Cryptotanshinone from the Salvia miltiorrhiza Bunge Attenuates Ethanol-Induced Liver Injury by Activation of AMPK/SIRT1 and Nrf2 Signaling Pathways


Abstract
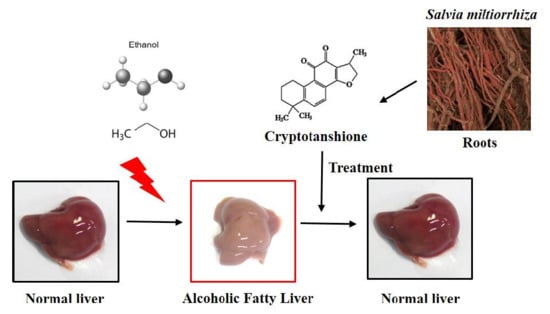
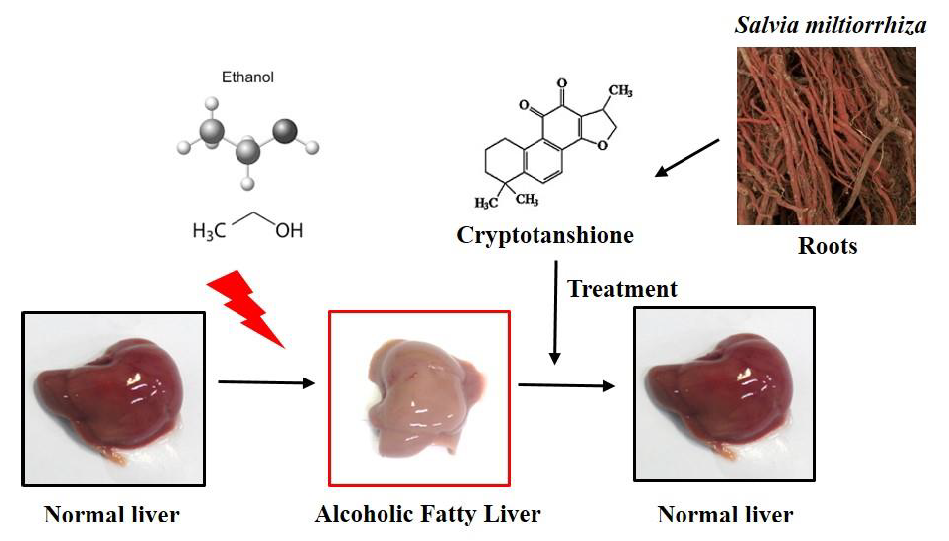{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
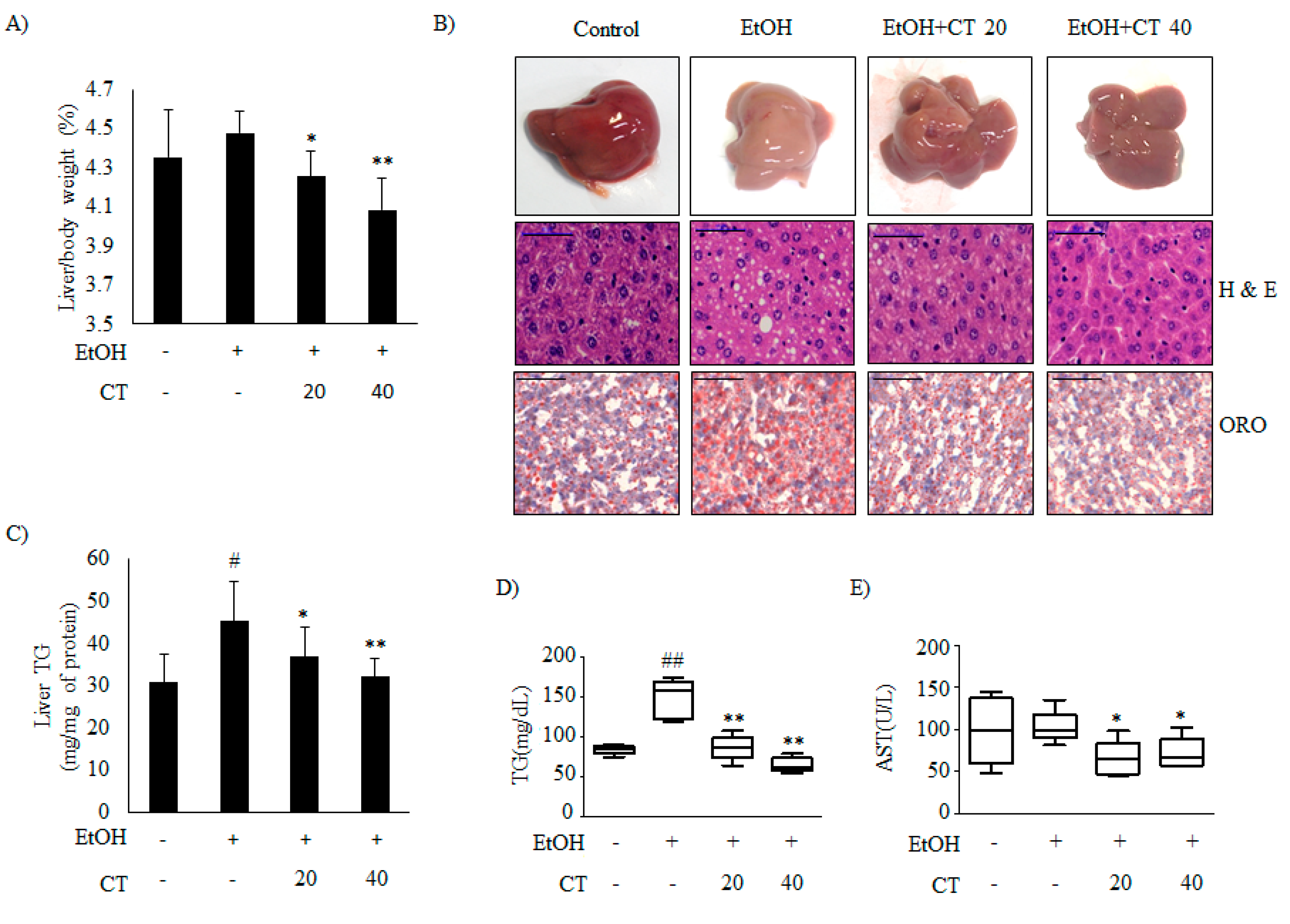
2.1. CT Countered Ethanol-Promoted Hepatic Steatosis in Chronic Ethanol-Fed Mice
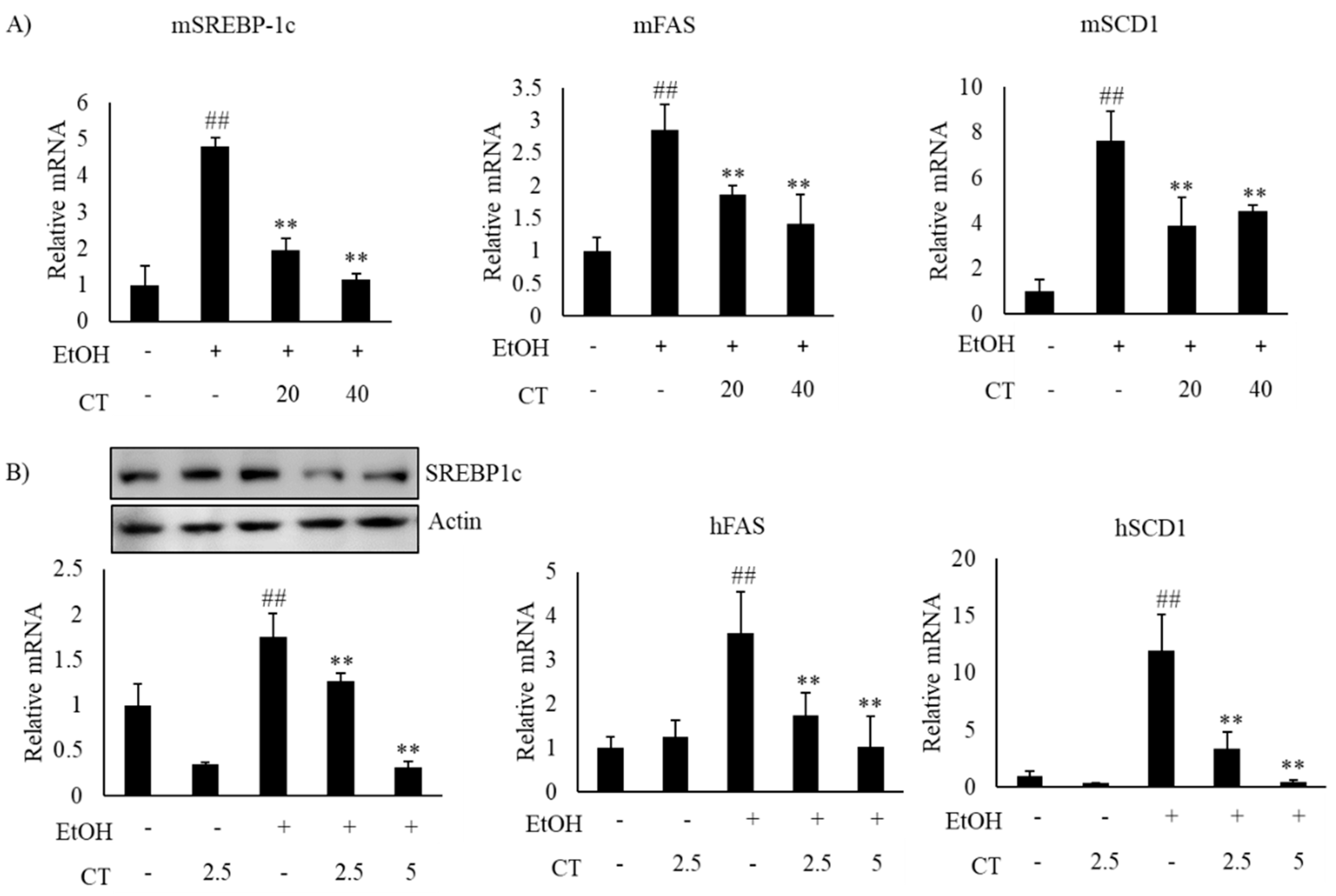
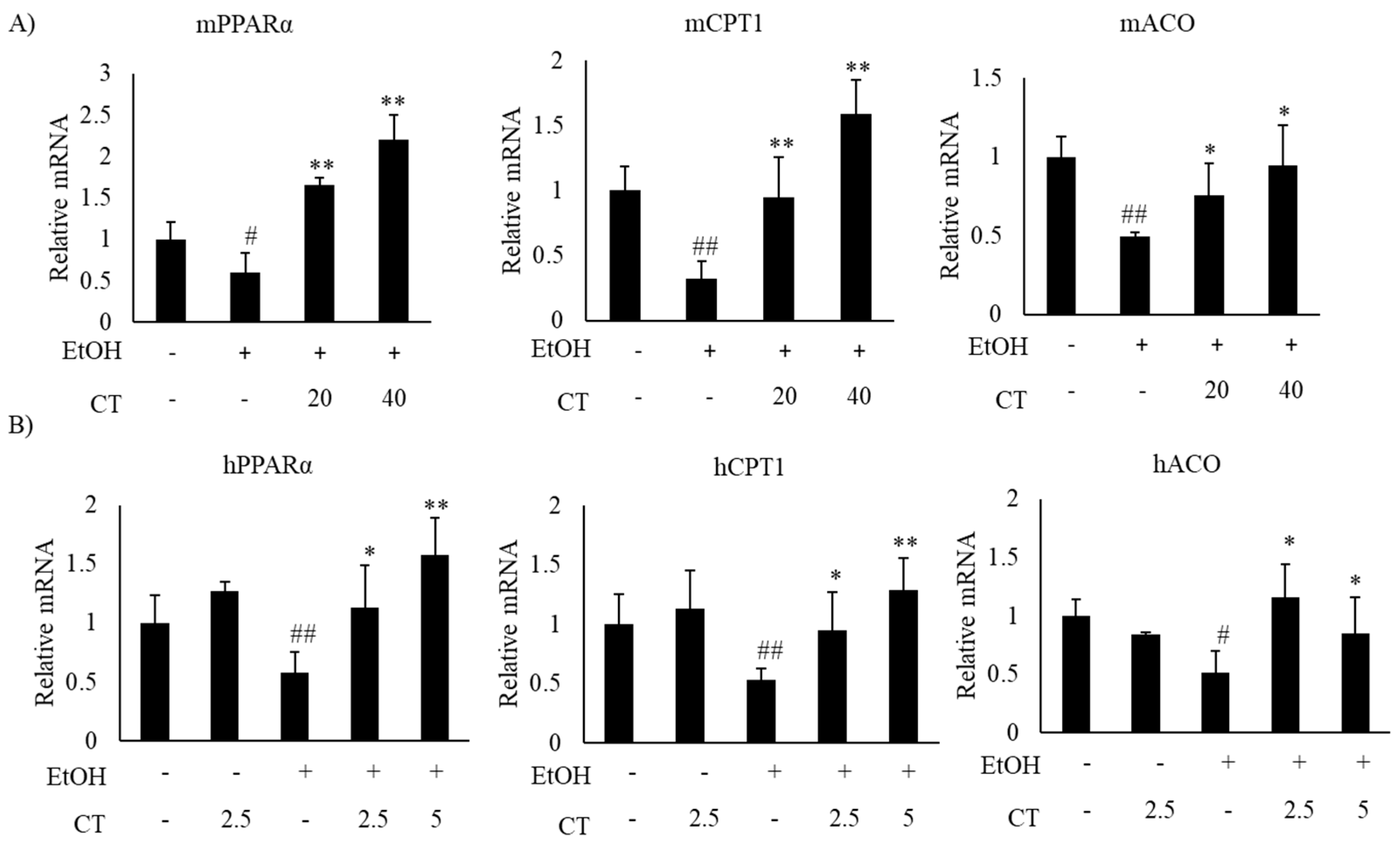
2.2. CT Mitigates Ethanol-Induced TG Accumulation by Regulating Lipogenesis and Fatty Acid Oxidation in Chronic Ethanol-Fed Mice and HepG2 Cells
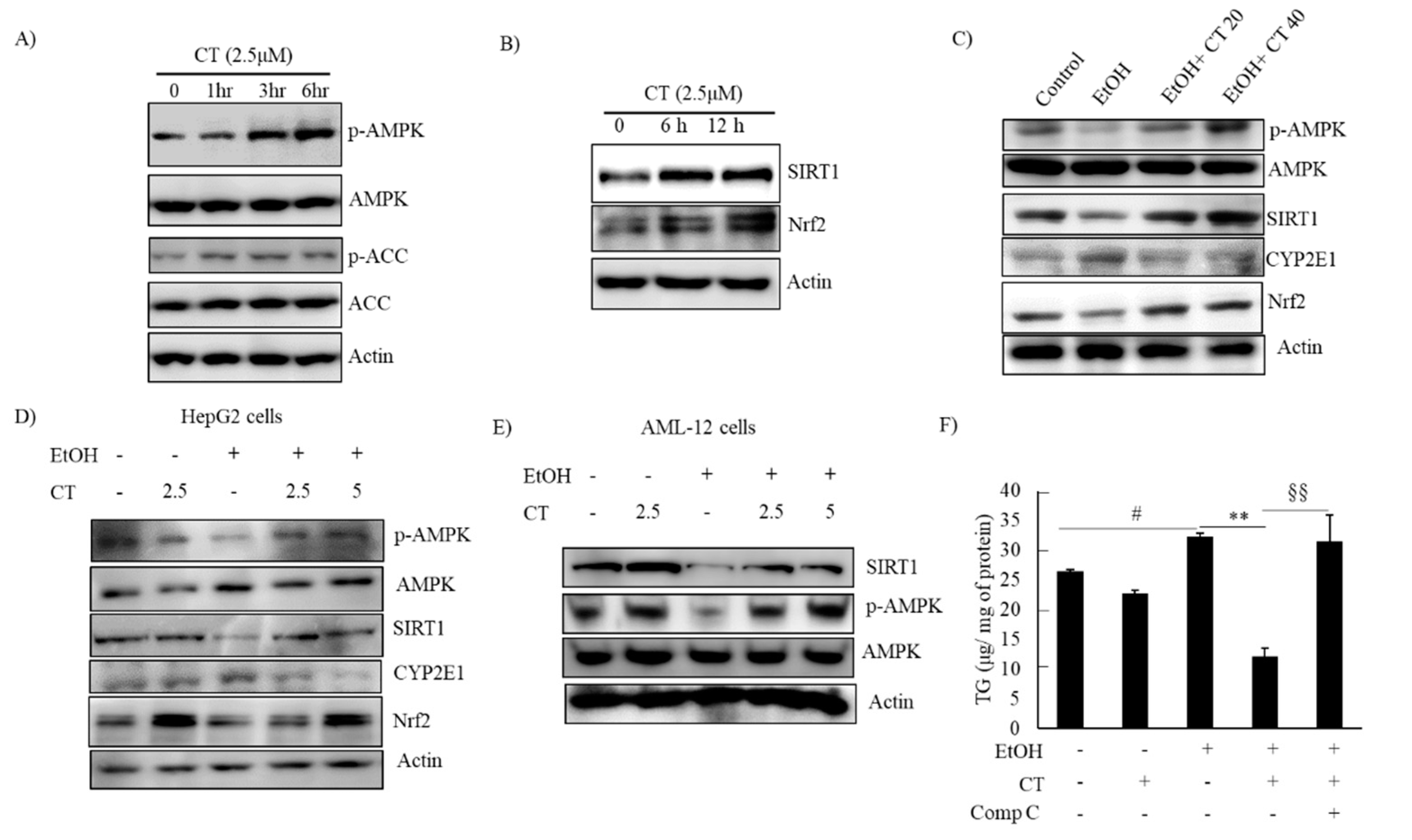
2.3. CT Activated AMPK/SIRT1 Signaling in Ethanol-Treated Mice and HepG2 Cells
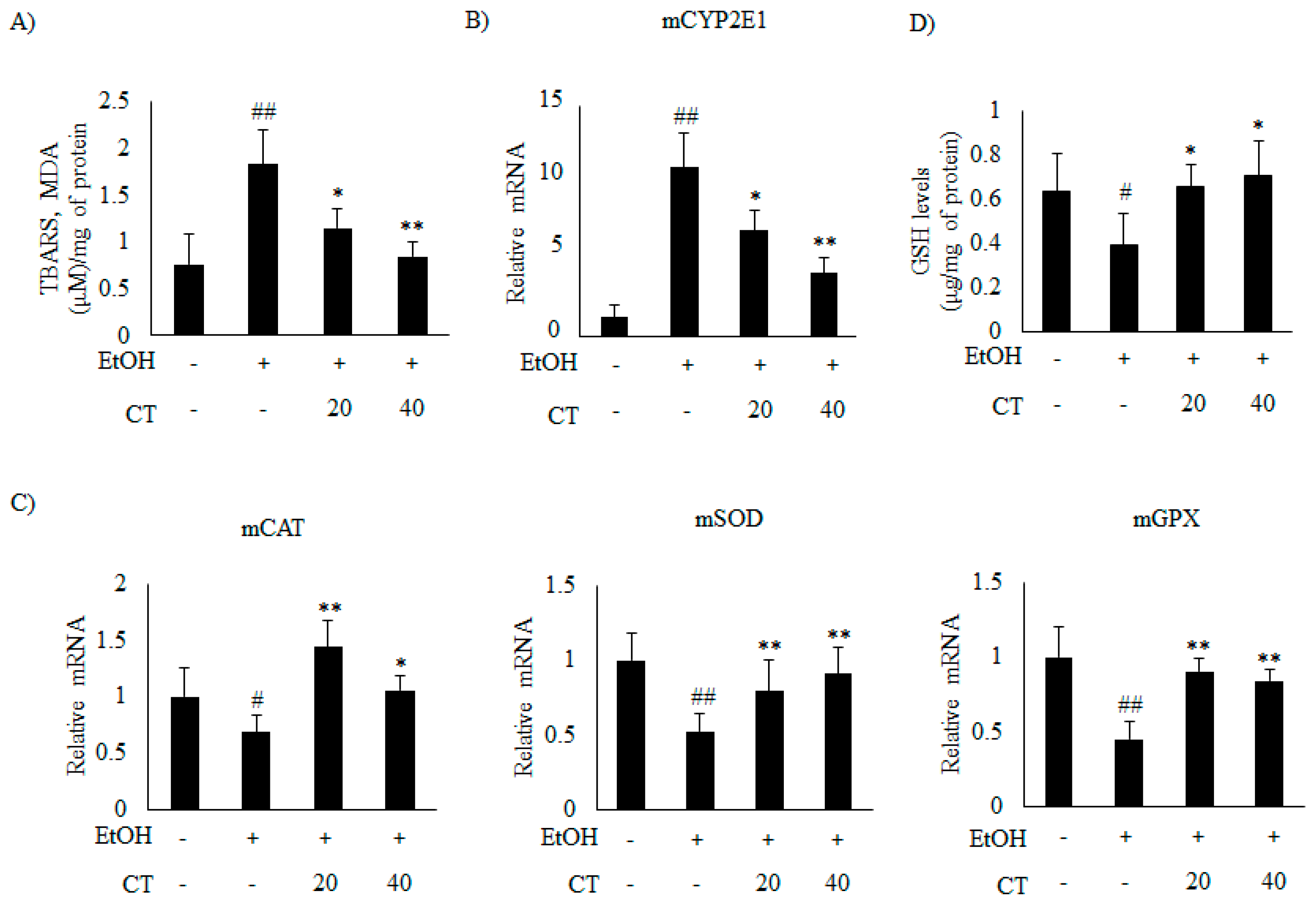
2.4. CT Prevents Ethanol-Induced Oxidative Stress in Chronic Ethanol-Fed Mice and HepG2 Cells
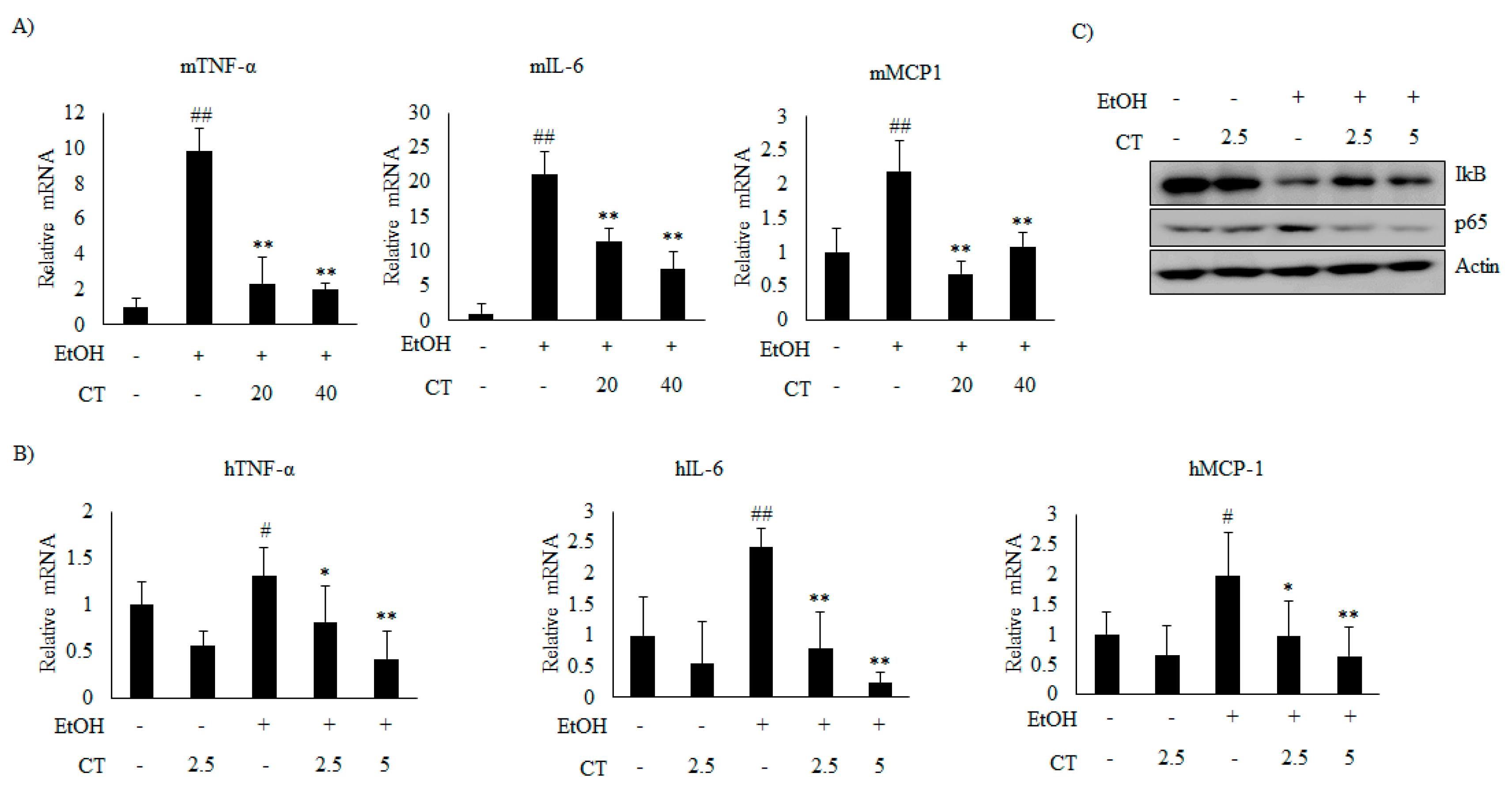
2.5. CT Prevents Ethanol-Induced Inflammation in Chronic Ethanol-Fed Mice and HepG2 Cells
3. Discussion
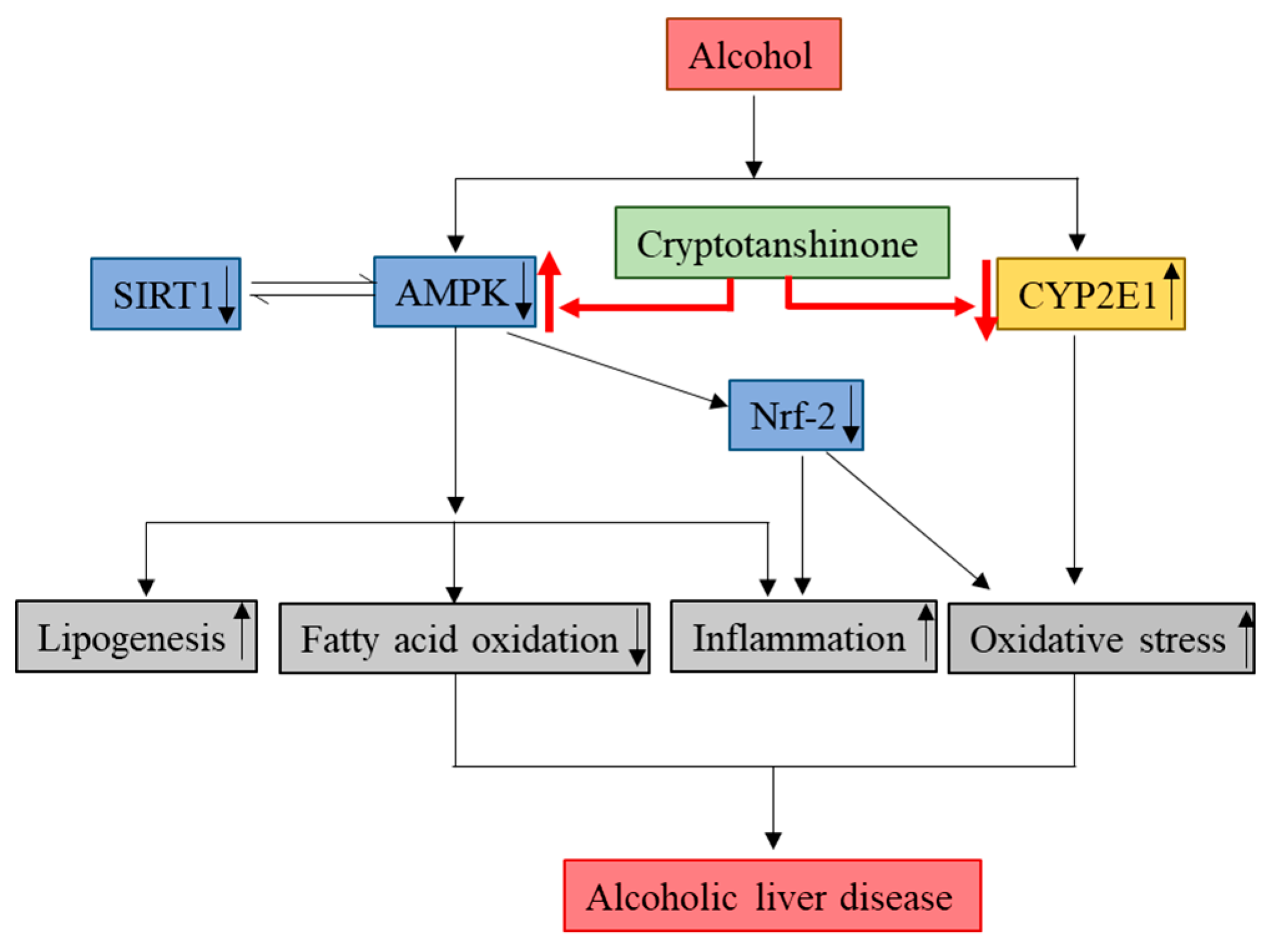
4. Conclusions
5. Materials and Methods
5.1. Reagents
5.2. Cell Culture
5.3. Cytotoxicity Assay
5.4. Animal Experiments
5.5. TG Measurement
5.6. Histopathological Analysis
5.7. Biochemical Analysis
5.8. Quantitative Polymerase Chain Reaction (qPCR)
5.9. Measurement of Hepatic Lipid Peroxidation and GSH Level
5.10. Western Blot Analysis
5.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| ACO | Acyl-coenzyme A oxidase |
| ALD | Alcoholic liver disease |
| ALT | Alanine aminotransferase |
| AMPK | AMP-activated protein kinase |
| AST | Aspartate aminotransferase |
| CYP2E1 | Cytochrome P450 2E1 |
| CPT1α | Carnitine palmitoyltransferase-1α |
| CAT | Catalase |
| CT | Cryptotanshinone |
| EtoH | Ethanol |
| FAS | Fatty acid synthase |
