. 2012, 106, 385–394. [Google Scholar] [CrossRef]
- Strassburg, H.M.; Koch, J.; Mayr, J.; Sperl, W.; Boltshauser, E. Acute flaccid paralysis as initial symptom in 4 patients with novel E1alpha mutations of the pyruvate dehydrogenase complex. Neuropediatrics 2006, 37, 137–141. [Google Scholar] [CrossRef]
- Livingstone, I.R.; Gardner-Medwin, D.; Pennington, R.J. Familial intermittent ataxia with possible X-linked recessive inheritance. Two patients with abnormal pyruvate metabolism and a response to acetazolamide. J. Neurol. Sci. 1984, 64, 89–97. [Google Scholar] [CrossRef]
- Bindoff, L.A.; Birch-Machin, M.A.; Farnsworth, L.; Gardner-Medwin, D.; Lindsay, J.G.; Turnbull, D.M. Familial intermittent ataxia due to a defect of the E1 component of pyruvate dehydrogenase complex. J. Neurol. Sci. 1989, 93, 311–318. [Google Scholar] [CrossRef]
- Egel, R.T.; Hoganson, G.E.; Katerji, M.A.; Borenstein, M.J. Zonisamide Ameliorates Symptoms of Secondary Paroxysmal Dystonia. Pediatric Neurol. 2010, 43, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Head, R.A.; de Goede, C.G.E.L.; Newton, R.W.N.; Walter, J.H.; McShane, M.A.; Brown, R.M.; Brown, G.K. Pyruvate dehydrogenase deficiency presenting as dystonia in childhood. Dev. Med. Child Neurol. 2004, 46, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Head, R.A.; Brown, R.M.; Zolkipli, Z.; Shahdadpuri, R.; King, M.D.; Clayton, P.T.; Brown, G.K. Clinical and genetic spectrum of pyruvate dehydrogenase deficiency: Dihydrolipoamide acetyltransferase (E2) deficiency. Ann. Neurol. 2005, 58, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Magner, M.; Vinšová, K.; Tesařová, M.; Hájková, Z.; Hansíková, H.; Wenchich, L.; Ješina, P.; Smolka, V.; Adam, T.; Vaněčková, M.; et al. Two patients with clinically distinct manifestation of pyruvate dehydrogenase deficiency due to mutations in PDHA1 gene. Prague Med. Rep. 2011, 112, 18–28. [Google Scholar] [PubMed]
- McWilliam, C.A.; Ridout, C.K.; Brown, R.M.; McWilliam, R.C.; Tolmie, J.; Brown, G.K. Pyruvate dehydrogenase E2 deficiency: A potentially treatable cause of episodic dystonia. Eur. J. Paediatric Neurol. 2010, 14, 349–353. [Google Scholar] [CrossRef]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The Pyruvate Dehydrogenase Complexes: Structure-based Function and Regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef]
- Quinonez, S.C.; Thoene, J.G. Dihydrolipoamide Dehydrogenase Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Imbard, A.; Boutron, A.; Vequaud, C.; Zater, M.; de Lonlay, P.; de Baulny, H.O.; Barnerias, C.; Miné, M.; Marsac, C.; Saudubray, J.-M.; et al. Molecular characterization of 82 patients with pyruvate dehydrogenase complex deficiency. Structural implications of novel amino acid substitutions in E1 protein. Mol. Genet. Metab. 2011, 104, 507–516. [Google Scholar] [CrossRef]
- Okajima, K.; Warman, M.L.; Byrne, L.C.; Kerr, D.S. Somatic mosaicism in a male with an exon skipping mutation in PDHA1 of the pyruvate dehydrogenase complex results in a milder phenotype. Mol. Genet. Metab. 2006, 87, 162–168. [Google Scholar] [CrossRef]
- Singer, B.H.; Iyer, R.K.; Kerr, D.S.; Ahmad, A. Deletion at chromosomal band Xp22.12-Xp22.13 involving PDHA1 in a patient with congenital lactic acidosis. Mol. Genet. Metab. 2010, 101, 87–89. [Google Scholar] [CrossRef]
- Steller, J.; Gargus, J.J.; Gibbs, L.H.; Hasso, A.N.; Kimonis, V.E. Mild phenotype in a male with pyruvate dehydrogenase complex deficiency associated with novel hemizygous in-frame duplication of the E1α subunit gene (PDHA1). Neuropediatrics 2014, 45, 56–60. [Google Scholar] [CrossRef]
- Korenke, G.C.; Nuoffer, J.-M.; Alhaddad, B.; Mayr, H.; Prokisch, H.; Haack, T.B. Paroxysmal Dyskinesia in ECHS1 Defect with Globus Pallidus Lesions. Neuropediatrics 2016, 47, PS01–PS10. [Google Scholar] [CrossRef]
- Haack, T.B.; Jackson, C.B.; Murayama, K.; Kremer, L.S.; Schaller, A.; Kotzaeridou, U.; de Vries, M.C.; Schottmann, G.; Santra, S.; Büchner, B.; et al. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann. Clin. Transl. Neurol. 2015, 2, 492–509. [Google Scholar] [CrossRef]
- Galosi, S.; Nardecchia, F.; Leuzzi, V. Treatable Inherited Movement Disorders in Children: Spotlight on Clinical and Biochemical Features. Mov. Disord. Clin. Pract. 2020, 7, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Sabouraud, P.; Riquet, A.; Spitz, M.-A.; Deiva, K.; Nevsimalova, S.; Mignot, C.; Lesca, G.; Bednarek, N.; Doummar, D.; Pietrement, C.; et al. Relapsing encephalopathy with cerebellar ataxia are caused by variants involving p.Arg756 in ATP1A3. Eur. J. Paediatr. Neurol. 2019, 23, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Graziola, F.; Cusmai, R.; Fusco, L.; Nicita, F.; Elia, M.; Travaglini, L.; Bertini, E.; Curatolo, P.; Vigevano, F.; et al. ATP1A3-related epileptic encephalopathy responding to ketogenic diet. Brain Dev. 2018, 40, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Marzin, P.; Mignot, C.; Dorison, N.; Dufour, L.; Ville, D.; Kaminska, A.; Panagiotakaki, E.; Dienpendaele, A.-S.; Penniello, M.-J.; Nougues, M.-C.; et al. Early-onset encephalopathy with paroxysmal movement disorders and epileptic seizures without hemiplegic attacks: About three children with novel ATP1A3 mutations. Brain Dev. 2018, 40, 768–774. [Google Scholar] [CrossRef]
- Brashear, A.; Sweadner, K.J.; Cook, J.F.; Swoboda, K.J.; Ozelius, L. ATP1A3-Related Neurologic Disorders. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Dard, R.; Mignot, C.; Durr, A.; Lesca, G.; Sanlaville, D.; Roze, E.; Mochel, F. Relapsing encephalopathy with cerebellar ataxia related to an ATP1A3 mutation. Dev. Med. Child Neurol. 2015, 57, 1183–1186. [Google Scholar] [CrossRef]
- Paciorkowski, A.R.; McDaniel, S.S.; Jansen, L.A.; Tully, H.; Tuttle, E.; Ghoneim, D.H.; Tupal, S.; Gunter, S.A.; Vasta, V.; Zhang, Q.; et al. Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 2015, 56, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Younes, T.B.; Benrhouma, H.; Klaa, H.; Rouissi, A.; Chaabouni, M.; Kraoua, I.; Youssef-Turki, I.B. Early Life Epilepsy and Episodic Apnea Revealing an ATP1A3 Mutation: Report of a Pediatric Case and Literature Review. Neuropediatrics 2018, 49, 339–341. [Google Scholar] [CrossRef]
- Ishihara, N.; Inagaki, H.; Miyake, M.; Kawamura, Y.; Yoshikawa, T.; Kurahashi, H. A case of early onset life-threatening epilepsy associated with a novel ATP1A3 gene variant. Brain Dev. 2019, 41, 285–291. [Google Scholar] [CrossRef]
