Implementing the CRISPR/Cas9 Technology in Eucalyptus Hairy Roots Using Wood-Related Genes

 , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Results
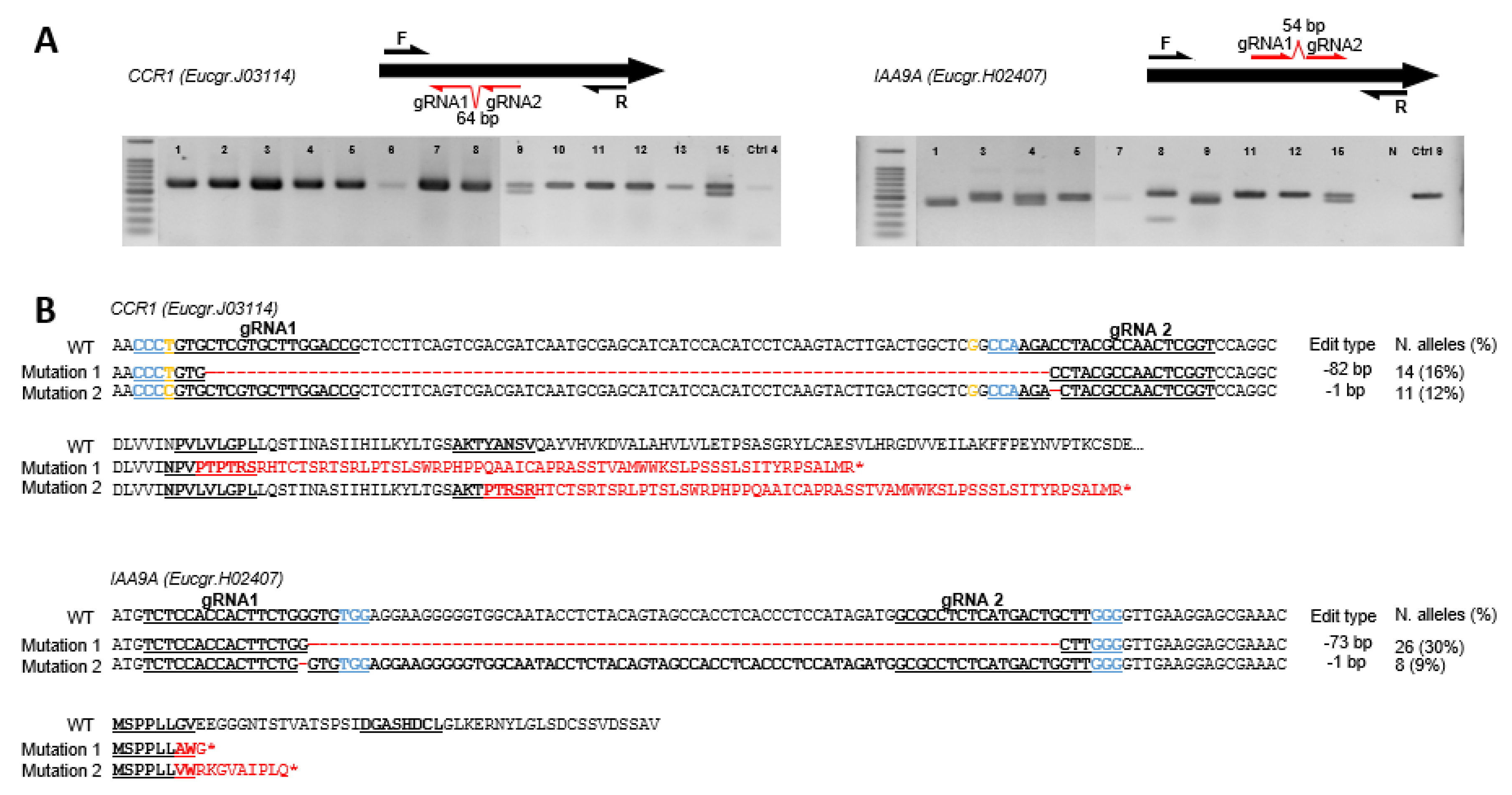
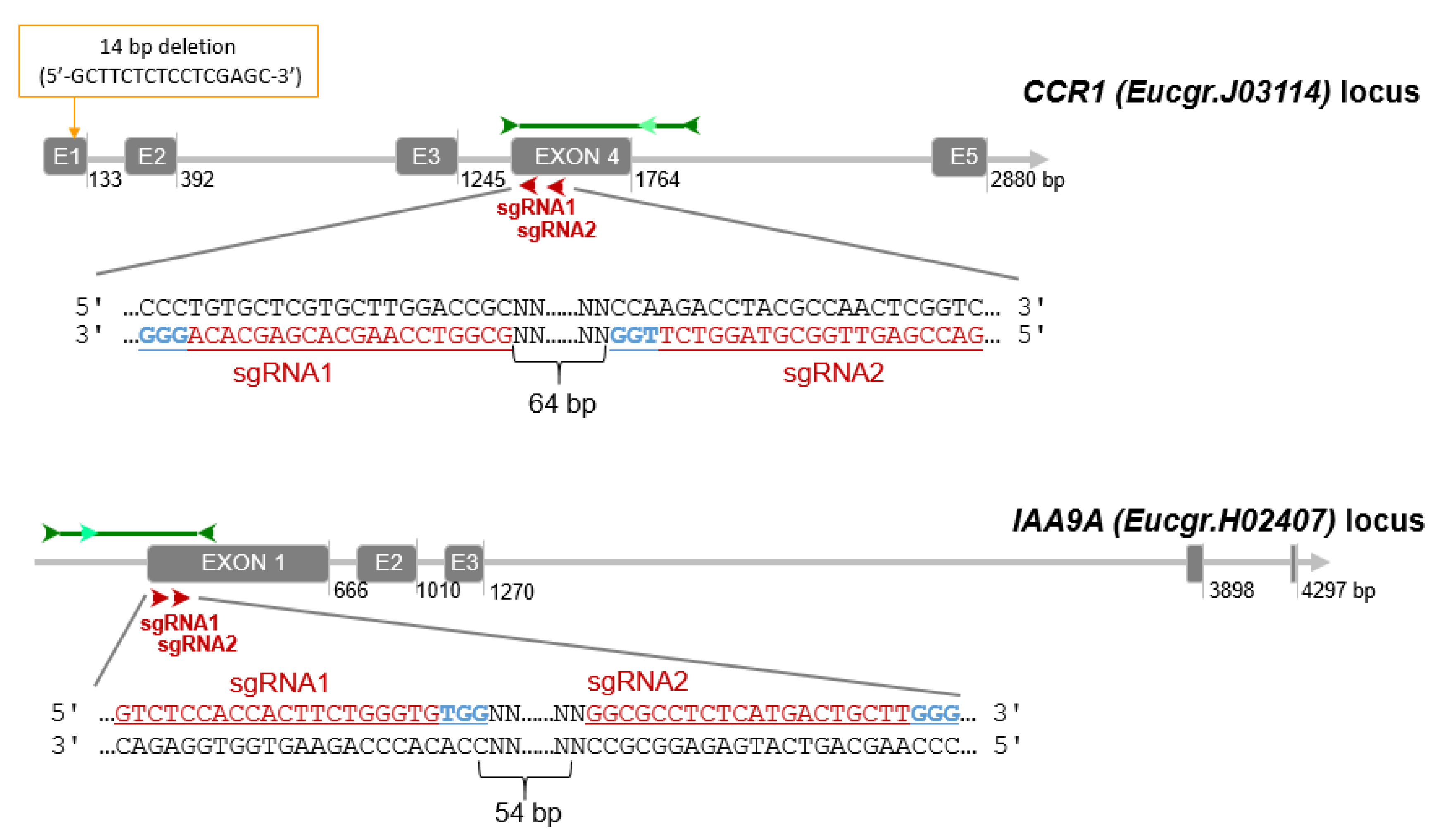
2.1. Genotyping Revealed High Knock-Down Rate in CCR1 but High Knock-out Rate in IAA9A
2.1.1. High Knock-Down Rate for CCR1 Editing
2.1.2. High Knock-Out Rate in IAA9A Lines
2.1.3. Mutation Spectra Vary Among sgRNA Targets
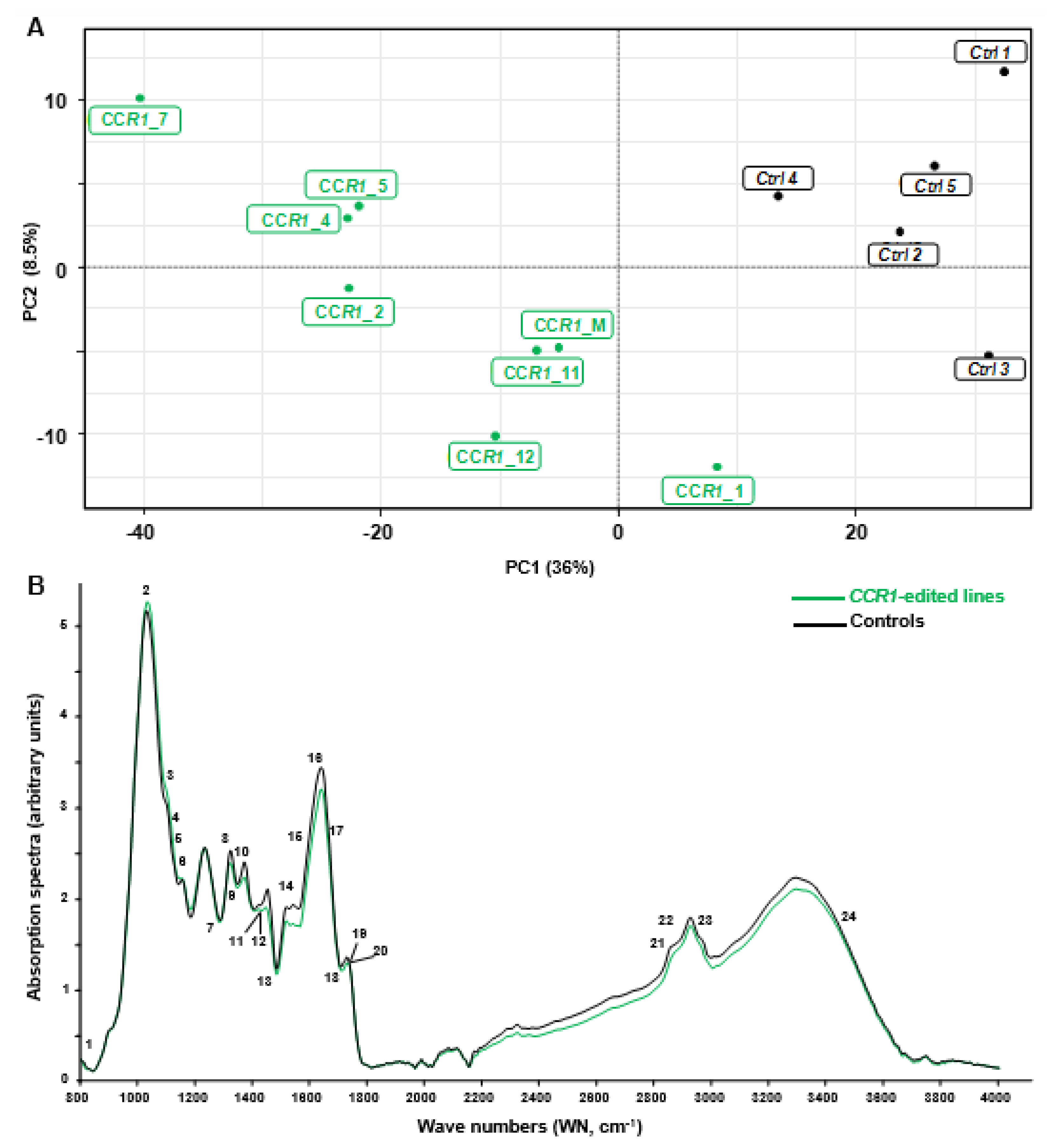
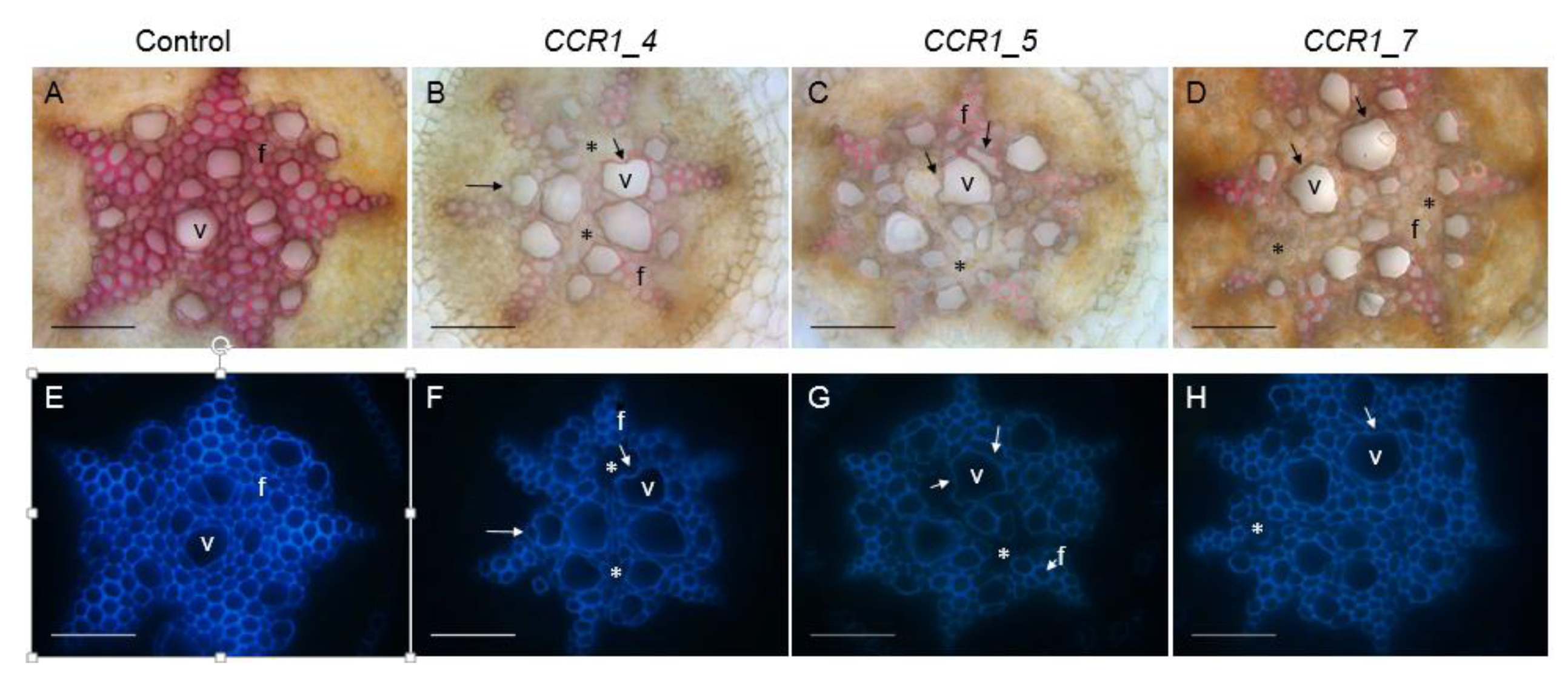
2.2. Phenotyping Revealed Expected Alterations of Lignification in CCR1-Edited Lines
2.2.1. Combination of FTIR Spectroscopy and Multivariate Analyses of CCR1-Edited Hairy Root Lines
2.2.2. Histochemical Characterization of CCR1-Edited Hairy Root Lines
3. Discussion
4. Materials and Methods
4.1. Plant Material
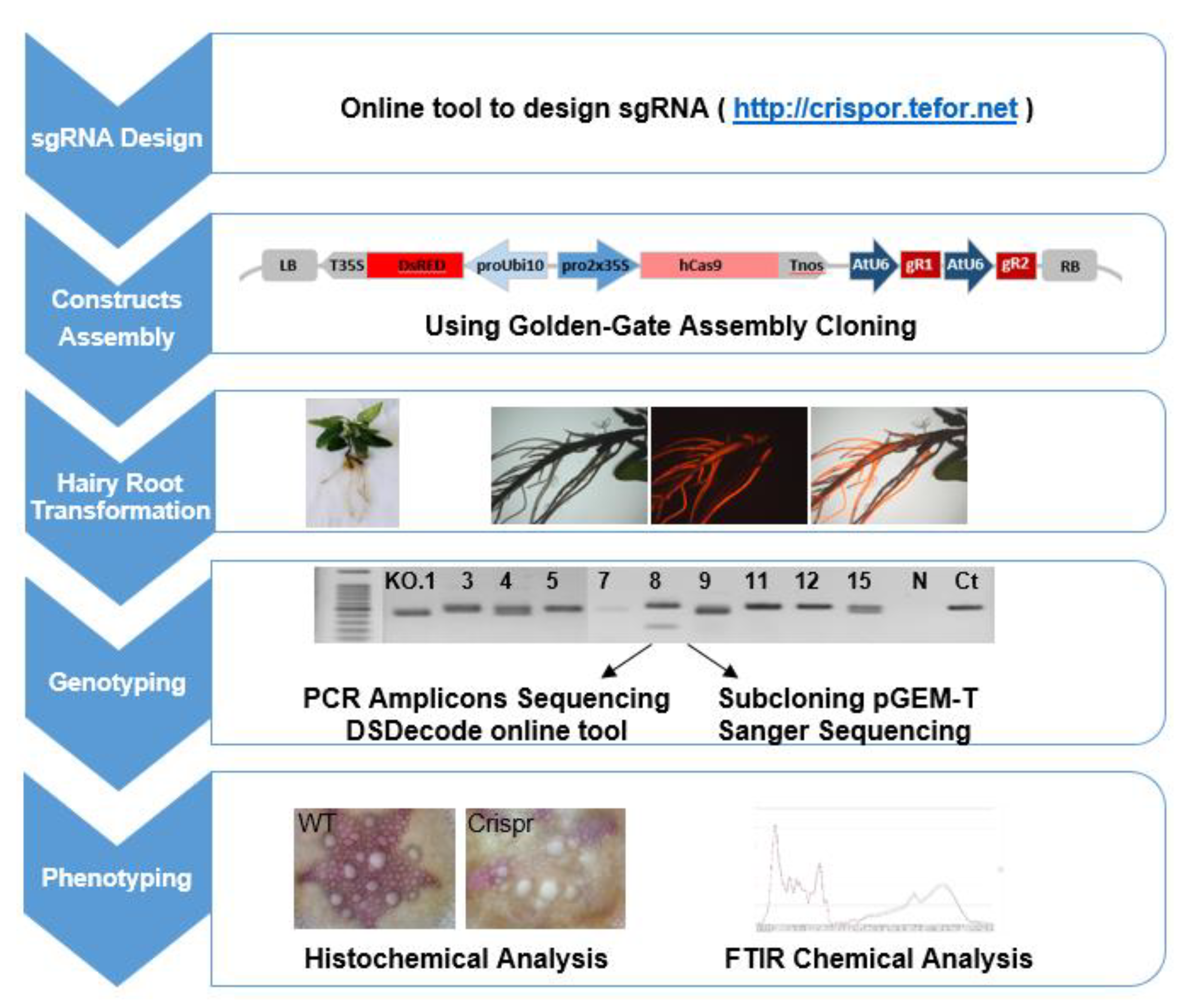
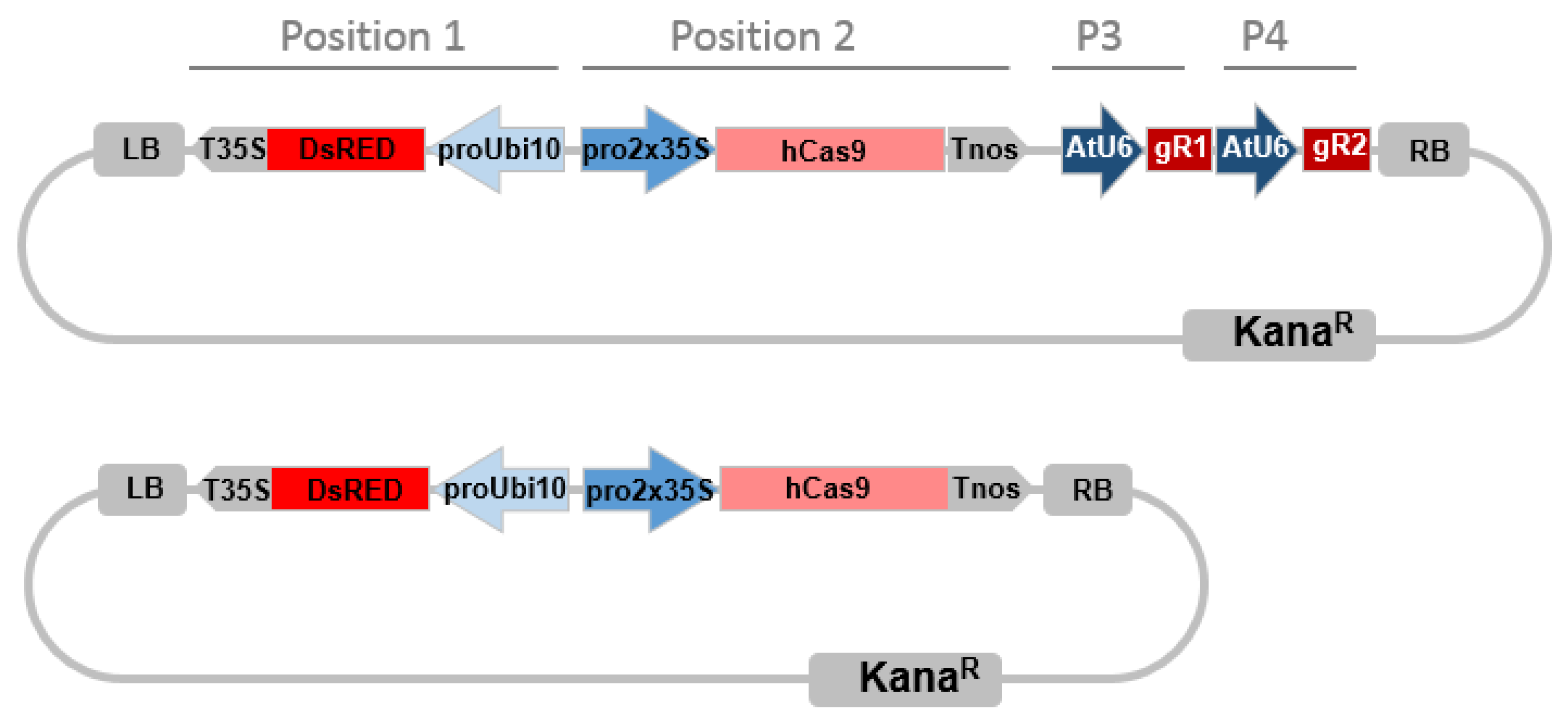
4.2. CRISPR/Cas9 Targeted Mutagenesis System Selection and Pipeline
4.3. CRISPR/Cas9 Target Site Selection and sgRNAs Design
4.4. CRISPR/Cas9 Constructs Assembly
4.5. Agrobacterium Rhizogenes-Mediated Transformation
4.6. DNA Isolation, PCR Amplification and Mutation Identification
4.7. FTIR Analyses
4.8. Histochemical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | Bovine serum albumin |
| CaMV | Cauliflower mosaic virus |
| CCR | Cinnamoyl CoA Reductase |
| CRISPR | The Clustered Regularly Interspaced Short Palindromic Repeats |
| CTAB | Cetyl Trimethylammonium Bromide |
| DSDecode | Degenerate Sequence Decoding |
| FT-IR | Fourier Transformed Infra-red spectroscopy |
| Kana | Kanamycin |
| ORF | Open reading frame |
| PAM | Protospacer Adjacent Motif |
| PLS-DA | Partial Least Square Analysis |
| sgRNA | Single guide RNA |
| SNP | Single Nucleotide Polymorphism |
References
- Myburg, A.A.; Potts, B.M.; Marques, C.M.; Kirst, M.; Gion, J.-M.; Grattapaglia, D.; Grima-Pettenatti, J. Eucalypts. In Forest Trees; Springer: Berlin, Germany, 2007; pp. 115–160. [Google Scholar]
- Paiva, J.A.; Rodrigues, J.C.; Fevereiro, P.; Neves, L.; Araújo, C.; Marques, C.; Freitas, A.T.; Bergès, H.; Grima-Pettenati, J. Building up resources and knowledge to unravel transcriptomics dynamics underlying Eucalyptus globulus xylogenesis. BMC Proc. 2011, 5, O52. [Google Scholar] [CrossRef]
- Holladay, J.E.; White, J.F.; Bozell, J.J.; Johnson, D. Top Value-Added Chemicals from Biomass-Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin; U.S. Department of Energy: Richland, WA, USA, 2007; pp. 3552–3559. [Google Scholar]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The genome of Eucalyptus grandis. Nature 2014, 510, 356–362. [Google Scholar] [CrossRef]