| GPX | Glutathione peroxidase |
| GSH | Glutathione |
| H&E | Haematoxylin and eosin |
| IκB | I Kappa B-alpha |
| IL6 | Interleukin 6 |
| MCP-1 | Monocyte chemotactic protein-1 |
| NF-κB | Nuclear factor-kappa B |
| NRF2 | Nuclear factor erythroid-derived 2-related factor 2 |
| ORO | Oil Red O |
| PPAR α | Peroxisome proliferator-activated receptor α |
| qPCR | Quantitative polymerase chain reaction |
| ROS | Reactive oxygen species |
| SCD1 | Stearoyl-CoA desaturase-1 |
| SIRT1 | Sirtuin 1 |
| SOD | Superoxide dismutase |
| SREBP-1c | Sterol regulatory element-binding protein-1c |
| TBARS | Thiobarbituric acid reactive substances |
| TG | Triglyceride |
| TNFα | Tumor necrosis factor-α |
References
- Lei, X.L.; Chiou, G.C. Studies on cardiovascular actions of Salvia miltiorrhiza. Am. J. Chin. Med. 1986, 14, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Luo, H.W.; Niwa, M.; Ji, J. A new platelet aggregation inhibitor from Salvia miltiorrhiza. Planta Med. 1989, 55, 390–391. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.B.; Wu, P.; Huang, Y.F. Protective mechanisms of radix salviae miltiorrhizae against chronic alcoholic liver injury in mice. Zhongguo Zhong Xi Yi Jie He Za Zhi 2005, 25, 425–428. [Google Scholar] [PubMed]
- Brunetti, G.; Serra, S.; Vacca, G.; Lobina, C.; Morazzoni, P.; Bombardelli, E.; Colombo, G.; Gessa, G.L.; Carai, M.A. IDN 5082, a standardized extract of Salvia miltiorrhiza, delays acquisition of alcohol drinking behavior in rats. J. Ethnopharmacol. 2003, 85, 93–97. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, T.; Huang, K.; Shen, L.; Tao, Y.; Liu, C. Salvia Miltiorrhiza Ameliorates Liver Fibrosis by Activating Hepatic Natural Killer Cells in Vivo and in Vitro. Front. Pharmacol. 2018, 9, 762. [Google Scholar] [CrossRef]