- Sweney, M.T.; Newcomb, T.M.; Swoboda, K.J. The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, Alternating Hemiplegia of Childhood, Rapid-onset Dystonia-Parkinsonism, CAPOS and beyond. Pediatr. Neurol. 2015, 52, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Rosewich, H.; Ohlenbusch, A.; Huppke, P.; Schlotawa, L.; Baethmann, M.; Carrilho, I.; Fiori, S.; Lourenço, C.M.; Sawyer, S.; Steinfeld, R.; et al. The expanding clinical and genetic spectrum of ATP1A3-related disorders. Neurology 2014, 82, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Roubergue, A.; Roze, E.; Vuillaumier-Barrot, S.; Fontenille, M.-J.; Méneret, A.; Vidailhet, M.; Fontaine, B.; Doummar, D.; Philibert, B.; Riant, F.; et al. The multiple faces of the ATP1A3-related dystonic movement disorder. Mov. Disord. 2013, 28, 1457–1459. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.; Stephen, C.D.; Udani, V.; Sankhla, C.S.; Barad, N.H.; Lang, A.E.; Bhatia, K.P. Paroxysmal Asymmetric Dystonic Arm Posturing-A Less Recognized but Characteristic Manifestation of ATP1A3-related disease. Mov. Disord. Clin. Pract. 2019, 6, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Ozelius, L.J. Clinical spectrum of disease associated with ATP1A3 mutations. Lancet Neurol. 2012, 11, 741–743. [Google Scholar] [CrossRef]
- Demos, M.K.; van Karnebeek, C.D.; Ross, C.J.; Adam, S.; Shen, Y.; Zhan, S.H.; Shyr, C.; Horvath, G.; Suri, M.; Fryer, A.; et al. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J. Rare Dis. 2014, 9, 15. [Google Scholar] [CrossRef]
- De Gusmao, C.M.; Dy, M.; Sharma, N. Beyond Dystonia-Parkinsonism: Chorea and Ataxia with ATP1A3 Mutations. Mov. Disord. Clin. Pract. 2016, 3, 402–404. [Google Scholar] [CrossRef]
- Duat Rodriguez, A.; Prochazkova, M.; Santos Santos, S.; Rubio Cabezas, O.; Cantarin Extremera, V.; Gonzalez-Gutierrez-Solana, L. Early Diagnosis of CAPOS Syndrome Before Acute-Onset Ataxia-Review of the Literature and a New Family. Pediatr. Neurol. 2017, 71, 60–64. [Google Scholar] [CrossRef]
- Schirinzi, T.; Graziola, F.; Nicita, F.; Travaglini, L.; Stregapede, F.; Valeriani, M.; Curatolo, P.; Bertini, E.; Vigevano, F.; Capuano, A. Childhood Rapid-Onset Ataxia: Expanding the Phenotypic Spectrum of ATP1A3 Mutations. Cerebellum 2018, 17, 489–493. [Google Scholar] [CrossRef]
- Pivovarov, A.S.; Calahorro, F.; Walker, R.J. Na+/K+-pump and neurotransmitter membrane receptors. Invert. Neurosci. 2018, 19, 1. [Google Scholar] [CrossRef]
- Bøttger, P.; Tracz, Z.; Heuck, A.; Nissen, P.; Romero-Ramos, M.; Lykke-Hartmann, K. Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J. Comp. Neurol. 2011, 519, 376–404. [Google Scholar] [CrossRef] [PubMed]
- Viollet, L.; Glusman, G.; Murphy, K.J.; Newcomb, T.M.; Reyna, S.P.; Sweney, M.; Nelson, B.; Andermann, F.; Andermann, E.; Acsadi, G.; et al. Alternating Hemiplegia of Childhood: Retrospective Genetic Study and Genotype-Phenotype Correlations in 187 Subjects from the US AHCF Registry. PLoS ONE 2015, 10, e0127045. [Google Scholar] [CrossRef]
- Boelman, C.; Lagman-Bartolome, A.M.; MacGregor, D.L.; McCabe, J.; Logan, W.J.; Minassian, B.A. Identical ATP1A3 mutation causes alternating hemiplegia of childhood and rapid-onset dystonia parkinsonism phenotypes. Pediatr. Neurol. 2014, 51, 850–853. [Google Scholar] [CrossRef]
- Vijiaratnam, N.; Bhatia, K.P.; Lang, A.E.; Raskind, W.H.; Espay, A.J. ADCY5-Related Dyskinesia: Improving Clinical Detection of an Evolving Disorder. Mov. Disord. Clin. Pract. 2019, 6, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.; Antelmi, E.; Mencacci, N.E.; Batla, A.; Eriksson, S.H.; Walker, M.C.; Bronstein, A.M.; Bhatia, K.P. Oculomotor apraxia and disrupted sleep with nocturnal ballistic bouts in ADCY5-related disease. Parkinsonism Relat. Disord. 2018, 54, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Carecchio, M.; Mencacci, N.E.; Iodice, A.; Pons, R.; Panteghini, C.; Zorzi, G.; Zibordi, F.; Bonakis, A.; Dinopoulos, A.; Jankovic, J.; et al. ADCY5-related movement disorders: Frequency, disease course and phenotypic variability in a cohort of paediatric patients. Parkinsonism Relat. Disord. 2017, 41, 37–43. [Google Scholar] [CrossRef]
- Méneret, A.; Gras, D.; McGovern, E.; Roze, E. Caffeine and the Dyskinesia Related to Mutations in the ADCY5 Gene. Ann. Intern. Med. 2019. [Google Scholar] [CrossRef]
- Chang, F.C.F.; Westenberger, A.; Dale, R.C.; Smith, M.; Pall, H.S.; Perez-Dueñas, B.; Grattan-Smith, P.; Ouvrier, R.A.; Mahant, N.; Hanna, B.C.; et al. Phenotypic insights into ADCY5-associated disease. Mov. Disord. 2016, 31, 1033–1040. [Google Scholar] [CrossRef]
- Meijer, I.A.; Miravite, J.; Kopell, B.H.; Lubarr, N. Deep Brain Stimulation in an Additional Patient With ADCY5-Related Movement Disorder. J. Child Neurol. 2017, 32, 438–439. [Google Scholar] [CrossRef]
- Abela, L.; Kurian, M.A. Postsynaptic movement disorders: Clinical phenotypes, genotypes, and disease mechanisms. J. Inherit. Metab. Dis. 2018, 41, 1077–1091. [Google Scholar] [CrossRef]
- Barrett, M.J.; Williams, E.S.; Chambers, C.; Dhamija, R. Autosomal recessive inheritance of ADCY5-related generalized dystonia and myoclonus. Neurol. Genet. 2017, 3, 193. [Google Scholar] [CrossRef] [PubMed]
- Bohlega, S.A.; Abou-Al-Shaar, H.; AlDakheel, A.; Alajlan, H.; Bohlega, B.S.; Meyer, B.F.; Monies, D.; Cupler, E.J.; Al-Saif, A.M. Autosomal recessive ADCY5-Related dystonia and myoclonus: Expanding the genetic spectrum of ADCY5-Related movement disorders. Parkinsonism Relat. Disord. 2019, 64, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-W.; Hong, J.-H.; Choi, I.Y.; Che, Y.; Lee, J.-K.; Yang, S.-D.; Song, C.-W.; Kang, H.S.; Lee, J.-H.; Noh, J.S.; et al. Impaired D2 dopamine receptor function in mice lacking type 5 adenylyl cyclase. J. Neurosci. 2002, 22, 7931–7940. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Z.; Friedman, J.R.; Chen, D.-H.; Chan, G.C.-K.; Bloss, C.S.; Hisama, F.M.; Topol, S.E.; Carson, A.R.; Pham, P.H.; Bonkowski, E.S.; et al. Gain-of-function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann. Neurol. 2014, 75, 542–549. [Google Scholar] [CrossRef]
- Doyle, T.B.; Hayes, M.P.; Chen, D.H.; Raskind, W.H.; Watts, V.J. Functional characterization of AC5 gain-of-function variants: Impact on the molecular basis of ADCY5-related dyskinesia. Biochem. Pharmacol. 2019, 163, 169–177. [Google Scholar] [CrossRef]