- Carocha, V.; Soler, M.; Hefer, C.; Cassan-Wang, H.; Fevereiro, P.; Myburg, A.A.; Paiva, J.A.P.; Grima-Pettenati, J. Genome-wide analysis of the lignin toolbox of Eucalyptus grandis. New Phytol. 2015, 206, 1297–1313. [Google Scholar] [CrossRef]
- Soler, M.; Camargo, E.L.O.; Carocha, V.; Cassan-Wang, H.; San Clemente, H.; Savelli, B.; Hefer, C.A.; Paiva, J.A.P.; Myburg, A.A.; Grima-Pettenati, J. The Eucalyptus grandis R2R3-MYB transcription factor family: Evidence for woody growth-related evolution and function. New Phytol. 2015, 206, 1364–1377. [Google Scholar] [CrossRef]
- Hussey, S.G.; Saïdi, M.N.; Hefer, C.A.; Myburg, A.A.; Grima-Pettenati, J. Structural, evolutionary and functional analysis of the NAC domain protein family in Eucalyptus. New Phytol. 2015, 206, 1337–1350. [Google Scholar] [CrossRef]
- Yu, H.; Soler, M.; Mila, I.; San Clemente, H.; Savelli, B.; Dunand, C.; Paiva, J.A.P.; Myburg, A.A.; Bouzayen, M.; Grima-Pettenati, J.; et al. Genome-Wide Characterization and Expression Profiling of the AUXIN RESPONSE FACTOR (ARF) Gene Family in Eucalyptus grandis. PLoS ONE 2014, 9, e108906. [Google Scholar] [CrossRef]
- Yu, H.; Soler, M.; San Clemente, H.; Mila, I.; Paiva, J.A.P.; Myburg, A.A.; Bouzayen, M.; Grima-Pettenati, J.; Cassan-Wang, H. Comprehensive Genome-Wide Analysis of the Aux/IAA Gene Family in Eucalyptus: Evidence for the Role of EgrIAA4 in Wood Formation. Plant Cell Physiol. 2015, 56, 700–714. [Google Scholar] [CrossRef]
- Tournier, V.; Grat, S.; Marque, C.; El Kayal, W.; Penchel, R.; de Andrade, G.; Boudet, A.-M.; Teulières, C. An Efficient Procedure to Stably Introduce Genes into an Economically Important Pulp Tree (Eucalyptus grandis × Eucalyptus urophylla). Transgenic Res. 2003, 12, 403–411. [Google Scholar] [CrossRef]
- Girijashankar, V. Genetic transformation of Eucalyptus. Physiol. Mol. Biol. Plants 2011, 17, 9–23. [Google Scholar] [CrossRef][Green Version]
- De la Torre, F.; Rodríguez, R.; Jorge, G.; Villar, B.; Álvarez-Otero, R.; Grima-Pettenati, J.; Gallego, P.P. Genetic transformation of Eucalyptus globulus using the vascular-specific EgCCR as an alternative to the constitutive CaMV35S promoter. Plant Cell Tissue Organ Cult. 2014, 117, 77–84. [Google Scholar] [CrossRef]
- Cao, P.B.; Ployet, R.; Nguyen, C.; Dupas, A.; Ladouce, N.; Martinez, Y.; Grima-Pettenati, J.; Marque, C.; Mounet, F.; Teulières, C. Wood architecture and composition are deeply remodeled in frost sensitive Eucalyptus overexpressing CBF/DREB1 transcription factors. Int. J. Mol. Sci. 2020, 21, 3019. [Google Scholar] [CrossRef]