- Cao, E.H.; Liu, X.Q.; Wang, J.J.; Xu, N.F. Effect of natural antioxidant tanshinone II-A on DNA damage by lipid peroxidation in liver cells. Free Radic. Biol. Med. 1996, 20, 801–806. [Google Scholar] [CrossRef]
- Park, E.J.; Zhao, Y.Z.; Kim, Y.C.; Sohn, D.H. PF2401-SF, standardized fraction of Salvia miltiorrhiza and its constituents, tanshinone I, tanshinone IIA, and cryptotanshinone, protect primary cultured rat hepatocytes from bile acid-induced apoptosis by inhibiting JNK phosphorylation. Food Chem. Toxicol 2007, 45, 1891–1898. [Google Scholar] [CrossRef]
- Li, X.; Lian, L.H.; Bai, T.; Wu, Y.L.; Wan, Y.; Xie, W.X.; Jin, X.; Nan, J.X. Cryptotanshinone inhibits LPS-induced proinflammatory mediators via TLR4 and TAK1 signaling pathway. Int. Immunopharmacol. 2011, 11, 1871–1876. [Google Scholar] [CrossRef]
- Tang, S.; Shen, X.Y.; Huang, H.Q.; Xu, S.W.; Yu, Y.; Zhou, C.H.; Chen, S.R.; Le, K.; Wang, Y.H.; Liu, P.Q. Cryptotanshinone suppressed inflammatory cytokines secretion in RAW264.7 macrophages through inhibition of the NF-kappaB and MAPK signaling pathways. Inflammation 2011, 34, 111–118. [Google Scholar] [CrossRef]
- Yin, H.Q.; Choi, Y.J.; Kim, Y.C.; Sohn, D.H.; Ryu, S.Y.; Lee, B.H. Salvia miltiorrhiza Bunge and its active component cryptotanshinone protects primary cultured rat hepatocytes from acute ethanol-induced cytotoxicity and fatty infiltration. Food Chem. Toxicol. 2009, 47, 98–103. [Google Scholar] [CrossRef]
- Mann, R.E.; Smart, R.G.; Govoni, R. The epidemiology of alcoholic liver disease. Alcohol Res. Health 2003, 27, 209–219. [Google Scholar] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Hwang, C.E.; Lee, C.K.; Lee, J.H.; Kim, G.M.; Jeong, S.H.; Shin, J.H.; Kim, J.S.; Cho, K.M. Characteristics and antioxidant effect of garlic in the fermentation of Cheonggukjang by Bacillus amyloliquefaciens MJ1-4. J. Microbiol. Biotechnol. 