- Iwamoto, T.; Okumura, S.; Iwatsubo, K.; Kawabe, J.-I.; Ohtsu, K.; Sakai, I.; Hashimoto, Y.; Izumitani, A.; Sango, K.; Ajiki, K.; et al. Motor dysfunction in type 5 adenylyl cyclase-null mice. J. Biol. Chem. 2003, 278, 16936–16940. [Google Scholar] [CrossRef]
- Carapito, R.; Paul, N.; Untrau, M.; Gentil, M.L.; Ott, L.; Alsaleh, G.; Jochem, P.; Radosavljevic, M.; Caignec, C.L.; David, A.; et al. A de novo ADCY5 mutation causes early-onset autosomal dominant chorea and dystonia. Mov. Disord. 2015, 30, 423–427. [Google Scholar] [CrossRef]
- Mucha, B.E.; Hennekam, R.C.; Sisodiya, S.; Campeau, P.M. TBC1D24-Related Disorders. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Steel, D.; Heim, J.; Kruer, M.C.; Sanchis-Juan, A.; Raymond, L.F.; Eunson, P.; Kurian, M.A. Biallelic mutations of TBC1D24 in exercise-induced paroxysmal dystonia. Mov. Disord. 2020, 35, 372–373. [Google Scholar] [CrossRef]
- Lüthy, K.; Mei, D.; Fischer, B.; De Fusco, M.; Swerts, J.; Paesmans, J.; Parrini, E.; Lubarr, N.; Meijer, I.A.; Mackenzie, K.M.; et al. TBC1D24-TLDc-related epilepsy exercise-induced dystonia: Rescue by antioxidants in a disease model. Brain 2019, 142, 2319–2335. [Google Scholar] [CrossRef]
- Guerrini, R.; Bonanni, P.; Nardocci, N.; Parmeggiani, L.; Piccirilli, M.; De Fusco, M.; Aridon, P.; Ballabio, A.; Carrozzo, R.; Casari, G. Autosomal recessive rolandic epilepsy with paroxysmal exercise-induced dystonia and writer’s cramp: Delineation of the syndrome and gene mapping to chromosome 16p12-11.2. Ann. Neurol. 1999, 45, 344–352. [Google Scholar] [CrossRef]
- Falace, A.; Filipello, F.; La Padula, V.; Vanni, N.; Madia, F.; De Pietri Tonelli, D.; de Falco, F.A.; Striano, P.; Dagna Bricarelli, F.; Minetti, C.; et al. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am. J. Hum. Genet. 2010, 87, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Sarret, C.; Oliver Petit, I.; Tonduti, D. Allan-Herndon-Dudley Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Fuchs, O.; Pfarr, N.; Pohlenz, J.; Schmidt, H. Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev. Med. Child Neurol. 2009, 51, 240–244. [Google Scholar] [CrossRef] [PubMed]
- García-de Teresa, B.; González-del Angel, A.; Reyna-Fabián, M.E.; Ruiz-Reyes, M.D.L.L.; Calzada-León, R.; Perez-Enriquez, B.; Alcántara-Ortigoza, M.A. Deletion of exon 1 of the SLC16A2 gene: A common occurrence in patients with Allan-Herndon-Dudley syndrome. Thyroid 2015, 25, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, S.; Peeters, R.P.; Moran, C.; Stoupa, A.; Auriol, F.; Tonduti, D.; Dica, A.; Paone, L.; Rozenkova, K.; Malikova, J.; et al. Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: An international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 695–706. [Google Scholar] [CrossRef]
- Meisler, M.H.; Helman, G.; Hammer, M.F.; Fureman, B.E.; Gaillard, W.D.; Goldin, A.L.; Hirose, S.; Ishii, A.; Kroner, B.L.; Lossin, C.; et al. SCN8A encephalopathy: Research progress and prospects. Epilepsia 2016, 57, 1027–1035. [Google Scholar] [CrossRef]
- Zhang, Z.-B.; Tian, M.-Q.; Gao, K.; Jiang, Y.-W.; Wu, Y. De novo KCNMA1 mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Mov. Disord. 2015, 30, 1290–1292. [Google Scholar] [CrossRef]
- Du, W.; Bautista, J.F.; Yang, H.; Diez-Sampedro, A.; You, S.-A.; Wang, L.; Kotagal, P.; Lüders, H.O.; Shi, J.; Cui, J.; et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet. 2005, 37, 733–738. [Google Scholar] [CrossRef]
- Bailey, C.S.; Moldenhauer, H.J.; Park, S.M.; Keros, S.; Meredith, A.L. KCNMA1-linked channelopathy. J. Gen. Physiol. 2019, 151, 1173–1189. [Google Scholar] [CrossRef]
- Zhou, Y.; Yuan, Y.; Liu, Z.; Zeng, S.; Chen, Z.; Shen, L.; Jiang, H.; Xia, K.; Tang, B.; Wang, J. Genetic and clinical analyses of spinocerebellar ataxia type 8 in mainland China. J. Neurol. 2019, 266, 2979–2986. [Google Scholar] [CrossRef]
- Myers, K.A.; Scheffer, I.E. DEPDC5 as a potential therapeutic target for epilepsy. Expert Opin. Ther. Targets 2017, 21, 591–600. [Google Scholar] [CrossRef]
- Dill, P.; Wagner, M.; Somerville, A.; Thöny, B.; Blau, N.; Weber, P. Child neurology: Paroxysmal stiffening, upward gaze, and hypotonia: Hallmarks of sepiapterin reductase deficiency. Neurology 2012, 78, e29–e32. [Google Scholar] [CrossRef]
- Kostić, V.S.; Petrović, I.N. Brain Calcification and Movement Disorders. Curr. Neurol. Neurosci. Rep. 2017, 17, 2. [Google Scholar] [CrossRef]
- Alemdar, M.; Selek, A.; Işeri, P.; Efendi, H.; Komsuoğlu, S.S. Fahr’s disease presenting with paroxysmal non-kinesigenic dyskinesia: A case report. Parkinsonism Relat. Disord. 2008, 14, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Montilla-Uzcátegui, V.; Araujo-Unda, H.; Daza-Restrepo, A.; Sáenz-Farret, M.; Micheli, F. Paroxysmal Nonkinesigenic Dyskinesias Responsive to Carbamazepine in Fahr Syndrome: A Case Report. Clin. Neuropharmacol. 2016, 39, 262–264. [Google Scholar] [CrossRef]
- Niccolini, F.; Mencacci, N.E.; Yousaf, T.; Rabiner, E.A.; Salpietro, V.; Pagano, G.; Balint, B.; Efthymiou, S.; Houlden, H.; Gunn, R.N.; et al. PDE10A and ADCY5 mutations linked to molecular and microstructural basal ganglia pathology. Mov. Disord. 2018, 33, 1961–1965. [Google Scholar] [CrossRef] [PubMed]
- Micheli, F.; Tschopp, L.; Cersosimo, M.G. Oxcarbazepine-responsive paroxysmal kinesigenic dyskinesia in Wilson disease. Clin. Neuropharmacol. 2011, 34, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Morales-Briceño, H.; Chang, F.C.F.; Wong, C.; Mallawaarachchi, A.; Wolfe, N.; Pellegrino da Silva, R.; Hakonarson, H.; Sandaradura, S.A.; Guo, Y.; Christodoulou, J.; et al. Paroxysmal dyskinesias with drowsiness and thalamic lesions in GABA transaminase deficiency. Neurology 2019, 92, 94–97. [Google Scholar] [CrossRef]
- De La Casa-Fages, B.; Pérez-Sánchez, J.R.; Grandas, F. Paroxysmal Kinesigenic Dystonia in a Lesch-Nyhan Disease Variant. Mov. Disord. Clin. Pract. 2014, 1, 123–124. [Google Scholar] [CrossRef]
- Temudo, T.; Martins, E.; Poças, F.; Cruz, R.; Vilarinho, L. Maple syrup disease presenting as paroxysmal dystonia. Ann. Neurol. 2004, 56, 749–750. [Google Scholar] [CrossRef]
- Efthymiou, S.; Salpietro, V.; Bettencourt, C.; Houlden, H. Paroxysmal Movement Disorder and Epilepsy Caused by a De Novo Truncating Mutation in KAT6A. J. Pediatr. Genet. 2018, 7, 114–116. [Google Scholar] [CrossRef]