- Plasencia, A.; Soler, M.; Dupas, A.; Ladouce, N.; Silva-Martins, G.; Martinez, Y.; Lapierre, C.; Franche, C.; Truchet, I.; Grima-Pettenati, J. Eucalyptus hairy roots, a fast, efficient and versatile tool to explore function and expression of genes involved in wood formation. Plant Biotechnol. J. 2016, 14, 1381–1393. [Google Scholar] [CrossRef]
- Travis, J. Making the cut. Science 2015, 350, 1456–1457. [Google Scholar] [CrossRef]
- Elorriaga, E.; Klocko, A.L.; Ma, C.; Strauss, S.H. CRISPR-Cas nuclease mutagenesis for genetic containment of genetically engineered forest trees. Mosaic 2015, 42, 46. [Google Scholar]
- Bortesi, L.; Fischer, R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol. Adv. 2015, 33, 41–52. [Google Scholar] [CrossRef]
- Ma, X.; Zhu, Q.; Chen, Y.; Liu, Y.-G. CRISPR/Cas9 Platforms for Genome Editing in Plants: Developments and Applications. Mol. Plant 2016, 9, 961–974. [Google Scholar] [CrossRef]
- Bewg, W.P.; Ci, D.; Tsai, C.-J. Genome Editing in Trees: From Multiple Repair Pathways to Long-Term Stability. Front. Plant Sci. 2018, 9, 1732. [Google Scholar] [CrossRef]
- Fan, D.; Liu, T.; Li, C.; Jiao, B.; Li, S.; Hou, Y.; Luo, K. Efficient CRISPR/Cas9-mediated Targeted Mutagenesis in Populus in the First Generation. Sci. Rep. 2015, 5, 12217. [Google Scholar] [CrossRef]
- Tsai, C.-J.; Xue, L.-J. CRISPRing into the woods. GM Crops Food 2015, 6, 206–215. [Google Scholar] [CrossRef]
- Zhou, X.; Jacobs, T.B.; Xue, L.-J.; Harding, S.A.; Tsai, C.-J. Exploiting SNPs for biallelic CRISPR mutations in the outcrossing woody perennial Populus reveals 4-coumarate: CoA ligase specificity and redundancy. New Phytol. 2015, 208, 298–301. [Google Scholar] [CrossRef]
- Xu, R.-F.; Li, H.; Qin, R.-Y.; Li, J.; Qiu, C.-H.; Yang, Y.-C.; Ma, H.; Li, L.; Wei, P.-C.; Yang, J.-B. Generation of inheritable and “transgene clean” targeted genome-modified rice in later generations using the CRISPR/Cas9 system. Sci. Rep. 2015, 5, 11491. [Google Scholar] [CrossRef]
- Wang, L.; Ran, L.; Hou, Y.; Tian, Q.; Li, C.; Liu, R.; Fan, D.; Luo, K. The transcription factor MYB115 contributes to the regulation of proanthocyanidin biosynthesis and enhances fungal resistance in poplar. New Phytol. 2017, 215, 351–367. [Google Scholar] [CrossRef]
- Wan, S.; Li, C.; Ma, X.; Luo, K. PtrMYB57 contributes to the negative regulation of anthocyanin and proanthocyanidin biosynthesis in poplar. Plant Cell Rep. 2017, 36, 1263–1276. [Google Scholar] [CrossRef]
