High Fat Diet Suppresses Peroxisome Proliferator-Activated Receptors and Reduces Dopaminergic Neurons in the Substantia Nigra


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
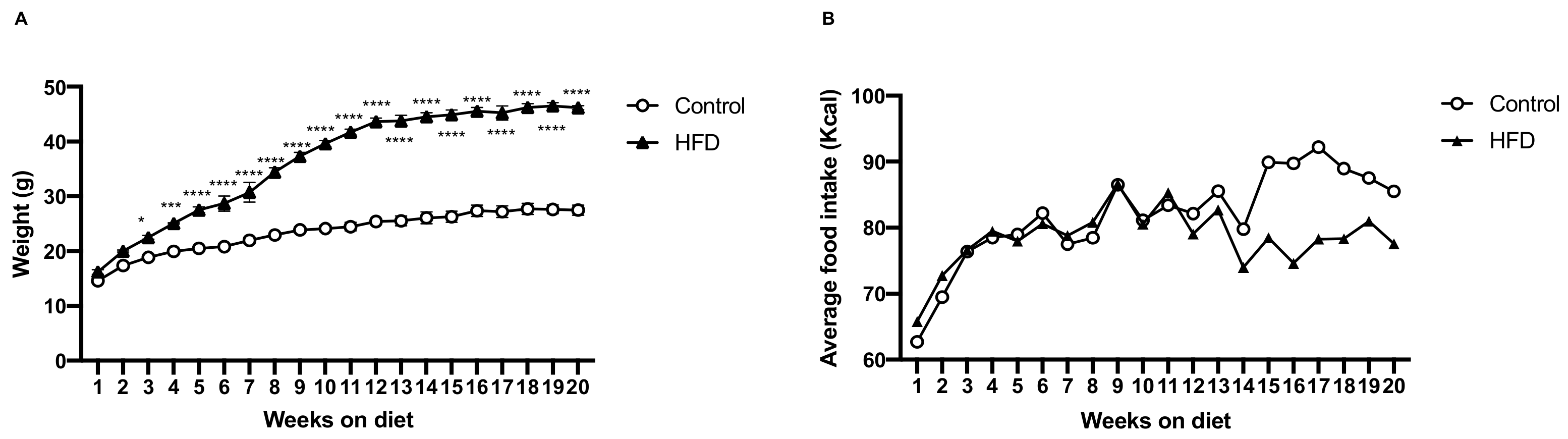
2.1. HFD Induces More Body Weight Gain
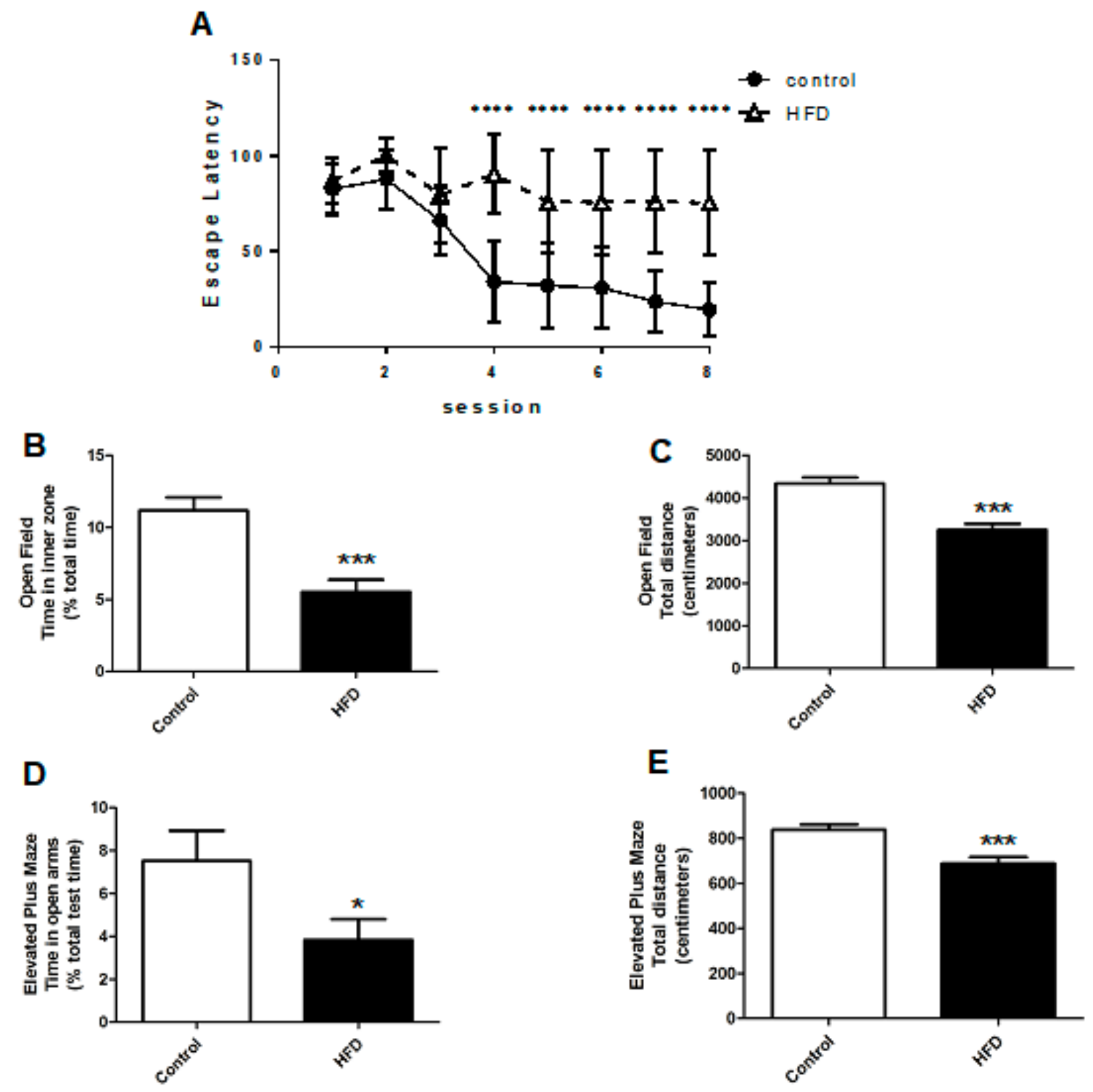
2.2. HFD Causes Cognitive Impairment, Increased Anxiety, and Decreased Locomotor Function
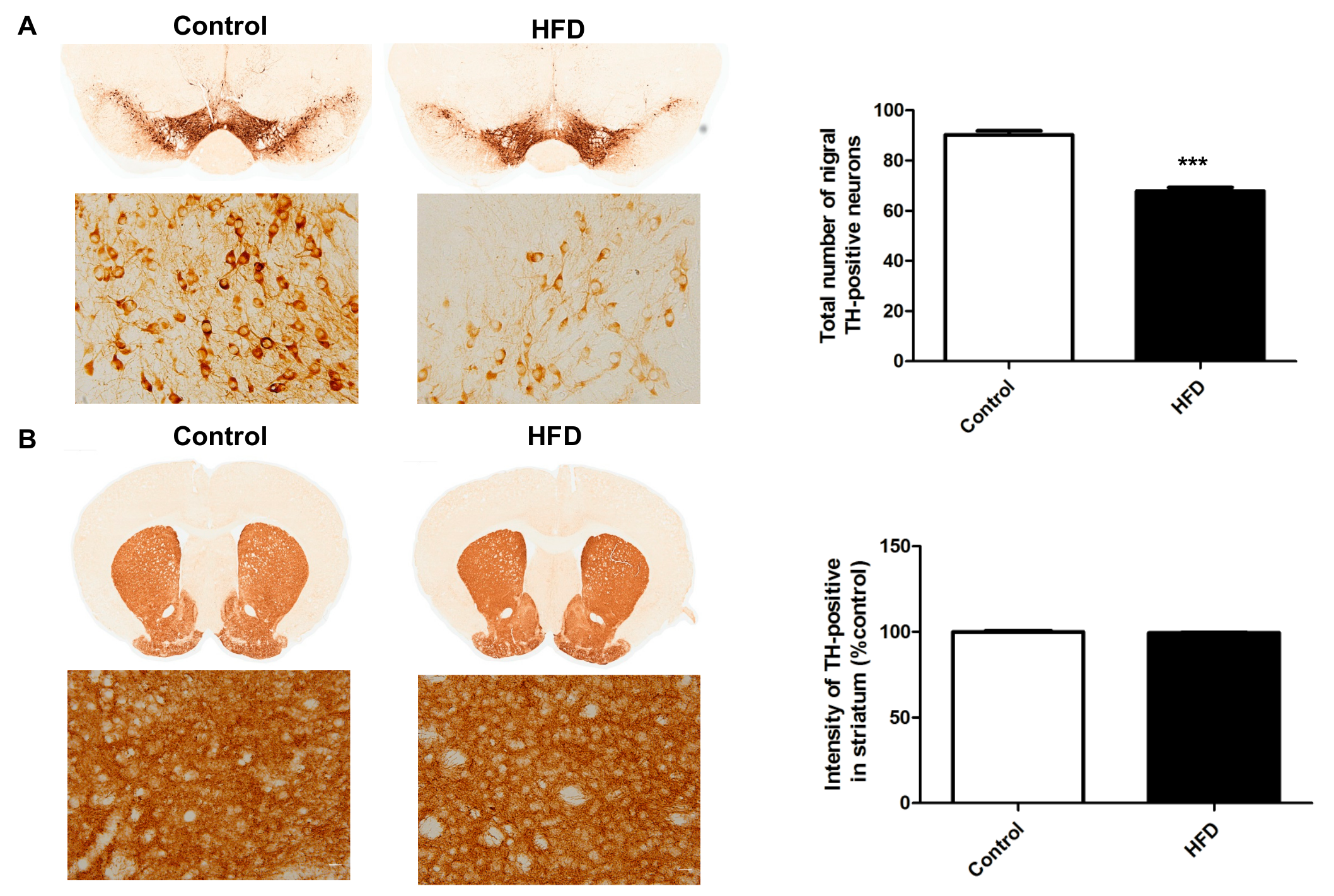
2.3. HFD Causes Decreased Dopaminergic Neurons in the SN
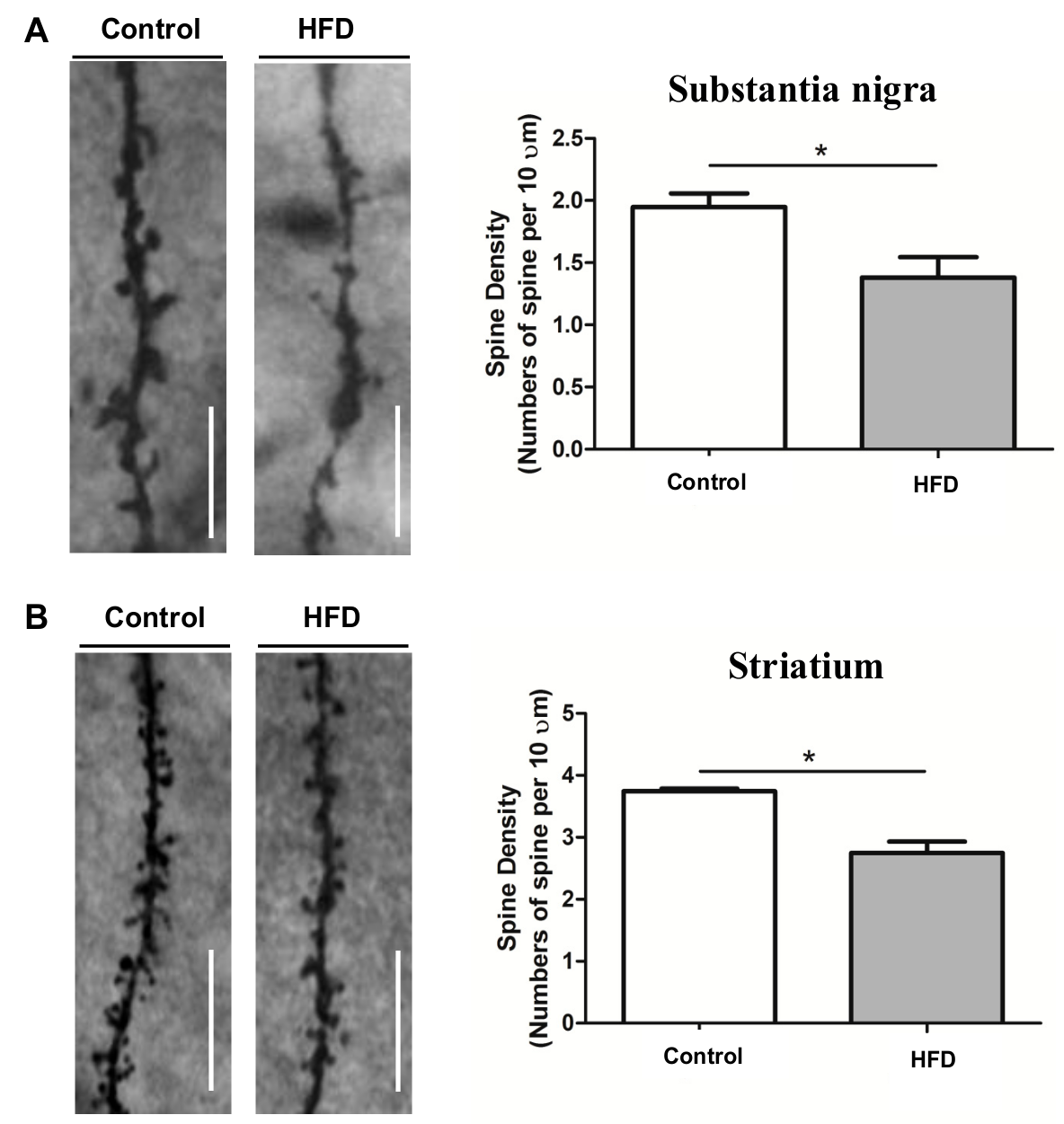
2.4. HFD Causes Reduced Dendritic Spine Density in the SN and Striatum
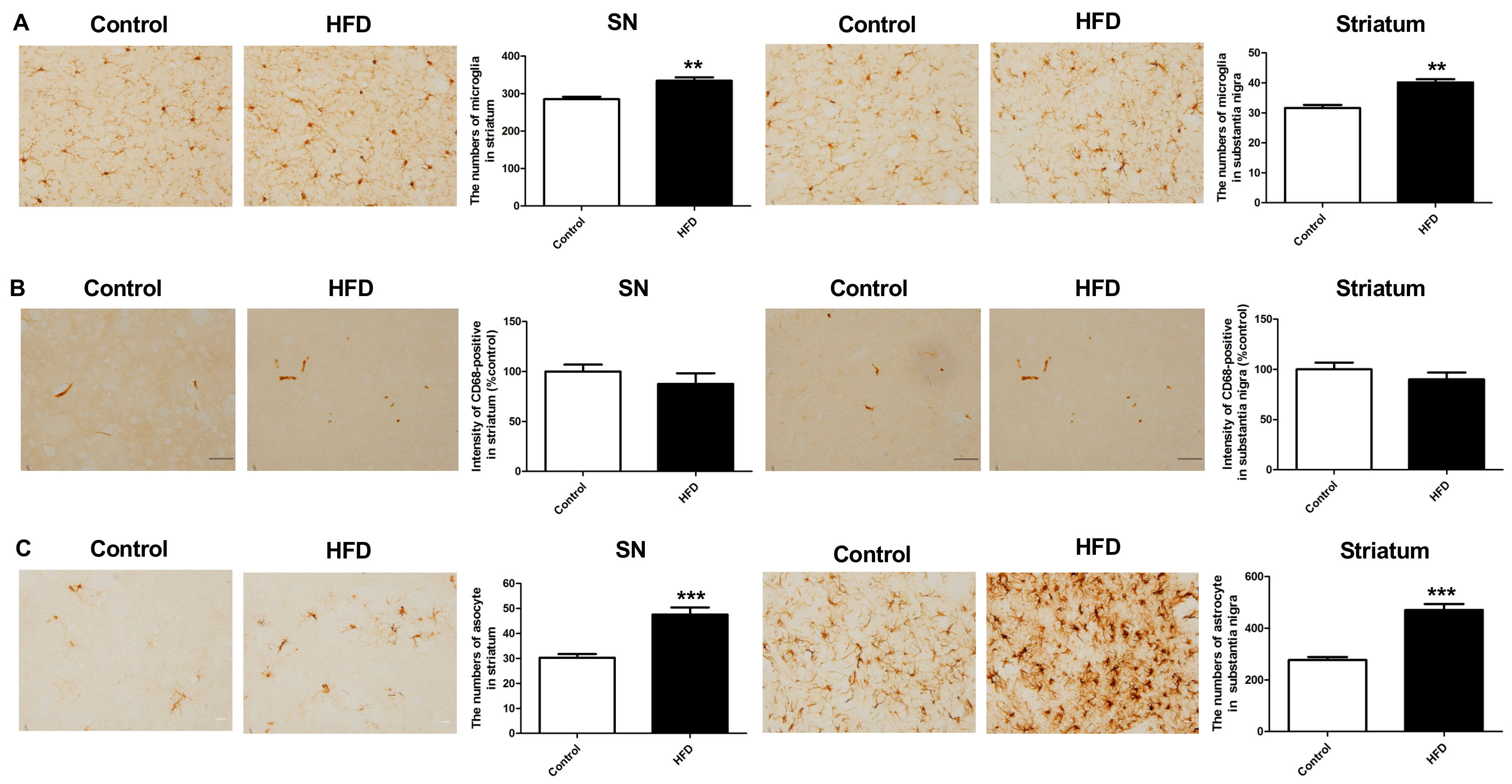
2.5. HFD Results in Increased Neuroinflammation in the Nigrostriatal Pathway
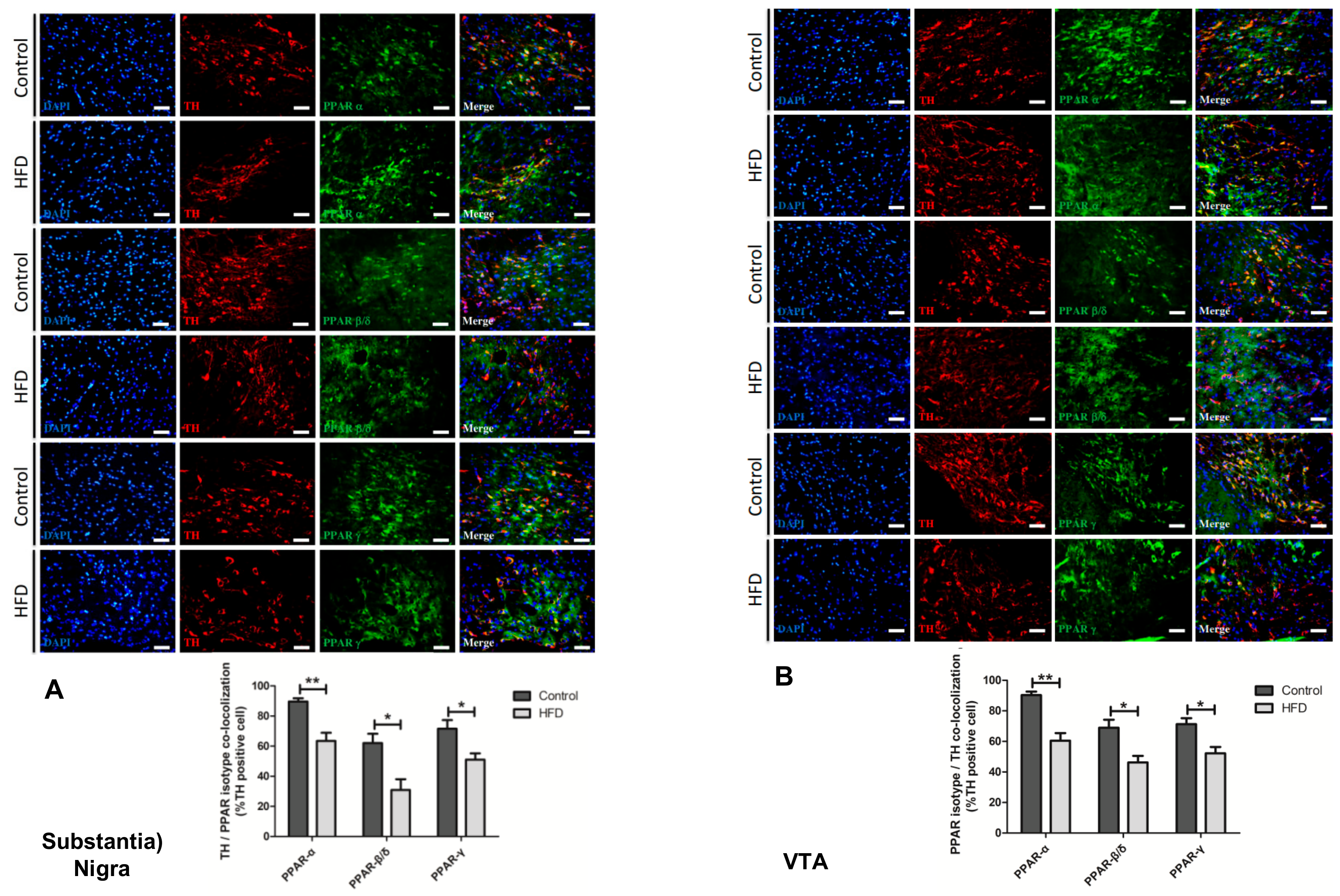
2.6. HFD Causes Reduction in All PPAR Isotype Levels in the Dopaminergic Neurons of the SN and VTA
3. Discussion
4. Materials and Methods
4.1. Animals and Diets
4.2. Neurobehavioral Tests
4.2.1. Morris Water Maze
4.2.2. Open-Field Test (OFT)
4.2.3. Elevated Plus Maze (EPM)
4.3. Brain Tissue Preparation
4.4. TH Staining
4.5. Immunohistochemistry for Neuroinflammation
4.6. Golgi Staining for Dendritic Spines
4.7. Immunofluorescence Staining for PPARs
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| AD | Alzheimer disease |
| HFD | High fat diet |
| DA | Dopamine |
| DIO | Diet-induced obesity |
| EPM | Elevated plus maze |
| OFT | Open field test |
| PD | Parkinson disease |
| PPAR | Peroxisome proliferator-activated receptor |
| SN | Substantia nigra |
| TH | Tyrosine hydroxylase |
| VTA | Ventral tegmental area |
References
- Scott, K.M.; McGee, M.A.; Wells, J.E.; Oakley Browne, M.A. Obesity and mental disorders in the adult general population. J. Psychosom. Res. 2008, 64, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.L.; White, K.S. The Role of Anxiety in Binge Eating Behavior: A Critical Examination of Theory and Empirical Literature. Health Psychol. Res. 2013, 1, e19. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metabolism 2016, 65, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Mazon, J.N.; de Mello, A.H.; Ferreira, G.K.; Rezin, G.T. The impact of obesity on neurodegenerative diseases. Life Sci. 2017, 182, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Tatton, W.G. Etiology and pathogenesis of parkinson’s disease. Annu. Rev. Neurosci. 1999, 22, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Sethi, K. Nonmotor manifestations of Parkinson’s disease. Ann. Neurol. 2008, 64 (Suppl. 2), S65–S80. [Google Scholar] [CrossRef]
- Knab, A.M.; Lightfoot, J.T. Does the difference between physically active and couch potato lie in the dopamine system? Int. J. Biol. Sci. 2010, 6, 133–150. [Google Scholar] [CrossRef]
- Bjorklund, A.; Dunnett, S.B. Dopamine neuron systems in the brain: An update. Trends Neurosci. 2007, 30, 194–202. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef]
- Papagno, C.; Trojano, L. Cognitive and behavioral disorders in Parkinson’s disease: An update. I: Cognitive impairments. Neurol. Sci. 2018, 39, 215–223. [Google Scholar] [CrossRef]
- Krishna, S.; Keralapurath, M.M.; Lin, Z.; Wagner, J.J.; de La Serre, C.B.; Harn, D.A.; Filipov, N.M. Neurochemical and electrophysiological deficits in the ventral hippocampus and selective behavioral alterations caused by high-fat diet in female C57BL/6 mice. Neuroscience 2015, 297, 170–181. [Google Scholar] [CrossRef]
- Krishna, S.; Lin, Z.; de La Serre, C.B.; Wagner, J.J.; Harn, D.H.; Pepples, L.M.; Djani, D.M.; Weber, M.T.; Srivastava, L.; Filipov, N.M. Time-dependent behavioral, neurochemical, and metabolic dysregulation in female C57BL/6 mice caused by chronic high-fat diet intake. Physiol. Behav. 2016, 157, 196–208. [Google Scholar] [CrossRef]
- Rollins, C.P.E.; Gallino, D.; Kong, V.; Ayranci, G.; Devenyi, G.A.; Germann, J.; Chakravarty, M.M. Contributions of a high-fat diet to Alzheimer’s disease-related decline: A longitudinal behavioural and structural neuroimaging study in mouse models. Neuroimage Clin. 2019, 21, 101606. [Google Scholar] [CrossRef]