2014, 24, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, J.; Bataller, R. Alcoholic liver disease: Pathogenesis and new targets for therapy. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.C.; Huang, H.P.; Lee, Y.J.; Tang, Y.H.; Wang, C.J. Hepatoprotective effect of mulberry water extracts on ethanol-induced liver injury via anti-inflammation and inhibition of lipogenesis in C57BL/6J mice. Food Chem. Toxicol. 2013, 62, 786–796. [Google Scholar] [CrossRef]
- Hardie, D.G. Minireview: The AMP-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 2003, 144, 5179–5183. [Google Scholar] [CrossRef]
- Carling, D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Rui, B.B.; Chen, H.; Jang, L.; Li, Z.; Yang, J.M.; Xu, W.P.; Wei, W. Melatonin Upregulates the Activity of AMPK and Attenuates Lipid Accumulation in Alcohol-induced Rats. Alcohol Alcohol. 2016, 51, 11–19. [Google Scholar] [CrossRef]
- Tomita, K.; Tamiya, G.; Ando, S.; Kitamura, N.; Koizumi, H.; Kato, S.; Horie, Y.; Kaneko, T.; Azuma, T.; Nagata, H.; et al. AICAR, an AMPK activator, has protective effects on alcohol-induced fatty liver in rats. Alcohol Clin. Exp. Res. 2005, 29, 240S–245S. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Jogasuria, A.; Taylor, C.; Wu, J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 88–100. [Google Scholar] [CrossRef]
- Nagappan, A.; Jung, D.Y.; Kim, J.H.; Lee, H.; Jung, M.H. Gomisin N Alleviates Ethanol-Induced Liver Injury through Ameliorating Lipid Metabolism and Oxidative Stress. Int. J. Mol. Sci. 2018, 19, 2601. [Google Scholar] [CrossRef]
- Cederbaum, A.I.; Wu, D.; Mari, M.; Bai, J. CYP2E1-dependent toxicity and oxidative stress in HepG2 cells. Free Radic. Biol. Med. 2001, 31, 1539–1543. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Introduction-serial review: Alcohol, oxidative stress and cell injury. Free Radic. Biol. Med. 2001, 31, 1524–1526. [Google Scholar] [CrossRef]