- Xu, C.; Fu, X.; Liu, R.; Guo, L.; Ran, L.; Li, C.; Tian, Q.; Jiao, B.; Wang, B.; Luo, K. PtoMYB170 positively regulates lignin deposition during wood formation in poplar and confers drought tolerance in transgenic Arabidopsis. Tree Physiol. 2017, 37, 1713–1726. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, X.; Ran, L.; Li, C.; Fan, D.; Luo, K. PtoMYB156 is involved in negative regulation of phenylpropanoid metabolism and secondary cell wall biosynthesis during wood formation in poplar. Sci. Rep. 2017, 7, 41209. [Google Scholar] [CrossRef]
- Shen, Y.; Li, Y.; Xu, D.; Yang, C.; Li, C.; Luo, K. Molecular cloning and characterization of a brassinosteriod biosynthesis-related gene PtoDWF4 from Populus tomentosa. Tree Physiol. 2018, 38, 1424–1436. [Google Scholar] [CrossRef]
- Elorriaga, E.; Klocko, A.L.; Ma, C.; Strauss, S.H. Variation in Mutation Spectra among CRISPR/Cas9 Mutagenized Poplars. Front. Plant Sci. 2018, 9, 594. [Google Scholar] [CrossRef]
- Brooks, C.; Nekrasov, V.; Lippman, Z.B.; Van Eck, J. Efficient Gene Editing in Tomato in the First Generation Using the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-Associated9 System. Plant Physiol. 2014, 166, 1292–1297. [Google Scholar] [CrossRef]
- Reyes-Rivera, J.; Terrazas, T. Lignin Analysis by HPLC and FTIR. In Xylem; de Lucas, M., Etchhells, J.P., Eds.; Springer: New York, NY, USA, 2017; Volume 1544, pp. 193–211. ISBN 978-1-4939-6720-9. [Google Scholar]
- Bjarnestad, S.; Dahlman, O. Chemical Compositions of Hardwood and Softwood Pulps Employing Photoacoustic Fourier Transform Infrared Spectroscopy in Combination with Partial Least-Squares Analysis. Anal. Chem. 2002, 74, 5851–5858. [Google Scholar] [CrossRef]
- Sammons, R.J.; Harper, D.P.; Labbé, N.; Bozell, J.J.; Elder, T.; Rials, T.G. Characterization of Organosolv Lignins using Thermal and FT-IR Spectroscopic Analysis. Bioresources 2013, 8, 2752–2767. [Google Scholar] [CrossRef]
- Szymanska-Chargot, M.; Zdunek, A. Use of FT-IR Spectra and PCA to the Bulk Characterization of Cell Wall Residues of Fruits and Vegetables Along a Fraction Process. Food Biophys. 2013, 8, 29–42. [Google Scholar] [CrossRef]
- Casas, A.; Alonso, M.V.; Oliet, M.; Rojo, E.; Rodríguez, F. FTIR analysis of lignin regenerated from Pinus radiata and Eucalyptus globulus woods dissolved in imidazolium-based ionic liquids. J. Chem. Technol. Biotechnol. 2012, 87, 472–480. [Google Scholar] [CrossRef]
- Popescu, M.; Zanoaga, M.; Mamunya, Y.; Myshak, V.; Vasile, C. Two dimensional infrared correlation spectroscopy studies of wood-plastic composites with a copolyamide as matrix. J. Optoelectron. Adv. Mater. 2007, 9, 923. [Google Scholar]