- Graves, T.D.; Rajakulendran, S.; Zuberi, S.M.; Morris, H.R.; Schorge, S.; Hanna, M.G.; Kullmann, D.M. Nongenetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology 2010, 75, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Martindale, J.; Shanmugarajah, P.; Grünewald, R.A.; Sarrigiannis, P.G.; Beauchamp, N.; Garrard, K.; Warburton, R.; Sanders, D.S.; Friend, D.; et al. Causes of progressive cerebellar ataxia: Prospective evaluation of 1500 patients. J. Neurol. Neurosurg. Psychiatry 2017, 88, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Demos, M.K.; Macri, V.; Farrell, K.; Nelson, T.N.; Chapman, K.; Accili, E.; Armstrong, L. A novel KCNA1 mutation associated with global delay and persistent cerebellar dysfunction. Mov. Disord. 2009, 24, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.; Golumbek, P.; Cellini, E.; Doccini, V.; Guerrini, R.; Wallgren-Pettersson, C.; Thuresson, A.-C.; Gurnett, C.A. De novo KCNA1 variants in the PVP motif cause infantile epileptic encephalopathy and cognitive impairment similar to recurrent KCNA2 variants. Am. J. Med. Genet. A 2018, 176, 1748–1752. [Google Scholar] [CrossRef] [PubMed]
- Eunson, L.H.; Rea, R.; Zuberi, S.M.; Youroukos, S.; Panayiotopoulos, C.P.; Liguori, R.; Avoni, P.; McWilliam, R.C.; Stephenson, J.B.; Hanna, M.G.; et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann. Neurol. 2000, 48, 647–656. [Google Scholar] [CrossRef]
- Verdura, E.; Fons, C.; Schlüter, A.; Ruiz, M.; Fourcade, S.; Casasnovas, C.; Castellano, A.; Pujol, A. Complete loss of KCNA1 activity causes neonatal epileptic encephalopathy and dyskinesia. J. Med. Genet. 2020, 57, 132–137. [Google Scholar] [CrossRef]
- Lacinová, L. Voltage-dependent calcium channels. Gen. Physiol. Biophys. 2005, 24 (Suppl. 1), 1–78. [Google Scholar]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef]
- Epi4K Consortium De Novo Mutations in SLC1A2 and CACNA1A Are Important Causes of Epileptic Encephalopathies. Am. J. Hum. Genet. 2016, 99, 287–298. [CrossRef]
- Travaglini, L.; Nardella, M.; Bellacchio, E.; D’Amico, A.; Capuano, A.; Frusciante, R.; Di Capua, M.; Cusmai, R.; Barresi, S.; Morlino, S.; et al. Missense mutations of CACNA1A are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Eur. J. Paediatr. Neurol. 2017, 21, 450–456. [Google Scholar] [CrossRef]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef]
- Li, J.; You, Y.; Yue, W.; Jia, M.; Yu, H.; Lu, T.; Wu, Z.; Ruan, Y.; Wang, L.; Zhang, D. Genetic Evidence for Possible Involvement of the Calcium Channel Gene CACNA1A in Autism Pathogenesis in Chinese Han Population. PLoS ONE 2015, 10, e0142887. [Google Scholar] [CrossRef]
- Reinson, K.; Õiglane-Shlik, E.; Talvik, I.; Vaher, U.; Õunapuu, A.; Ennok, M.; Teek, R.; Pajusalu, S.; Murumets, Ü.; Tomberg, T.; et al. Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am. J. Med. Genet. A 2016, 170, 2173–2176. [Google Scholar] [CrossRef] [PubMed]
- Mantuano, E.; Romano, S.; Veneziano, L.; Gellera, C.; Castellotti, B.; Caimi, S.; Testa, D.; Estienne, M.; Zorzi, G.; Bugiani, M.; et al. Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J. Neurol. Sci. 2010, 291, 30–36. [Google Scholar] [CrossRef]
- Jiang, X.; Raju, P.K.; D’Avanzo, N.; Lachance, M.; Pepin, J.; Dubeau, F.; Mitchell, W.G.; Bello-Espinosa, L.E.; Pierson, T.M.; Minassian, B.A.; et al. Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia 2019, 60, 1881–1894. [Google Scholar] [CrossRef]
- Jen, J.; Wan, J.; Graves, M.; Yu, H.; Mock, A.F.; Coulin, C.J.; Kim, G.; Yue, Q.; Papazian, D.M.; Baloh, R.W. Loss-of-function EA2 mutations are associated with impaired neuromuscular transmission. Neurology 2001, 57, 1843–1848. [Google Scholar] [CrossRef]
- Burgess, D.L.; Jones, J.M.; Meisler, M.H.; Noebels, J.L. Mutation of the Ca2+ channel beta subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell 1997, 88, 385–392. [Google Scholar] [CrossRef]
- Coste de Bagneaux, P.; von Elsner, L.; Bierhals, T.; Campiglio, M.; Johannsen, J.; Obermair, G.J.; Hempel, M.; Flucher, B.E.; Kutsche, K. A homozygous missense variant in CACNB4 encoding the auxiliary calcium channel beta4 subunit causes a severe neurodevelopmental disorder and impairs channel and non-channel functions. PLoS Genet. 2020, 16, e1008625. [Google Scholar] [CrossRef]
- Barresi, S.; Niceta, M.; Alfieri, P.; Brankovic, V.; Piccini, G.; Bruselles, A.; Barone, M.R.; Cusmai, R.; Tartaglia, M.; Bertini, E.; et al. Mutations in the IRBIT domain of ITPR1 are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Clin. Genet. 2017, 91, 86–91. [Google Scholar] [CrossRef]
- Dentici, M.L.; Barresi, S.; Nardella, M.; Bellacchio, E.; Alfieri, P.; Bruselles, A.; Pantaleoni, F.; Danieli, A.; Iarossi, G.; Cappa, M.; et al. Identification of novel and hotspot mutations in the channel domain of ITPR1 in two patients with Gillespie syndrome. Gene 2017, 628, 141–145. [Google Scholar] [CrossRef]
- Van de Leemput, J.; Chandran, J.; Knight, M.A.; Holtzclaw, L.A.; Scholz, S.; Cookson, M.R.; Houlden, H.; Gwinn-Hardy, K.; Fung, H.-C.; Lin, X.; et al. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007, 3, e108. [Google Scholar] [CrossRef]
- Pode-Shakked, N.; Korman, S.H.; Pode-Shakked, B.; Landau, Y.; Kneller, K.; Abraham, S.; Shaag, A.; Ulanovsky, I.; Daas, S.; Saraf-Levy, T.; et al. Clues and challenges in the diagnosis of intermittent maple syrup urine disease. Eur. J. Med. Genet. 2020, 103901. [Google Scholar] [CrossRef]
- Dhawan, S.R.; Saini, A.G.; Vyas, S.; Attri, S.V. Teaching NeuroImages: When MRI is a clue in episodic ataxia. Neurology 2019, 93, e2074–e2075. [Google Scholar] [CrossRef]
- Saini, A.G.; Attri, S.; Sankhyan, N.; Singhi, P. Hypomorphic citrullinaemia due to mutated ASS1 with episodic ataxia. BMJ Case Rep. 2018, 2018. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Craig, K.; Elliott, H.R.; Keers, S.M.; Lambert, C.; Pyle, A.; Graves, T.D.; Woodward, C.; Sweeney, M.G.; Davis, M.B.; Hanna, M.G.; et al. Episodic ataxia and hemiplegia caused by the 8993T->C mitochondrial DNA mutation. J. Med. Genet. 2007, 44, 797–799. [Google Scholar] [CrossRef]
- van der Knaap, M.S.; van der Voorn, P.; Barkhof, F.; Van Coster, R.; Krägeloh-Mann, I.; Feigenbaum, A.; Blaser, S.; Vles, J.S.H.; Rieckmann, P.; Pouwels, P.J.W. A new leukoencephalopathy with brainstem and spinal cord involvement and high lactate. Ann. Neurol. 2003, 53, 252–258. [Google Scholar] [CrossRef]