- Arteel, G.E. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology 2003, 124, 778–790. [Google Scholar] [CrossRef]
- Caro, A.A.; Cederbaum, A.I. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 27–42. [Google Scholar] [CrossRef]
- Butura, A.; Nilsson, K.; Morgan, K.; Morgan, T.R.; French, S.W.; Johansson, I.; Schuppe-Koistinen, I.; Ingelman-Sundberg, M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol. 2009, 50, 572–583. [Google Scholar] [CrossRef]
- Lu, Y.; Zhuge, J.; Wang, X.; Bai, J.; Cederbaum, A.I. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology 2008, 47, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, M.; Hagbjork, A.L.; Wan, Y.J.; Fu, P.C.; Clot, P.; Albano, E.; Ingelman-Sundberg, M.; French, S.W. Modulation of experimental alcohol-induced liver disease by cytochrome P450 2E1 inhibitors. Hepatology 1995, 21, 1610–1617. [Google Scholar] [PubMed]
- Hou, D.X.; Korenori, Y.; Tanigawa, S.; Yamada-Kato, T.; Nagai, M.; He, X.; He, J. Dynamics of Nrf2 and Keap1 in ARE-mediated NQO1 expression by wasabi 6-(methylsulfinyl)hexyl isothiocyanate. J. Agric. Food Chem. 2011, 59, 11975–11982. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Zhou, C.; Shi, Y.; Lu, H.; Xu, R.; He, X. Nuclear transcription factor Nrf2 suppresses prostate cancer cells growth and migration through upregulating ferroportin. Oncotarget 2016, 7, 78804–78812. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Chen, B.; Deng, Y.; Lu, X.; Yu, G. Baicalin increases the antioxidant capacity via promoting the nuclear translocation of NF-E2-related factor 2 (Nrf2) in N2a/APPswe cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2015, 31, 1597–1601. [Google Scholar] [PubMed]