- Faix, O. Classification of lignins from different botanical origins by FT-IR spectroscopy. Holzforsch. Int. J. Biol. Chem. Phys. Technol. Wood 1991, 45, 21–28. [Google Scholar] [CrossRef]
- Largo-Gosens, A.; Hernández-Altamirano, M.; García-Calvo, L.; Alonso-Simón, A.; Álvarez, J.; Acebes, J.L. Fourier transform mid infrared spectroscopy applications for monitoring the structural plasticity of plant cell walls. Front. Plant Sci. 2014, 5, 303. [Google Scholar] [CrossRef]
- Adamafio, N.; Kyeremeh, K.; Datsomor, A.; Osei-Owusu, J. Others Cocoa pod ash pre-treatment of wawa (Triplochiton scleroxylon) and sapele (Entandrophragma cylindricum) sawdust: Fourier transform infrared spectroscopic characterization of lignin. Asian J. Sci. Res. 2013, 6, 812–818. [Google Scholar] [CrossRef]
- Shi, J.; Xing, D.; Lia, J. FTIR Studies of the Changes in Wood Chemistry from Wood Forming Tissue under Inclined Treatment. Energy Proced. 2012, 16, 758–762. [Google Scholar] [CrossRef]
- Stark, N.M.; Yelle, D.J.; Agarwal, U.P. Techniques for Characterizing Lignin. In Lignin in Polymer Composites; Elsevier: Amsterdam, The Netherlands, 2016; pp. 49–66. ISBN 978-0-323-35565-0. [Google Scholar]
- Mouille, G.; Robin, S.; Lecomte, M.; Pagant, S.; Höfte, H. Classification and identification of Arabidopsis cell wall mutants using Fourier-Transform InfraRed (FT-IR) microspectroscopy. Plant J. 2003, 35, 393–404. [Google Scholar] [CrossRef]
- Li, X.; Sun, C.; Zhou, B.; He, Y. Determination of Hemicellulose, Cellulose and Lignin in Moso Bamboo by Near Infrared Spectroscopy. Sci. Rep. 2015, 5, 17210. [Google Scholar] [CrossRef]
- Jia, H.; Wang, N. Targeted Genome Editing of Sweet Orange Using Cas9/sgRNA. PLoS ONE 2014, 9, e93806. [Google Scholar] [CrossRef]
- Jia, H.; Xu, J.; Orbović, V.; Zhang, Y.; Wang, N. Editing Citrus Genome via SaCas9/sgRNA System. Front. Plant Sci. 2017, 8, 2135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, S.; Li, D.; Zhang, Q.; Li, L.; Zhong, C.; Liu, Y.; Huang, H. Optimized paired-sgRNA/Cas9 cloning and expression cassette triggers high-efficiency multiplex genome editing in kiwifruit. Plant Biotechnol. J. 2018, 16, 1424–1433. [Google Scholar] [CrossRef] [PubMed]
- Breitler, J.-C.; Dechamp, E.; Campa, C.; Zebral Rodrigues, L.A.; Guyot, R.; Marraccini, P.; Etienne, H. CRISPR/Cas9-mediated efficient targeted mutagenesis has the potential to accelerate the domestication of Coffea canephora. Plant Cell Tissue Organ Cult. 2018, 134, 383–394. [Google Scholar] [CrossRef]