- Synofzik, M.; Schicks, J.; Lindig, T.; Biskup, S.; Schmidt, T.; Hansel, J.; Lehmann-Horn, F.; Schöls, L. Acetazolamide-responsive exercise-induced episodic ataxia associated with a novel homozygous DARS2 mutation. J. Med. Genet. 2011, 48, 713–715. [Google Scholar] [CrossRef]
- Debray, F.-G.; Lambert, M.; Gagne, R.; Maranda, B.; Laframboise, R.; MacKay, N.; Robinson, B.H.; Mitchell, G.A. Pyruvate dehydrogenase deficiency presenting as intermittent isolated acute ataxia. Neuropediatrics 2008, 39, 20–23. [Google Scholar] [CrossRef]
- Di Re, J.; Wadsworth, P.A.; Laezza, F. Intracellular Fibroblast Growth Factor 14: Emerging Risk Factor for Brain Disorders. Front. Cell. Neurosci. 2017, 11, 103. [Google Scholar] [CrossRef]
- Yan, H.; Pablo, J.L.; Pitt, G.S. FGF14 regulates presynaptic Ca2+ channels and synaptic transmission. Cell Rep. 2013, 4, 66–75. [Google Scholar] [CrossRef]
- Wang, Q.; Bardgett, M.E.; Wong, M.; Wozniak, D.F.; Lou, J.; McNeil, B.D.; Chen, C.; Nardi, A.; Reid, D.C.; Yamada, K.; et al. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron 2002, 35, 25–38. [Google Scholar] [CrossRef]
- Van Swieten, J.C.; Brusse, E.; de Graaf, B.M.; Krieger, E.; van de Graaf, R.; de Koning, I.; Maat-Kievit, A.; Leegwater, P.; Dooijes, D.; Oostra, B.A.; et al. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected]. Am. J. Hum. Genet. 2003, 72, 191–199. [Google Scholar] [CrossRef]
- Groth, C.L.; Berman, B.D. Spinocerebellar Ataxia 27: A Review and Characterization of an Evolving Phenotype. Tremor Other Hyperkinet. Mov. (N Y) 2018, 8, 534. [Google Scholar] [CrossRef]
- Paucar, M.; Lundin, J.; Alshammari, T.; Bergendal, Å.; Lindefeldt, M.; Alshammari, M.; Solders, G.; Di Re, J.; Savitcheva, I.; Granberg, T.; et al. Broader phenotypic traits and widespread brain hypometabolism in spinocerebellar ataxia 27. J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Miura, S.; Kosaka, K.; Fujioka, R.; Uchiyama, Y.; Shimojo, T.; Morikawa, T.; Irie, A.; Taniwaki, T.; Shibata, H. Spinocerebellar ataxia 27 with a novel nonsense variant (Lys177X) in FGF14. Eur. J. Med. Genet. 2019, 62, 172–176. [Google Scholar] [CrossRef]
- Coebergh, J.A.; Fransen van de Putte, D.E.; Snoeck, I.N.; Ruivenkamp, C.; van Haeringen, A.; Smit, L.M. A new variable phenotype in spinocerebellar ataxia 27 (SCA 27) caused by a deletion in the FGF14 gene. Eur. J. Paediatr. Neurol. 2014, 18, 413–415. [Google Scholar] [CrossRef]
- Choquet, K.; La Piana, R.; Brais, B. A novel frameshift mutation in FGF14 causes an autosomal dominant episodic ataxia. Neurogenetics 2015, 16, 233–236. [Google Scholar] [CrossRef]
- Amado, A.; Blanco, M.O.; Repáraz-Andrade, A. Spinocerebellar Ataxia 27: Clinical Phenotype of Twin Sisters with FGF14 Deletion. Neuropediatrics 2017, 48, 131. [Google Scholar] [CrossRef]
- Schesny, M.; Joncourt, F.; Tarnutzer, A.A. Acetazolamide-Responsive Episodic Ataxia Linked to Novel Splice Site Variant in FGF14 Gene. Cerebellum 2019, 18, 649–653. [Google Scholar] [CrossRef]
- Piarroux, J.; Riant, F.; Humbertclaude, V.; Remerand, G.; Hadjadj, J.; Rejou, F.; Coubes, C.; Pinson, L.; Meyer, P.; Roubertie, A. FGF14-related episodic ataxia: Delineating the phenotype of Episodic Ataxia type 9. Ann. Clin. Transl. Neurol. 2020. [Google Scholar] [CrossRef]
- Sesh, S.; Radhakrishnan, A. Episodic ataxia in a child with senataxin mutation. Neurol. India 2018, 66, 842–844. [Google Scholar] [CrossRef]
- Valente, E.M.; Silhavy, J.L.; Brancati, F.; Barrano, G.; Krishnaswami, S.R.; Castori, M.; Lancaster, M.A.; Boltshauser, E.; Boccone, L.; Al-Gazali, L.; et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006, 38, 623–625. [Google Scholar] [CrossRef]
- Brancati, F.; Barrano, G.; Silhavy, J.L.; Marsh, S.E.; Travaglini, L.; Bielas, S.L.; Amorini, M.; Zablocka, D.; Kayserili, H.; Al-Gazali, L.; et al. CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am. J. Hum. Genet. 2007, 81, 104–113. [Google Scholar] [CrossRef]
- Hamed, M.; Shetty, A.; Dzwiniel, T.; Buller, M.; Koskinen, L.; Suchowersky, O. Episodic Ataxia Secondary to CEP290 Compound Heterozygous Mutations: A Case Report. Mov. Disord. Clin. Pract. 2020, 7, 104–106. [Google Scholar] [CrossRef]
- Bramswig, N.C.; Bertoli-Avella, A.M.; Albrecht, B.; Al Aqeel, A.I.; Alhashem, A.; Al-Sannaa, N.; Bah, M.; Bröhl, K.; Depienne, C.; Dorison, N.; et al. Genetic variants in components of the NALCN-UNC80-UNC79 ion channel complex cause a broad clinical phenotype (NALCN channelopathies). Hum. Genet. 2018, 137, 753–768. [Google Scholar] [CrossRef]
- Campbell, J.; FitzPatrick, D.R.; Azam, T.; Gibson, N.A.; Somerville, L.; Joss, S.K.; Deciphering Developmental Disorders Study; Urquhart, D.S. NALCN Dysfunction as a Cause of Disordered Respiratory Rhythm with Central Apnea. Pediatrics 2018, 141, S485–S490. [Google Scholar] [CrossRef]
- Chong, J.X.; McMillin, M.J.; Shively, K.M.; Beck, A.E.; Marvin, C.T.; Armenteros, J.R.; Buckingham, K.J.; Nkinsi, N.T.; Boyle, E.A.; Berry, M.N.; et al. De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am. J. Hum. Genet. 2015, 96, 462–473. [Google Scholar] [CrossRef]
- Wang, Y.; Koh, K.; Ichinose, Y.; Yasumura, M.; Ohtsuka, T.; Takiyama, Y. A de novo mutation in the NALCN gene in an adult patient with cerebellar ataxia associated with intellectual disability and arthrogryposis. Clin. Genet. 2016, 90, 556–557. [Google Scholar] [CrossRef]
- Aoyagi, K.; Rossignol, E.; Hamdan, F.F.; Mulcahy, B.; Xie, L.; Nagamatsu, S.; Rouleau, G.A.; Zhen, M.; Michaud, J.L. A Gain-of-Function Mutation in NALCN in a Child with Intellectual Disability, Ataxia, and Arthrogryposis. Hum. Mutat. 2015, 36, 753–757. [Google Scholar] [CrossRef]
- Brew, H.M.; Gittelman, J.X.; Silverstein, R.S.; Hanks, T.D.; Demas, V.P.; Robinson, L.C.; Robbins, C.A.; McKee-Johnson, J.; Chiu, S.Y.; Messing, A.; et al. Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J. Neurophysiol. 2007, 98, 1501–1525. [Google Scholar] [CrossRef] [PubMed]
- Syrbe, S.; Hedrich, U.B.S.; Riesch, E.; Djémié, T.; Müller, S.; Møller, R.S.; Maher, B.; Hernandez-Hernandez, L.; Synofzik, M.; Caglayan, H.S.; et al. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 2015, 47, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.L.; Hedrich, U.B.S.; Shinde, D.N.; Krey, I.; Teichmann, A.-C.; Hentschel, J.; Schubert, J.; Chamberlin, A.C.; Huether, R.; Lu, H.-M.; et al. A recurrent mutation in KCNA2 as a novel cause of hereditary spastic paraplegia and ataxia. Ann. Neurol. 2016, 80. [Google Scholar] [CrossRef]