- Lamle, J.; Marhenke, S.; Borlak, J.; von Wasielewski, R.; Eriksson, C.J.; Geffers, R.; Manns, M.P.; Yamamoto, M.; Vogel, A. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology 2008, 134, 1159–1168. [Google Scholar] [CrossRef]
- Wu, K.C.; Liu, J.; Klaassen, C.D. Role of Nrf2 in preventing ethanol-induced oxidative stress and lipid accumulation. Toxicol. Appl. Pharmacol. 2012, 262, 321–329. [Google Scholar] [CrossRef]
- Gu, L.; Tao, X.; Xu, Y.; Han, X.; Qi, Y.; Xu, L.; Yin, L.; Peng, J. Dioscin alleviates BDL- and DMN-induced hepatic fibrosis via Sirt1/Nrf2-mediated inhibition of p38 MAPK pathway. Toxicol. Appl. Pharmacol. 2016, 292, 19–29. [Google Scholar] [CrossRef]
- Tang, L.; Chen, Q.; Meng, Z.; Sun, L.; Zhu, L.; Liu, J.; Hu, J.; Ni, Z.; Wang, X. Suppression of Sirtuin-1 Increases IL-6 Expression by Activation of the Akt Pathway During Allergic Asthma. Cell. Physiol. Biochem. 2017, 43, 1950–1960. [Google Scholar] [CrossRef]
- Meng, X.; Tan, J.; Li, M.; Song, S.; Miao, Y.; Zhang, Q. Sirt1: Role Under the Condition of Ischemia/Hypoxia. Cell. Mol. Neurobiol. 2017, 37, 17–28. [Google Scholar] [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Singh, S.; Aggarwal, B.B. Identification of a p65 peptide that selectively inhibits NF-kappa B activation induced by various inflammatory stimuli and its role in down-regulation of NF-kappaB-mediated gene expression and up-regulation of apoptosis. J. Biol. Chem. 2004, 279, 15096–15104. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Ren, W.; Li, W.; Zhao, S.; Mi, H.; Wang, R.; Zhang, Y.; Wu, W.; Nan, Y.; Yu, J. Activation of peroxisome proliferator activated receptor alpha ameliorates ethanol induced steatohepatitis in mice. Lipids Health Dis. 2011, 10, 246. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar]