- Boitard, C.; Etchamendy, N.; Sauvant, J.; Aubert, A.; Tronel, S.; Marighetto, A.; Laye, S.; Ferreira, G. Juvenile, but not adult exposure to high-fat diet impairs relational memory and hippocampal neurogenesis in mice. Hippocampus 2012, 22, 2095–2100. [Google Scholar] [CrossRef]
- Sivanathan, S.; Thavartnam, K.; Arif, S.; Elegino, T.; McGowan, P.O. Chronic high fat feeding increases anxiety-like behaviour and reduces transcript abundance of glucocorticoid signalling genes in the hippocampus of female rats. Behav. Brain Res. 2015, 286, 265–270. [Google Scholar] [CrossRef]
- Prasad, A.; Prasad, C. Short-term consumption of a diet rich in fat decreases anxiety response in adult male rats. Physiol. Behav. 1996, 60, 1039–1042. [Google Scholar] [CrossRef]
- McNeilly, A.D.; Stewart, C.A.; Sutherland, C.; Balfour, D.J. High fat feeding is associated with stimulation of the hypothalamic-pituitary-adrenal axis and reduced anxiety in the rat. Psychoneuroendocrinology 2015, 52, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Maniam, J.; Morris, M.J. Long-term postpartum anxiety and depression-like behavior in mother rats subjected to maternal separation are ameliorated by palatable high fat diet. Behav. Brain Res. 2010, 208, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Lee, M.J.; Han, J.; Kim, S.J.; Ryu, I.; Ju, X.; Ryu, M.J.; Chung, W.; Oh, E.; Kweon, G.R.; et al. A High-fat Diet Induces a Loss of Midbrain Dopaminergic Neuronal Function That Underlies Motor Abnormalities. Exp. Neurobiol. 2017, 26, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Friend, D.M.; Devarakonda, K.; O’Neal, T.J.; Skirzewski, M.; Papazoglou, I.; Kaplan, A.R.; Liow, J.S.; Guo, J.; Rane, S.G.; Rubinstein, M.; et al. Basal Ganglia Dysfunction Contributes to Physical Inactivity in Obesity. Cell Metab. 2017, 25, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Khang, R.; Park, C.; Shin, J.H. Dysregulation of parkin in the substantia nigra of db/db and high-fat diet mice. Neuroscience 2015, 294, 182–192. [Google Scholar] [CrossRef]