- Canafoglia, L.; Castellotti, B.; Ragona, F.; Freri, E.; Granata, T.; Chiapparini, L.; Gellera, C.; Scaioli, V.; Franceschetti, S.; DiFrancesco, J.C. Progressive myoclonus epilepsy caused by a gain-of-function KCNA2 mutation. Seizure 2019, 65, 106–108. [Google Scholar] [CrossRef]
- Corbett, M.A.; Bellows, S.T.; Li, M.; Carroll, R.; Micallef, S.; Carvill, G.L.; Myers, C.T.; Howell, K.B.; Maljevic, S.; Lerche, H.; et al. Dominant KCNA2 mutation causes episodic ataxia and pharmacoresponsive epilepsy. Neurology 2016, 87, 1975–1984. [Google Scholar] [CrossRef]
- Wolff, M.; Brunklaus, A.; Zuberi, S.M. Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia 2019, 60 (Suppl. 3), S59–S67. [Google Scholar] [CrossRef]
- Schwarz, N.; Bast, T.; Gaily, E.; Golla, G.; Gorman, K.M.; Griffiths, L.R.; Hahn, A.; Hukin, J.; King, M.; Korff, C.; et al. Clinical and genetic spectrum of SCN2A-associated episodic ataxia. Eur. J. Paediatr. Neurol. 2019, 23, 438–447. [Google Scholar] [CrossRef]
- Nobile, C.; Striano, P. PRRT2: A major cause of infantile epilepsy and other paroxysmal disorders of childhood. Prog. Brain Res. 2014, 213, 141–158. [Google Scholar] [CrossRef]
- Legris, N.; Chassin, O.; Nasser, G.; Riant, F.; Tournier-Lasserve, E.; Denier, C. Acute-Onset Ataxia and Transient Cerebellar Diffusion Restriction Associated with a PRRT2 Mutation. J. Stroke Cerebrovasc. Dis. 2019, 28, e3–e4. [Google Scholar] [CrossRef]
- Blakeley, J.; Jankovic, J. Secondary causes of paroxysmal dyskinesia. Adv. Neurol. 2002, 89, 401–420. [Google Scholar]
- Ciampi, E.; Uribe-San-Martín, R.; Godoy-Santín, J.; Cruz, J.P.; Cárcamo-Rodríguez, C.; Juri, C. Secondary paroxysmal dyskinesia in multiple sclerosis: Clinical-radiological features and treatment. Case report of seven patients. Mult. Scler. 2017, 23, 1791–1795. [Google Scholar] [CrossRef]
- Fröhlich, K.; Winder, K.; Linker, R.A.; Huhn, K.; Engelhorn, T.; Dörfler, A.; Lee, D.-H.; Schwab, S.; Seifert, F. Lesion correlates of secondary paroxysmal dyskinesia in multiple sclerosis. J. Neurol. 2018, 265, 2277–2283. [Google Scholar] [CrossRef]
- Pop, R.; Kipfer, S. Paroxysmal kinesigenic dyskinesia-like phenotype in multiple sclerosis. Mult. Scler. 2017, 23, 1795–1797. [Google Scholar] [CrossRef]
- Ostermann, P.O.; Westerberg, C.E. Paroxysmal attacks in multiple sclerosis. Brain 1975, 98, 189–202. [Google Scholar] [CrossRef]
- Pareés, I. Clinical commentary on “Paroxysmal kinesigenic dyskinesia-like phenotype in multiple sclerosis” and “Secondary paroxysmal dyskinesia in multiple sclerosis: Clinical-radiological features and treatment. Case report of seven patients”. Mult. Scler. 2017, 23, 1797–1798. [Google Scholar] [CrossRef]
- Mehanna, R.; Jankovic, J. Movement disorders in multiple sclerosis and other demyelinating diseases. J. Neurol. Sci. 2013, 328, 1–8. [Google Scholar] [CrossRef]
- Carecchio, M.; Massano, J.; Bhatia, K.P. Paroxysmal Dyskinesias. In Hyperkinetic Movement Disorders; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012; pp. 363–374. ISBN 978-1-4443-4618-3. [Google Scholar]
- Krogias, C.; Hoepner, R.; Müller, A.; Schneider-Gold, C.; Schröder, A.; Gold, R. Successful treatment of anti-Caspr2 syndrome by interleukin 6 receptor blockade through tocilizumab. JAMA Neurol. 2013, 70, 1056–1059. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Irani, S.R.; Cheeran, B.J.; Baker, I.W.S.; Cader, Z.M.; Vincent, A. Immunotherapy-responsive chorea as the presenting feature of LGI1-antibody encephalitis. Neurology 2012, 79, 195–196. [Google Scholar] [CrossRef]
- Chaudhry, N.; Puri, V.; Patidar, Y.; Khwaja, G.A. Pathological laughter associated with paroxysmal kinesigenic dyskinesia: A rare presentation of acute disseminated encephalomyelitis. Epilepsy Behav. Case Rep. 2013, 1, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Tijssen, M.A.J. Paroxysmal non-kinesigenic dyskinesia in antiphospholipid syndrome. Mov. Disord. 2005, 20, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, N.G.; De Marzi, R.; Zangaglia, R.; Minafra, B.; Pacchetti, C. Paroxysmal Dystonia with Axonal Neuropathy Resulting from Benignant Insulinoma: Case Report. Mov. Disord. Clin. Pract. 2015, 2, 69–71. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, C.; Zheng, H.; Yang, L.; Hu, Z. Chorea-ballism associated with ketotic hyperglycemia. Neurol. Sci. 2014, 35, 1851–1855. [Google Scholar] [CrossRef] [PubMed]
- Dure, L.S.; Mussell, H.G. Paroxysmal dyskinesia in a patient with pseudohypoparathyroidism. Mov. Disord. 1998, 13, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.-J.; Jung, J.-M.; Choi, J.-Y.; Kwon, D.-Y. Paroxysmal kinesigenic dyskinesia in pseudohypoparathyroidism: Is basal ganglia calcification a necessary finding? J. Neurol. Sci. 2015, 357, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Tabaee-Zadeh, M.J.; Frame, B.; Kapphahn, K. Kinesiogenic choreoathetosis and idiopathic hypoparathyroidism. N. Engl. J. Med. 1972, 286, 762–763. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alegre, P.; Ammache, Z.; Davis, P.H.; Rodnitzky, R.L. Moyamoya-induced paroxysmal dyskinesia. Mov. Disord. 2003, 18, 1051–1056. [Google Scholar] [CrossRef]
- Parker, H.L. Periodic ataxia. Collect Pap. Mayo Clin. Mayo Found 1946, 38, 642–645. [Google Scholar]
- Andermann, F.; Cosgrove, J.B.; Lloyd-Smith, D.; Walters, A.M. Paroxysmal dysarthria and ataxia in multiple sclerosis; a report of 2 unusual cases. Neurology 1959, 9, 211–215. [Google Scholar] [CrossRef]
- Marcel, C.; Anheim, M.; Flamand-Rouvière, C.; Heran, F.; Masnou, P.; Boulay, C.; Mari, I.; Tranchant, C.; Roze, E. Symptomatic paroxysmal dysarthria-ataxia in demyelinating diseases. J. Neurol. 2010, 257, 1369–1372. [Google Scholar] [CrossRef]
- Klaas, J.P.; Burkholder, D.B.; Singer, W.; Boes, C.J. Harry Lee Parker and paroxysmal dysarthria and ataxia. Neurology 2013, 80, 311–314. [Google Scholar] [CrossRef]