- Gao, Y.; Zhou, Z.; Ren, T.; Kim, S.J.; He, Y.; Seo, W.; Guillot, A.; Ding, Y.; Wu, R.; Shao, S.; et al. Alcohol inhibits T-cell glucose metabolism and hepatitis in ALDH2-deficient mice and humans: Roles of acetaldehyde and glucocorticoids. Gut 2019, 68, 1311–1322. [Google Scholar] [CrossRef]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef]
- Ji, C.; Kaplowitz, N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef]
- Khanal, T.; Choi, J.H.; Hwang, Y.P.; Chung, Y.C.; Jeong, H.G. Protective effects of saponins from the root of Platycodon grandiflorum against fatty liver in chronic ethanol feeding via the activation of AMP-dependent protein kinase. Food Chem. Toxicol. 2009, 47, 2749–2754. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, J.W.; Jung, Y.S.; Kwon, D.Y.; Park, H.K.; Ryu, C.S.; Kim, S.K.; Oh, G.T.; Kim, Y.C. Ethanol-induced liver injury and changes in sulfur amino acid metabolomics in glutathione peroxidase and catalase double knockout mice. J. Hepatol. 2009, 50, 1184–1191. [Google Scholar] [CrossRef]
- Dickinson, D.A.; Forman, H.J. Cellular glutathione and thiols metabolism. Biochem. Pharmacol. 2002, 64, 1019–1026. [Google Scholar] [CrossRef]
- Das, S.K.; Vasudevan, D.M. Alcohol-induced oxidative stress. Life Sci. 2007, 81, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.L.; Wan, J.B.; Wang, B.; He, C.W.; Ma, H.; Li, T.W.; Kang, J.X. Suppression of acute ethanol-induced hepatic steatosis by docosahexaenoic acid is associated with downregulation of stearoyl-CoA desaturase 1 and inflammatory cytokines. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Yang, W.; Xue, Q.; Gao, L.; Huo, J.; Ren, D.; Chen, X. Rutin ameliorates diabetic neuropathy by lowering plasma glucose and decreasing oxidative stress via Nrf2 signaling pathway in rats. Eur. J. Pharmacol. 