- Naef, L.; Pitman, K.A.; Borgland, S.L. Mesolimbic dopamine and its neuromodulators in obesity and binge eating. CNS Spectr. 2015, 20, 574–583. [Google Scholar] [CrossRef]
- Wilcox, C.E.; Braskie, M.N.; Kluth, J.T.; Jagust, W.J. Overeating Behavior and Striatal Dopamine with 6-[F]-Fluoro-L-m-Tyrosine PET. J. Obes. 2010, 2010. [Google Scholar] [CrossRef]
- Johnson, P.M.; Kenny, P.J. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat. Neurosci. 2010, 13, 635–641. [Google Scholar] [CrossRef]
- Vucetic, Z.; Carlin, J.L.; Totoki, K.; Reyes, T.M. Epigenetic dysregulation of the dopamine system in diet-induced obesity. J. Neurochem. 2012, 120, 891–898. [Google Scholar] [CrossRef]
- Wang, G.J.; Volkow, N.D.; Logan, J.; Pappas, N.R.; Wong, C.T.; Zhu, W.; Netusil, N.; Fowler, J.S. Brain dopamine and obesity. Lancet 2001, 357, 354–357. [Google Scholar] [CrossRef]
- Murray, S.; Tulloch, A.; Gold, M.S.; Avena, N.M. Hormonal and neural mechanisms of food reward, eating behaviour and obesity. Nat. Rev. Endocrinol. 2014, 10, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Buaud, B.; Esterle, L.; Vaysse, C.; Alfos, S.; Combe, N.; Higueret, P.; Pallet, V. A high-fat diet induces lower expression of retinoid receptors and their target genes GAP-43/neuromodulin and RC3/neurogranin in the rat brain. Br. J. Nutr. 2010, 103, 1720–1729. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.I.; Nomoto, M.; Fukuda, T. Regulation of GAP-43 protein and mRNA in nigrostriatal dopaminergic neurons after the partial destruction of dopaminergic terminals with intrastriatal 6-hydroxydopamine. Synapse 2001, 39, 16–22. [Google Scholar] [CrossRef]