- Jiang, Y.; Guo, L.; Ma, X.; Zhao, X.; Jiao, B.; Li, C.; Luo, K. The WRKY transcription factors PtrWRKY18 and PtrWRKY35 promote Melampsora resistance in Populus. Tree Physiol. 2017, 37, 665–675. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Q.; Zhu, Q.; Liu, W.; Chen, Y.; Qiu, R.; Wang, B.; Yang, Z.; Li, H.; Lin, Y.; et al. A Robust CRISPR/Cas9 System for Convenient, High-Efficiency Multiplex Genome Editing in Monocot and Dicot Plants. Mol. Plant 2015, 8, 1274–1284. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, J.; Wei, P.; Zhang, B.; Gou, F.; Feng, Z.; Mao, Y.; Yang, L.; Zhang, H.; Xu, N.; et al. The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol. J. 2014, 12, 797–807. [Google Scholar] [CrossRef]
- Bruegmann, T.; Deecke, K.; Fladung, M. Evaluating the Efficiency of gRNAs in CRISPR/Cas9 Mediated Genome Editing in Poplars. Int. J. Mol. Sci. 2019, 20, 3623. [Google Scholar] [CrossRef]
- Piquemal, J.; Lapierre, C.; Myton, K.; O’connell, A.; Schuch, W.; Grima-pettenati, J.; Boudet, A.-M. Down-regulation of Cinnamoyl-CoA Reductase induces significant changes of lignin profiles in transgenic tobacco plants. Plant J. 1998, 13, 71–83. [Google Scholar] [CrossRef]
- Leplé, J.-C.; Dauwe, R.; Morreel, K.; Storme, V.; Lapierre, C.; Pollet, B.; Naumann, A.; Kang, K.-Y.; Kim, H.; Ruel, K.; et al. Downregulation of Cinnamoyl-Coenzyme A Reductase in Poplar: Multiple-Level Phenotyping Reveals Effects on Cell Wall Polymer Metabolism and Structure. Plant Cell 2007, 19, 3669–3691. [Google Scholar] [CrossRef]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.-B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef]
- Ron, M.; Kajala, K.; Pauluzzi, G.; Wang, D.; Reynoso, M.A.; Zumstein, K.; Garcha, J.; Winte, S.; Masson, H.; Inagaki, S.; et al. Hairy Root Transformation Using Agrobacterium rhizogenes as a Tool for Exploring Cell Type-Specific Gene Expression and Function Using Tomato as a Model. Plant Physiol. 2014, 166, 455–469. [Google Scholar] [CrossRef]
- Sun, X.; Hu, Z.; Chen, R.; Jiang, Q.; Song, G.; Zhang, H.; Xi, Y. Targeted mutagenesis in soybean using the CRISPR-Cas9 system. Sci. Rep. 2015, 5, 10342. [Google Scholar] [CrossRef]
- Cai, Y.; Chen, L.; Liu, X.; Sun, S.; Wu, C.; Jiang, B.; Han, T.; Hou, W. CRISPR/Cas9-Mediated Genome Editing in Soybean Hairy Roots. PLoS ONE 2015, 10, e0136064. [Google Scholar] [CrossRef]
- Jacobs, T.B.; LaFayette, P.R.; Schmitz, R.J.; Parrott, W.A. Targeted genome modifications in soybean with CRISPR/Cas9. BMC Biotechnol. 2015, 15, 16. [Google Scholar] [CrossRef]