2016, 771, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.Q.; Kim, Y.S.; Choi, Y.J.; Kim, Y.C.; Sohn, D.H.; Ryu, S.Y.; Lee, B.H. Effects of tanshinone IIA on the hepatotoxicity and gene expression involved in alcoholic liver disease. Arch. Pharm. Res. 2008, 31, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Ki, S.H.; Park, O.; Zheng, M.; Morales-Ibanez, O.; Kolls, J.K.; Bataller, R.; Gao, B. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: Role of signal transducer and activator of transcription 3. Hepatology 2010, 52, 1291–1300. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagappan, A.; Kim, J.-H.; Jung, D.Y.; Jung, M.H. Cryptotanshinone from the Salvia miltiorrhiza Bunge Attenuates Ethanol-Induced Liver Injury by Activation of AMPK/SIRT1 and Nrf2 Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 265. https://doi.org/10.3390/ijms21010265
Nagappan A, Kim J-H, Jung DY, Jung MH. Cryptotanshinone from the Salvia miltiorrhiza Bunge Attenuates Ethanol-Induced Liver Injury by Activation of AMPK/SIRT1 and Nrf2 Signaling Pathways. International Journal of Molecular Sciences. 2020; 21(1):265. https://doi.org/10.3390/ijms21010265
Chicago/Turabian StyleNagappan, Arulkumar, Ji-Hyun Kim, Dae Young Jung, and Myeong Ho Jung. 2020. "Cryptotanshinone from the Salvia miltiorrhiza Bunge Attenuates Ethanol-Induced Liver Injury by Activation of AMPK/SIRT1 and Nrf2 Signaling Pathways" International Journal of Molecular Sciences 21, no. 1: 265. https://doi.org/10.3390/ijms21010265
APA StyleNagappan, A., Kim, J.-H., Jung, D. Y., & Jung, M. H. (2020). Cryptotanshinone from the Salvia miltiorrhiza Bunge Attenuates Ethanol-Induced Liver Injury by Activation of AMPK/SIRT1 and Nrf2 Signaling Pathways. International Journal of Molecular Sciences, 21(1), 265. https://doi.org/10.3390/ijms21010265