- Sharma, S.; Taliyan, R. High fat diet feeding induced insulin resistance exacerbates 6-OHDA mediated neurotoxicity and behavioral abnormalities in rats. Behav. Brain Res. 2018, 351, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Diano, S.; Liu, Z.W.; Jeong, J.K.; Dietrich, M.O.; Ruan, H.B.; Kim, E.; Suyama, S.; Kelly, K.; Gyengesi, E.; Arbiser, J.L.; et al. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat. Med. 2011, 17, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Truckenmuller, F.M.; Schell, H.; Kahle, P.J. Diet-induced obesity accelerates the onset of terminal phenotypes in alpha-synuclein transgenic mice. J. Neurochem. 2014, 131, 848–858. [Google Scholar] [CrossRef]
- Betarbet, R.; Turner, R.; Chockkan, V.; DeLong, M.R.; Allers, K.A.; Walters, J.; Levey, A.I.; Greenamyre, J.T. Dopaminergic neurons intrinsic to the primate striatum. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 6761–6768. [Google Scholar] [CrossRef]
- Maysami, S.; Haley, M.J.; Gorenkova, N.; Krishnan, S.; McColl, B.W.; Lawrence, C.B. Prolonged diet-induced obesity in mice modifies the inflammatory response and leads to worse outcome after stroke. J. Neuroinflamm. 2015, 12, 140. [Google Scholar] [CrossRef]
- Jayaraman, A.; Lent-Schochet, D.; Pike, C.J. Diet-induced obesity and low testosterone increase neuroinflammation and impair neural function. J. Neuroinflamm. 2014, 11, 162. [Google Scholar] [CrossRef]
- Sobesky, J.L.; Barrientos, R.M.; De May, H.S.; Thompson, B.M.; Weber, M.D.; Watkins, L.R.; Maier, S.F. High-fat diet consumption disrupts memory and primes elevations in hippocampal IL-1beta, an effect that can be prevented with dietary reversal or IL-1 receptor antagonism. Brain Behav. Immun. 2014, 42, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Guillemot-Legris, O.; Masquelier, J.; Everard, A.; Cani, P.D.; Alhouayek, M.; Muccioli, G.G. High-fat diet feeding differentially affects the development of inflammation in the central nervous system. J. Neuroinflamm. 2016, 13, 206. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Sarasua, S.; Moustafa, S.; Garcia-Aviles, A.; Lopez-Climent, M.F.; Gomez-Cadenas, A.; Olucha-Bordonau, F.E.; Sanchez-Perez, A.M. The effect of abscisic acid chronic treatment on neuroinflammatory markers and memory in a rat model of high-fat diet induced neuroinflammation. Nutr. Metab. (Lond.) 2016, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.B.; Koo, J.H.; Jang, Y.C.; Yang, C.H.; Lee, Y.; Cosio-Lima, L.M.; Cho, J.Y. Neuroprotective Effects of Endurance Exercise Against High-Fat Diet-Induced Hippocampal Neuroinflammation. J. Neuroendocrinol. 2016, 28. [Google Scholar] [CrossRef] [PubMed]
- De Luca, S.N.; Ziko, I.; Sominsky, L.; Nguyen, J.C.; Dinan, T.; Miller, A.A.; Jenkins, T.A.; Spencer, S.J. Early life overfeeding impairs spatial memory performance by reducing microglial sensitivity to learning. J. Neuroinflamm. 2016, 13, 112. [Google Scholar] [CrossRef]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef]
- Moody, L.; Xu, G.B.; Chen, H.; Pan, Y.X. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 141–152. [Google Scholar] [CrossRef]
- Ryan, K.K.; Li, B.; Grayson, B.E.; Matter, E.K.; Woods, S.C.; Seeley, R.J. A role for central nervous system PPAR-gamma in the regulation of energy balance. Nat. Med. 2011, 17, 623–626. [Google Scholar] [CrossRef]
- Cifani, C.; Micioni Di Bonaventura, M.V.; Pucci, M.; Giusepponi, M.E.; Romano, A.; Di Francesco, A.; Maccarrone, M.; D’Addario, C. Regulation of hypothalamic neuropeptides gene expression in diet induced obesity resistant rats: Possible targets for obesity prediction? Front. Neurosci. 2015, 9, 187. [Google Scholar] [CrossRef]
- Poon, K.; Alam, M.; Karatayev, O.; Barson, J.R.; Leibowitz, S.F. Regulation of the orexigenic neuropeptide, enkephalin, by PPARdelta and fatty acids in neurons of the hypothalamus and forebrain. J. Neurochem. 2015, 135, 918–931. [Google Scholar] [CrossRef]