- Rossi, S.; Studer, V.; Motta, C.; Centonze, D. Paroxysmal dysarthria-ataxia syndrome resolving after fingolimod treatment. J. Neurol. Sci. 2015, 350, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zeng, C.; Luo, T. Paroxysmal dysarthria and ataxia in multiple sclerosis and corresponding magnetic resonance imaging findings. J. Neurol. 2011, 258, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Piffer, S.; Turri, G.; Acler, M.; Richelli, S.; Cerini, R.; Fiaschi, A.; Monaco, S.; Bonetti, B. Paroxysmal dysarthria-ataxia in remitting-relapsing Bickerstaff’s-like encephalitis. J. Neurol. Sci. 2014, 341, 85–87. [Google Scholar] [CrossRef]
- Matsui, M.; Tomimoto, H.; Sano, K.; Hashikawa, K.; Fukuyama, H.; Shibasaki, H. Paroxysmal dysarthria and ataxia after midbrain infarction. Neurology 2004, 63, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Akman-Demir, F.G.; Eraksoy, M.; Gürvit, I.H.; Saruhan-Direskeneli, G.; Aral, O. Paroxysmal dysarthria and ataxia in a patient with Behçet’s disease. J. Neurol. 1995, 242, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, E.; Marazzi, M.R.; Trimboli, M.; Dalla Costa, D.; Erminio, C.; Nobili, L. Brainstem lesion causing paroxysmal ataxia, dysarthria, diplopia and hemifacial spasm (PADDHS). Epileptic Disord. 2019, 21, 389–390. [Google Scholar] [CrossRef]
- López Chiriboga, A.S.; Pittock, S. Episodic ataxia in CASPR2 autoimmunity. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e536. [Google Scholar] [CrossRef]
- Joubert, B.; Gobert, F.; Thomas, L.; Saint-Martin, M.; Desestret, V.; Convers, P.; Rogemond, V.; Picard, G.; Ducray, F.; Psimaras, D.; et al. Autoimmune episodic ataxia in patients with anti-CASPR2 antibody-associated encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e371. [Google Scholar] [CrossRef]
- Langer, J.E.; Lopes, M.B.S.; Fountain, N.B.; Pranzatelli, M.R.; Thiele, E.A.; Rust, R.S.; Goodkin, H.P. An unusual presentation of anti-Hu-associated paraneoplastic limbic encephalitis. Dev. Med. Child Neurol. 2012, 54, 863–866. [Google Scholar] [CrossRef]






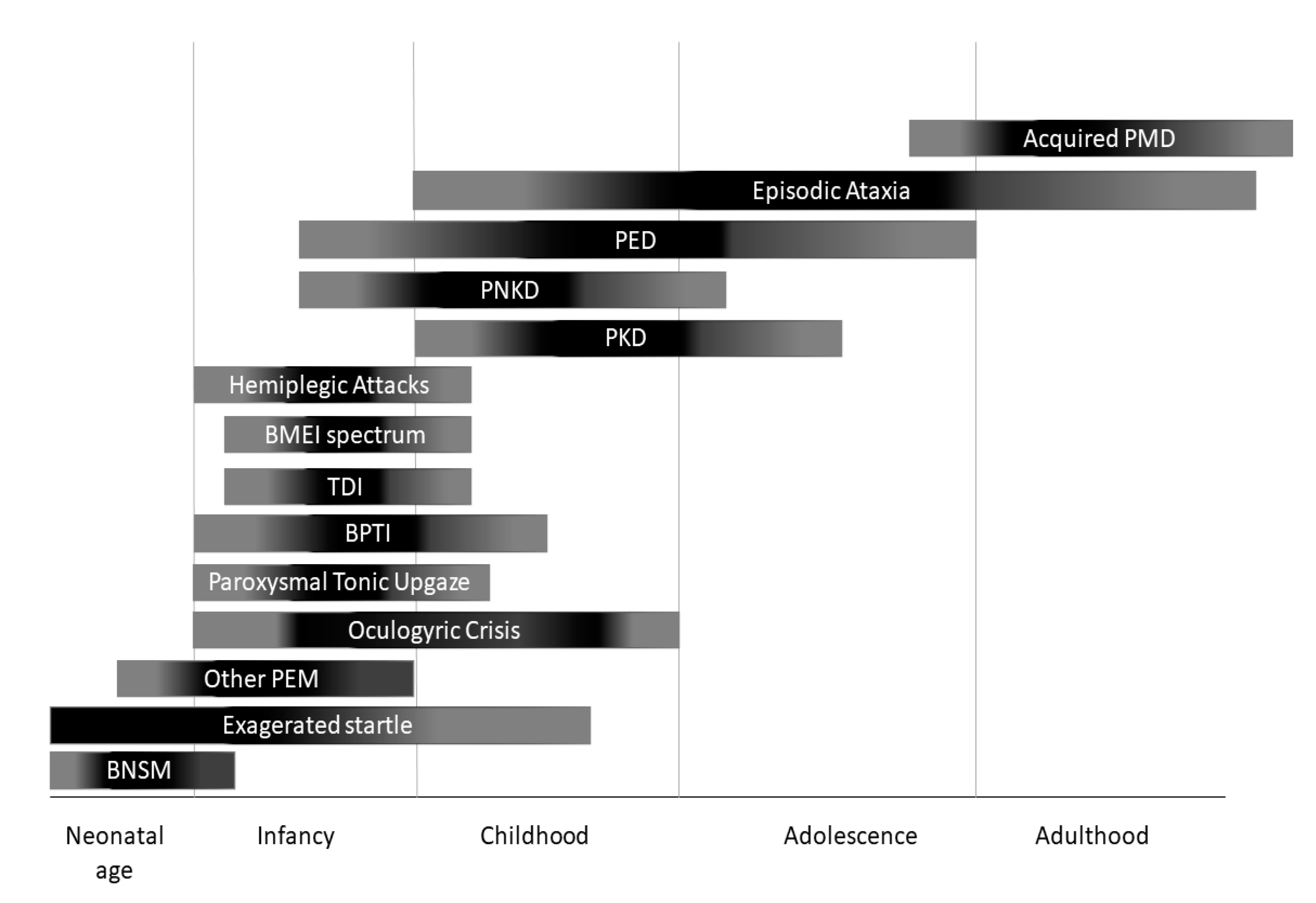{kind=link}
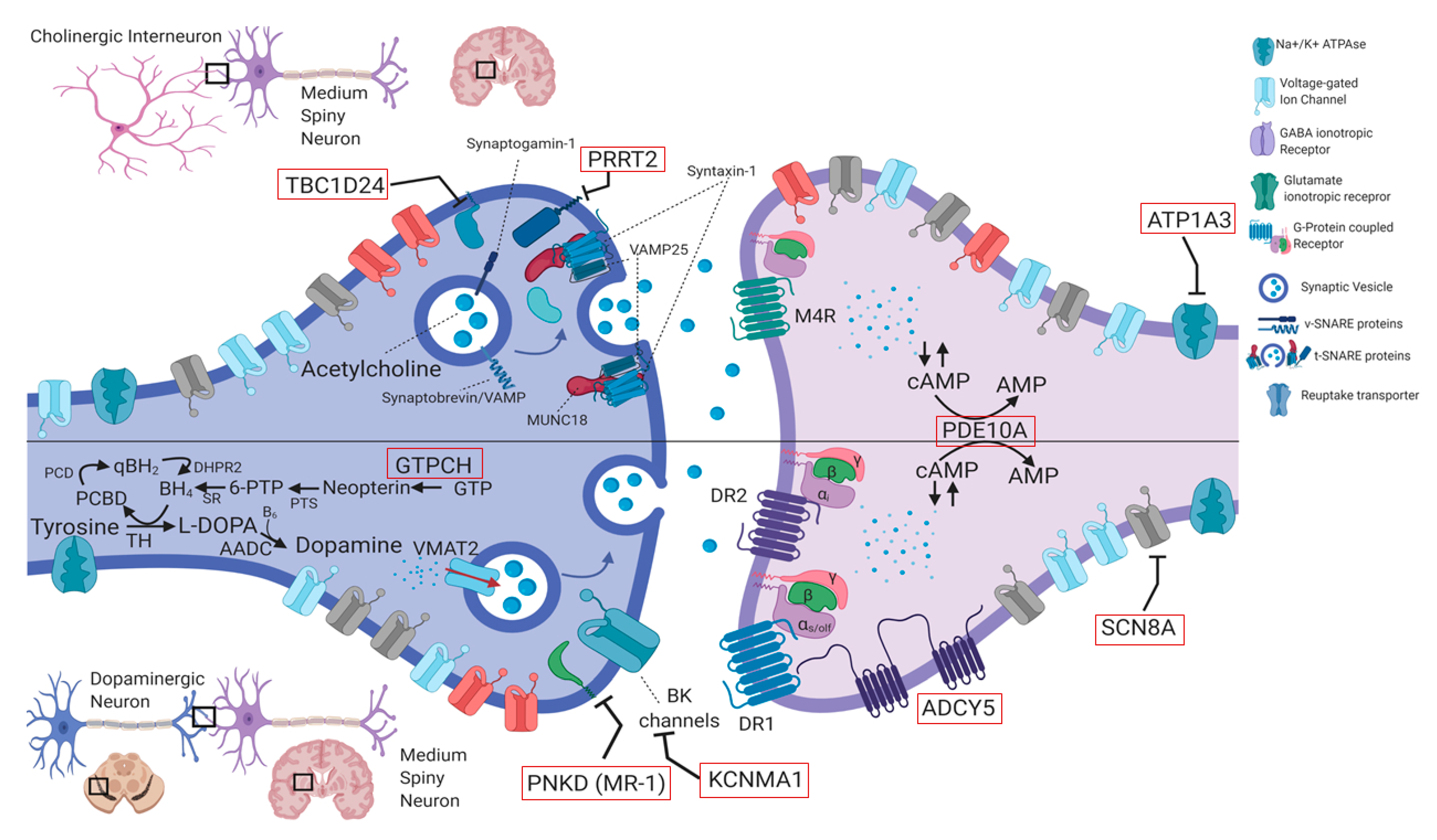{kind=link}
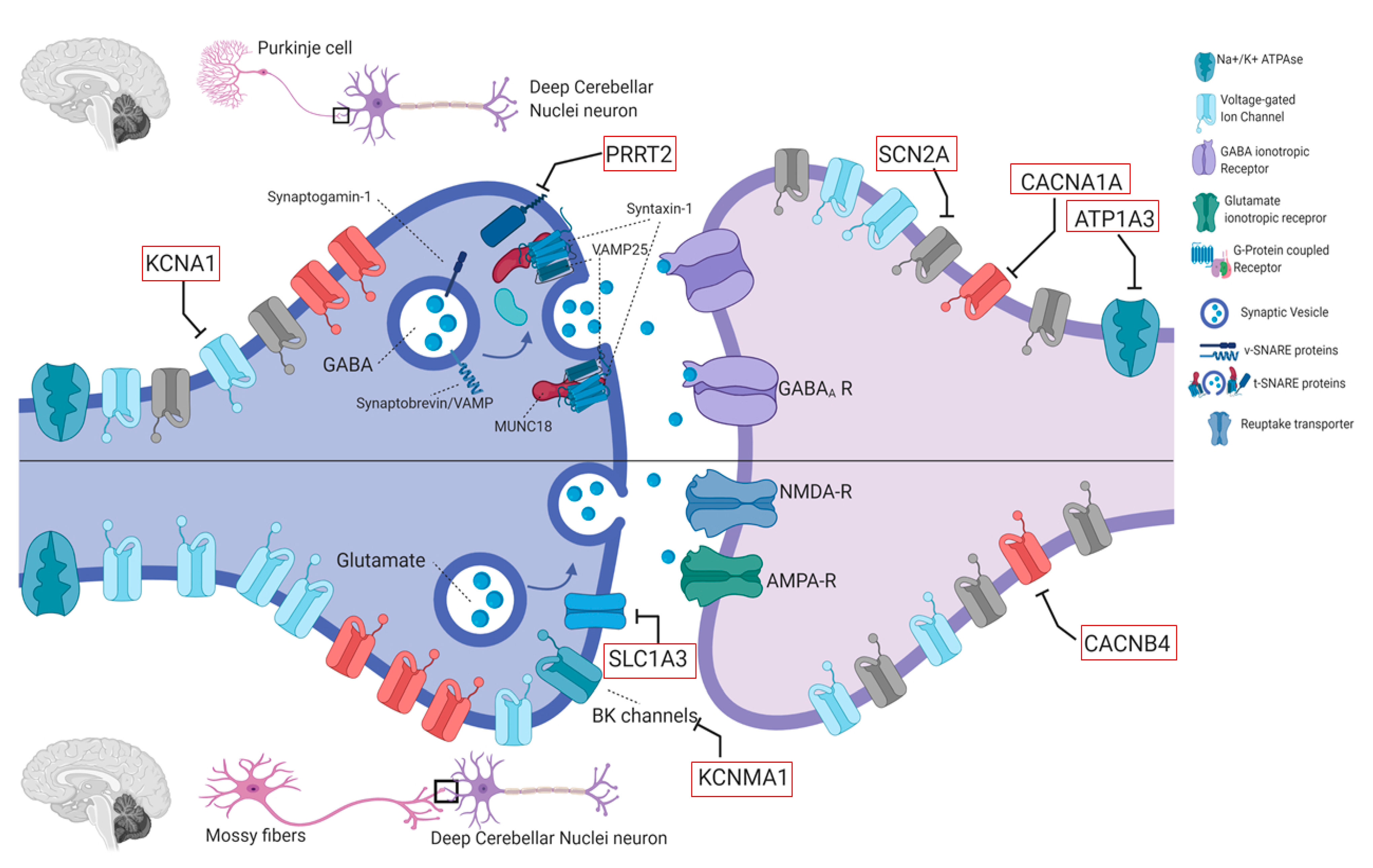{kind=link}
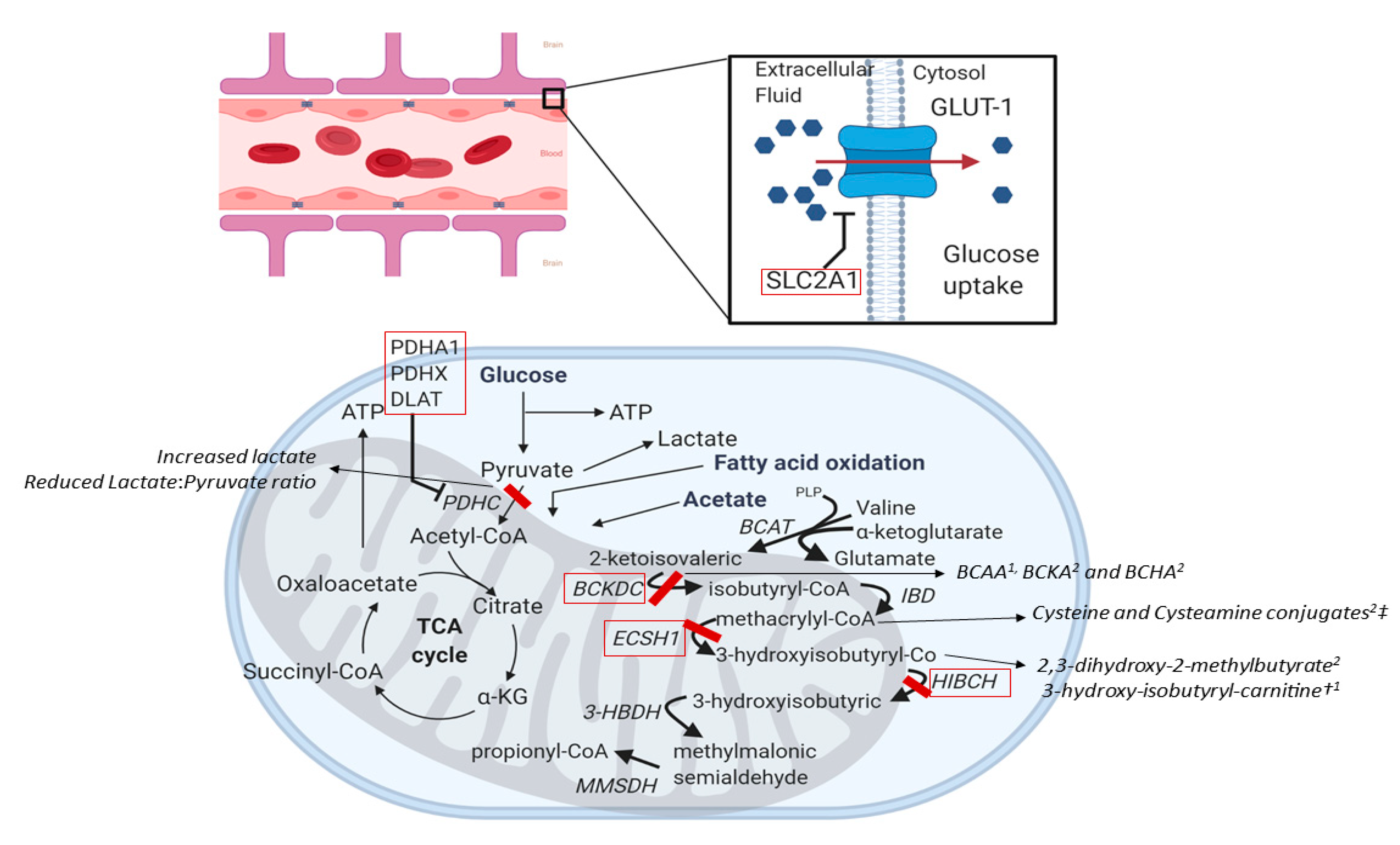{kind=link}
{kind=link}
| Gene | OMIM | Inheritance | Age at Onset | PMDs Subtype | Attack Duration | Isolated Versus Combined | Allelic Disorders | Other Possible Features | MRI | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|
| PRRT2 | 614386 | AD | <18 years | PKD | Very brief (<1 min) | I/C | BFIS, ICCA, FHM, EA | Normal | CBZ (PKD) ACZM (EA) | |
| PNKD | 609023 | AD | <18 years | PNKD | Long (>1 h) | I | Migraine (rare), PKD | BDZ (Attacks relief) | ||
| SLC2A1 (GLUT-1) | 138140 | AD | Variable | PED, EA, HA, PEM | Intermediate (5–40 min) | I/C | Classic GLUT1-DS, HSP, | Anaemia, hypotonia, spasticity, seizures, Developmental delay/ID, dystonia, ataxia | Ketogenic diet, triheptanoin | |
| PDH complex (PDHA1/PDHX /DLAT) | 300502/608769/608770 | AR | Infancy | PED/PNKD | Variable | I/C | Leigh syndrome | Developmantal delay/ID, Seizures, progressive dystonia | Pallidal hyperintensities, Callosal agenesis | Ketogenic diet |
| ECHS1 | 602292 | AR | Infancy | PED | Variable | I/C | Leigh syndrome | From Pallidal hyperintensities to Leigh-like abnormalities | Valine-restricted diet? detoxifying drugs? | |
| HIBCH | 610690 | AR | Infancy | PED | Variable | I/C | Leigh syndrome | ID, Seizures, progressive dystonia | From Pallidal hyperintensities to Leigh-like abnormalities | Valine-restricted diet? detoxifying drugs? |
| ATP1A3 | 182350 | AD | Variable | PNKD ([hemi] dystonic attacks), HA, PEM | Variable | C | EIEE, AHC, CAPOS, RECA, RDP | Seizures, dysautonomic paroxysms, nonparoxysmal dystonia, ataxia, parkinsonism | Flunarizine (HA prophylaxis), BDZ (HA relief) | |
| ADCY5 | 600293 | AD | Variable | PKD/PNKD/PED/PND | Brief (minutes) | C | PNKD | Axial hypotonia, nonparoxysmal dystonia and chorea | Caffeine? | |
| TBC1D24 | 613577 | AR | Childhood | PED | Variable | C | Deafness, DOORS syndrome, Rolandic Epilepsy, EIEE16, Myoclonic epilepsy | Sizures, Developmental delay/ID, myoclonus, ataxia, extraneurological abnormalities | ||
| SLC16A2 (MCT8) | 300095 | X linked | <1–2 months | PKD (triggered by passive movements) | Very brief (seconds to minutes) | C | Mental retardation | TRIAC? | ||
| SCN8A | 600702 | AD | Infancy | PKD | Brief | C | Epilepsy | Mental retardation | CBZ, oxcarbazepine | |
| KCNMA1 | 600150 | AD | Childhood | PNKD | Long (>1 h) | C | epilepsy, developmental delay, progressive HSP, ataxia | |||
| GCH1 | 600225 | AD | <18 years | PED | Variable | I/C | DRD | Non paroxysmal dystonia and parkinsonism | L-DOPA | |
| PDE10A | 610652 | AR/AD | Childhood | PNKD | NR | C | Chorea without paroxysms | Dystonia, Parkinsonism, marked fluctuations | Striatal hyperintensities (in AD cases) | |
| KCNA1 | 176260 | AD | Childhood (2–15) | EA1 | Minutes | I | EIEE, PKD, EDE (AR) | interictal Myokymia; progressive ataxia (20%), epilepsy (10%) | Normal ore cerebellar atrophy (10%) | CBZ, PHT, ACZM |
| CACNA1A | 601011 | AD | Childhood (0–20) | EA2/PTU/BPT | Variable (minutes to days) | I/C | FHM1, SCA6, CA | progressive ataxia, Developmental delay | Normal or cerebellar atrophy | ACZM, 4-APD, LEV |
| CACNB4 | 601949 | AD | Young-adult onset | EA5 | several hours | I | JME, IGE, CND (AR) | Epilepsy, permanent ataxia | Normal | ACZM |
| SLC1A3 (EAAT1) | 600111 | AD | infancy or childhood (rarely adulthood) | EA6 | several hours | I | Adult-onset progressive ataxia | Seizures (rare) HA | Nornmal; rarely cerebellar atrophy | ACZM |
| UBR4 | 609890 | AD | around age 2 years | EA8 | minutes to hours | I | nystagmus, myokymia, tremor | Clonazepam | ||
| FGF14 | 601515 | AD | late-childhood to early adulthood | EA9 | minutes | I/C | SCA27, CA | progressive ataxia, nystagmus, postural upper limb tremor, ID | ||
| BCKD Complex | 608348/248611 | AR | Variable | EA/PNKD | Minutes to hours | C | Classic MSUD | developmental delay, progressive psychomotor retardation, seizures, ataxia, | T2 hypersignal in in the brainstem, globus pallidus, thalami, and dentate nuclei | BCAA restricted diet |
| KCNA2 | 176262 | AD | Infancy or childhood | EA | Seconds to hours | C | EIEE32, SCA, PME | Epilepsy | ACZM (variable) | |
| SCN2A | 182390 | AD | infancy or childhood | EA | minutes to days | C | EIEE11, BFIS3 | Seizures +/− encephalopathy, developmental delay/ID | Normal or cerebellar atrophy | ACZM (variable) |