- Bernard, G.; Gagneul, D.; Alves Dos Santos, H.; Etienne, A.; Hilbert, J.-L.; Rambaud, C. Efficient Genome Editing Using CRISPR/Cas9 Technology in Chicory. Int. J. Mol. Sci. 2019, 20, 1155. [Google Scholar] [CrossRef]
- Shan, Q.; Wang, Y.; Li, J.; Zhang, Y.; Chen, K.; Liang, Z.; Zhang, K.; Liu, J.; Xi, J.J.; Qiu, J.-L.; et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 686–688. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Y.; Zhang, R.; Zhang, H.; Gao, C. CRISPR/Cas Genome Editing and Precision Plant Breeding in Agriculture. Annu. Rev. Plant Biol. 2019, 70, 667–697. [Google Scholar] [CrossRef]
- Belhaj, K.; Chaparro-Garcia, A.; Kamoun, S.; Nekrasov, V. Plant genome editing made easy: Targeted mutagenesis in model and crop plants using the CRISPR/Cas system. Plant Methods 2013, 9, 39. [Google Scholar] [CrossRef]
- Nekrasov, V.; Staskawicz, B.; Weigel, D.; Jones, J.D.G.; Kamoun, S. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 691–693. [Google Scholar] [CrossRef]
- Weber, E.; Engler, C.; Gruetzner, R.; Werner, S.; Marillonnet, S. A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLoS ONE 2011, 6, e16765. [Google Scholar] [CrossRef]
- Patron, N.J.; Orzaez, D.; Marillonnet, S.; Warzecha, H.; Matthewman, C.; Youles, M.; Raitskin, O.; Leveau, A.; Farré, G.; Rogers, C. Standards for plant synthetic biology: A common syntax for exchange of DNA parts. New Phytol. 2015, 208, 13–19. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Ployet, R.; Veneziano Labate, M.T.; Regiani Cataldi, T.; Christina, M.; Morel, M.; San Clemente, H.; Denis, M.; Favreau, B.; Tomazello Filho, M.; Laclau, J.; et al. A systems biology view of wood formation in Eucalyptus grandis trees submitted to different potassium and water regimes. New Phytol. 2019, 223, 766–782. [Google Scholar] [CrossRef]
- Lacombe, E.; Hawkins, S.; Van Doorsselaere, J.; Piquemal, J.; Goffner, D.; Poeydomenge, O.; Boudet, A.-M.; Grima-Pettenati, J. Cinnamoyl CoA reductase, the first committed enzyme of the lignin branch biosynthetic pathway: Cloning, expression and phylogenetic relationships. Plant J. 1997, 11, 429–441. [Google Scholar] [CrossRef]
- Verbylaitë, R.; Beiðys, P.; Rimas, V.; Kuusienë, S. Comparison of Ten DNA Extraction Protocols from Wood of European Aspen (Populus tremula L.). Balt. For. 2010, 16, 8. [Google Scholar]
- Beleites, C.; Sergo, V. HyperSpec: A Package to Handle Hyperspectral Data Sets in R. R Package Version 0.99-20200213.1. Available online: https://github.com/cbeleites/hyperSpec (accessed on 10 February 2020).