- Kocalis, H.E.; Turney, M.K.; Printz, R.L.; Laryea, G.N.; Muglia, L.J.; Davies, S.S.; Stanwood, G.D.; McGuinness, O.P.; Niswender, K.D. Neuron-specific deletion of peroxisome proliferator-activated receptor delta (PPARdelta) in mice leads to increased susceptibility to diet-induced obesity. PLoS ONE 2012, 7, e42981. [Google Scholar] [CrossRef] [PubMed]
- Stump, M.; Guo, D.F.; Lu, K.T.; Mukohda, M.; Liu, X.; Rahmouni, K.; Sigmund, C.D. Effect of selective expression of dominant-negative PPARgamma in pro-opiomelanocortin neurons on the control of energy balance. Physiol Genom. 2016, 48, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Sarruf, D.A.; Talukdar, S.; Sharma, S.; Li, P.; Bandyopadhyay, G.; Nalbandian, S.; Fan, W.; Gayen, J.R.; Mahata, S.K.; et al. Brain PPAR-gamma promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nat. Med. 2011, 17, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Galan-Rodriguez, B.; Suarez, J.; Gonzalez-Aparicio, R.; Bermudez-Silva, F.J.; Maldonado, R.; Robledo, P.; Rodriguez de Fonseca, F.; Fernandez-Espejo, E. Oleoylethanolamide exerts partial and dose-dependent neuroprotection of substantia nigra dopamine neurons. Neuropharmacology 2009, 56, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Aparicio, R.; Flores, J.A.; Tasset, I.; Tunez, I.; Fernandez-Espejo, E. Mice lacking the peroxisome proliferator-activated receptor alpha gene present reduced number of dopamine neurons in the substantia nigra without altering motor behavior or dopamine neuron decline over life. Neuroscience 2011, 186, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Impellizzeri, D.; Mazzon, E.; Paterniti, I.; Cuzzocrea, S. Neuroprotective activities of palmitoylethanolamide in an animal model of Parkinson’s disease. PLoS ONE 2012, 7, e41880. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, J.K.; Santiago, R.; Tonin, F.S.; Boschen, S.; da Silva, L.M.; Werner, M.F.; da Cunha, C.; Lima, M.M.; Vital, M.A. PPAR-alpha agonist fenofibrate protects against the damaging effects of MPTP in a rat model of Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 53, 35–44. [Google Scholar] [CrossRef]