| PMD Type | Clinial Criteria | Main Genetic Causes | Rare Genetic Causes | Acquired Causes |
|---|---|---|---|---|
| Paroxysmal Dyskinesias | ||||
| Paroxysmal kynesigenic dyskinesia | Onset between 1–20y Triggered by sudden movements (kinesigenic) Duration of attacks < 1 min Good response to antiepileptic drugs | PRRT2 | SCN8A, SLC16A2, SLC2A1, DEPDC5, CLCN2, PNKD, KCNMA1, KCNA1, CHRNA4, SACS, Wilson disease, Basal Ganglia Calcifications (SLC20A2, PDGFB) | Multiple sclerosis & other demyelinating diseases, Stroke (including vasculopathy), Autoimmune encephalopathies (antiNMDAr, CASPR2, VGKC, Hashimoto), Perinatal brain injury, CNS infections, Thyrotoxicosis, PSP, central pontine myelinolysis, Methylphenidate treatment, Functional Disorders |
| Paroxysmal non-kynesigenic dyskinesia | Onset of attack in infancy or early childhood Triggered by caffeine and alcohol intake Duration of attacks between 10 min–1 h (<4 h) Often responds to benzodiazepines | PNKD | PRRT2, SLC2A1, ATP1A3, ADCY5, TBC1D24, KCNMA1, PDE10A, KCNA1, BCKD complex, Neuroacanthocytosis, Lesch-Nyhan disease, GABA-Transaminase deficiency, Basal Ganglia Calcifications (SLC20A2, PDGFB) | Multiple sclerosis & other demyelinating diseases, Stroke (including vasculopathy), TIA, Hypo-/hyperglycemia, Systemic autoimmune disorders, Celiac disease, Perinatal brain injury, CNS infections, Parasagittal meningioma, Trauma, Functional Disorders |
| Paroxysmal exercise-induced dyskinesia | Onset of attack from childhood to adulthood Triggered by exercise (at least minutes of exercise) Duration of attacks between 5 min–30 min Treatment according with the underlying defect | SLC2A1 | PDH complex (PDHA1/PDHX/DLAT), HIBCH, ECSH1, GCH1, PARK2, ADCY5, TBC1D24 | |
| Paroxysmal nocturnal dyskinesia | Paroxysmal Bouts of non-epileptic dyskinesias occurring in sleep | ADCY5 | PRRT2 | Stroke, autoimmune encephalitis (NMDAr, IgLON5) |
| Other Paroxysmal Movement Disorders | ||||
| Benign Paroxysmal Toricollis of Infancy * | recurrent episodes of painless paroxysmal cervical dystonia (featuring latero-, retro- or torticollis) Onset 3–30 months Remission before 5y | CACNA1A | ||
| Hemiplegic attacks | Onset within 18 months episodes of hemiplegia, alternating in laterality Possible quadriplegic attacks (in isolation or as generalization of HA) emotional or environmental trigger factors | ATP1A3 | ATP1A2, SLC2A1, SCN4A, ADCY5, TBC1D24, TANGO2 | |
| Hyperkplexia | excessive startling to unexpected, auditory or tactile stimuli | GLRA1, GLRB, SLC6A5, ATAD1 | GM1 gangliosidosis, SCN8A, Pontocerebellar hypoplasia, Posterior fossa malformations | Perinatal injury, Postanoxic encephalopathy, trauma, Paraneoplastic, Multiple sclerosis, ALS, CNS Infections, Medulla compression, MSA |
| Paroxysmal abnormal eye movements | ||||
| Paroxysmal Tonic Upgaze | paroxysms of conjugate upward gaze down-beating saccades on attempts to downward gaze preserved horizontal eye movements unimpaired consciousness episode lasting hours (up to 48 h) | CACNA1A * | GRID2, Pelizaeus-Merzbacher Disease, Brain Malformations | Perinatal injury, hydrocephalus, brain tumors |
| Oculogyric Crisis | paroxysmal, tonic, conjugate, often upward ocular deviation lasting minutes to hours | Biogenic amine metabolism defects (GTPCH, TH, SPR, AADC, PTS, VMAT2, DAT) | NOTCH2NLC (NIID), DCTN1 (Perry syndrome), LYST (Chediak-Higashi), ATP13A2 (Kufor-Rakeb disease), ATP1A3 (RDP) Rett syndrome | Dopamine-receptor blocking agents, encephalitis Lethargica, Anti-NMDAr encephalitis |
| Saccadic eye-head gaze shifts | brief episodes of eye–head movements | SLC2A1 (GLUT1-DS) | ||
| Paroxysmal nystagmus | paroxysmal episodes of monocular and binocular nystagmus, often disconjugate | ATP1A3 | ||
| Episodic Ataxias | ||||
| Episodic ataxia | Onset in childhood or adolescence (genetic EA) or adulthood (acquired EA) Attacks of ataxia, dysarthria, nystagmus lasting seconds (acquired EA), minutes (EA1) or hours to days (EA2) | KCNA1, CACNA1A | CACNB4, CEP290, FGF14, KCNA2, NALCN, PRRT2, SLC1A3, SLC2A1, UBR4, SCN2A, few neurometabolic and mithocondrial disorders | Multiple sclerosis & other demyelinating diseases, Stroke and Behcet’s Disease (brainstem and red nuclei involvement); Paraneoplastic limbic encephalitis with anti-CASPR2 or anti-Hu antibodies |
| Type of DPMDs | Suggested Investigations |
|---|---|
| Benign neonatal sleep myoclonus | No investigation required |
| Paroxysmal Tonic Upgaze | Perform EEG to differentiate from versive seizures If interictal neurological abnormalities are present (developmental delay, hypotonus, hypokinesia, dystonia, nystagmus), consider CSF sampling for neurotransmitter analysis (to differentiate from OGC) and MRI (to exclude brain abnormalities) Consider CACNA1A testing |
| Benign Paroxysmal Toricollis of Infancy | Consider CACNA1A testing |
| Transient dystonia of infancy | Perform MRI to exclude focal lesions |
| Benign polymorphous movement disorders of infancy * | No investigation usually required Consider EEG if manifestations are difficult to interpret |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garone, G.; Capuano, A.; Travaglini, L.; Graziola, F.; Stregapede, F.; Zanni, G.; Vigevano, F.; Bertini, E.; Nicita, F. Clinical and Genetic Overview of Paroxysmal Movement Disorders and Episodic Ataxias. Int. J. Mol. Sci. 2020, 21, 3603. https://doi.org/10.3390/ijms21103603
Garone G, Capuano A, Travaglini L, Graziola F, Stregapede F, Zanni G, Vigevano F, Bertini E, Nicita F. Clinical and Genetic Overview of Paroxysmal Movement Disorders and Episodic Ataxias. International Journal of Molecular Sciences. 2020; 21(10):3603. https://doi.org/10.3390/ijms21103603
Chicago/Turabian StyleGarone, Giacomo, Alessandro Capuano, Lorena Travaglini, Federica Graziola, Fabrizia Stregapede, Ginevra Zanni, Federico Vigevano, Enrico Bertini, and Francesco Nicita. 2020. "Clinical and Genetic Overview of Paroxysmal Movement Disorders and Episodic Ataxias" International Journal of Molecular Sciences 21, no. 10: 3603. https://doi.org/10.3390/ijms21103603
APA StyleGarone, G., Capuano, A., Travaglini, L., Graziola, F., Stregapede, F., Zanni, G., Vigevano, F., Bertini, E., & Nicita, F. (2020). Clinical and Genetic Overview of Paroxysmal Movement Disorders and Episodic Ataxias. International Journal of Molecular Sciences, 21(10), 3603. https://doi.org/10.3390/ijms21103603