- Barnes, R.J.; Dhanoa, M.S.; Lister, S.J. Standard Normal Variate Transformation and De-Trending of Near-Infrared Diffuse Reflectance Spectra. Appl. Spectrosc. 1989, 43, 772–777. [Google Scholar] [CrossRef]
- Becker, R.A.; Chambers, J.M.; Wilks, A.R. The New S Language; Wadsworth & Brooks; Chapman & Hall: London, UK, 1988. [Google Scholar]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Edited Plants/Total Transgenic Plants | Plants with All Alleles Altered (no WT) | Plants with one WT Allele | WT/WT | |||
|---|---|---|---|---|---|---|---|
| Homoz. (A1/A1) | Biallelic (A1/A2) | Chimera (A1/A2/A3...) | Monoallelic (WT/A1) | Chimera (WT/A1/A2...) | |||
| CCR1 | 24/24 (100%) | 0 | 0 | 1 (4.2%) | 10 (41.2%) | 13 (54.2%) | 0 |
| IAA9A | 12/13 (92.3%) | 0 | 7 (53.8%) | 4 (30.8%) | 1 (7.7%) | 0 | 1 (7.7%) |
| Gene | Total Sequenced Subclones | Edited Subclones | Editions in sgRNA1 | Editions in sgRNA2 | Editions in sgRNA1&2 | Large Deletions |
|---|---|---|---|---|---|---|
| CCR1 | 278 | 89 (32.0%) | 46 (51.7%) | 65 (73.0%) | 22 (24.7%) | 19 (21.3%) |
| IAA9A | 95 | 88 (92.6%) | 84 (95.5%) | 79 (89.9%) | 75 (85.2%) | 27 (30.7%) |
| Gene | Edited Clones | Presumed Significant Modifications | Presumed Less Significant Modifications (15 bp Indel, Substitution without Shift) | |
|---|---|---|---|---|
| Reading Frame Shift 1 | ≥15 bp Indel | |||
| CCR1 | 89 | 60 (67.4%) | 22 (24.7%) | 27 (30.3%) |
| 62 (69.7%) | ||||
| IAA9A | 88 | 75 (85.2%) | 50 (56.8%) | 12 (13.6%) |
| 76 (86.4%) | ||||
| Gene | Total Edited Clones | Total Edition Types | Deletion | Insertion | Substitution | Expected Large Deletion | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Small Deletion (≤15 bp) | Large Deletion (>15 bp) | Small Insertion (≤15 bp) | Large Insertion (>15 bp) | Small Substitution (≤15 bp) | Large Substitution (>15 bp) | ||||||||||||||||
| sg1 | sg2 | sg1&2 | sg1 | sg2 | sg1&2 | sg1 | sg2 | sg1&2 | sg1 | sg2 | sg1&2 | sg1 | sg2 | sg1&2 | sg1 | sg2 | sg1&2 | ||||
| CCR1 | 89 | 43 | 12 | 40 | 0 | 19 | 21 | 19 | 3 | 1 | 0 | 0 | 0 | 0 | 24 | 5 | 0 | 0 | 0 | 0 | 19 (21.3%) |
| 52 (58.4%) | 21 (23.6%) | 4 (4.5%) | 0 | 29 (32.6%) | 0 | ||||||||||||||||
| IAA9A | 88 | 20 | 29 | 39 | 19 | 38 | 27 | 27 | 5 | 11 | 0 | 12 | 0 | 0 | 12 | 2 | 0 | 0 | 0 | 0 | 27 (30.7%) |
| 49 (55.7%) | 38 (43.2%) | 16 (18.2%) | 12 (13.6%) | 14 (15.9%) | 0 | ||||||||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Y.; Hu, G.; Dupas, A.; Medina, L.; Blandels, N.; San Clemente, H.; Ladouce, N.; Badawi, M.; Hernandez-Raquet, G.; Mounet, F.; et al. Implementing the CRISPR/Cas9 Technology in Eucalyptus Hairy Roots Using Wood-Related Genes. Int. J. Mol. Sci. 2020, 21, 3408. https://doi.org/10.3390/ijms21103408
Dai Y, Hu G, Dupas A, Medina L, Blandels N, San Clemente H, Ladouce N, Badawi M, Hernandez-Raquet G, Mounet F, et al. Implementing the CRISPR/Cas9 Technology in Eucalyptus Hairy Roots Using Wood-Related Genes. International Journal of Molecular Sciences. 2020; 21(10):3408. https://doi.org/10.3390/ijms21103408
Chicago/Turabian StyleDai, Ying, Guojian Hu, Annabelle Dupas, Luciano Medina, Nils Blandels, Hélène San Clemente, Nathalie Ladouce, Myriam Badawi, Guillermina Hernandez-Raquet, Fabien Mounet, and et al. 2020. "Implementing the CRISPR/Cas9 Technology in Eucalyptus Hairy Roots Using Wood-Related Genes" International Journal of Molecular Sciences 21, no. 10: 3408. https://doi.org/10.3390/ijms21103408
APA StyleDai, Y., Hu, G., Dupas, A., Medina, L., Blandels, N., San Clemente, H., Ladouce, N., Badawi, M., Hernandez-Raquet, G., Mounet, F., Grima-Pettenati, J., & Cassan-Wang, H. (2020). Implementing the CRISPR/Cas9 Technology in Eucalyptus Hairy Roots Using Wood-Related Genes. International Journal of Molecular Sciences, 21(10), 3408. https://doi.org/10.3390/ijms21103408