- Avagliano, C.; Russo, R.; De Caro, C.; Cristiano, C.; La Rana, G.; Piegari, G.; Paciello, O.; Citraro, R.; Russo, E.; De Sarro, G.; et al. Palmitoylethanolamide protects mice against 6-OHDA-induced neurotoxicity and endoplasmic reticulum stress: In vivo and in vitro evidence. Pharmacol. Res. 2016, 113, 276–289. [Google Scholar] [CrossRef]
- Barbiero, J.K.; Santiago, R.M.; Persike, D.S.; da Silva Fernandes, M.J.; Tonin, F.S.; da Cunha, C.; Lucio Boschen, S.; Lima, M.M.; Vital, M.A. Neuroprotective effects of peroxisome proliferator-activated receptor alpha and gamma agonists in model of parkinsonism induced by intranigral 1-methyl-4-phenyl-1,2,3,6-tetrahyropyridine. Behav. Brain Res. 2014, 274, 390–399. [Google Scholar] [CrossRef]
- Scheggi, S.; Melis, M.; De Felice, M.; Aroni, S.; Muntoni, A.L.; Pelliccia, T.; Gambarana, C.; De Montis, M.G.; Pistis, M. PPARalpha modulation of mesolimbic dopamine transmission rescues depression-related behaviors. Neuropharmacology 2016, 110, 251–259. [Google Scholar] [CrossRef]
- Lee, Y.; Cho, J.H.; Lee, S.; Lee, W.; Chang, S.C.; Chung, H.Y.; Moon, H.R.; Lee, J. Neuroprotective effects of MHY908, a PPAR alpha/gamma dual agonist, in a MPTP-induced Parkinson’s disease model. Brain Res. 2019, 1704, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Mounsey, R.B.; Martin, H.L.; Nelson, M.C.; Evans, R.M.; Teismann, P. The effect of neuronal conditional knock-out of peroxisome proliferator-activated receptors in the MPTP mouse model of Parkinson’s disease. Neuroscience 2015, 300, 576–584. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martin, H.L.; Mounsey, R.B.; Sathe, K.; Mustafa, S.; Nelson, M.C.; Evans, R.M.; Teismann, P. A peroxisome proliferator-activated receptor-delta agonist provides neuroprotection in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neuroscience 2013, 240, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Wu, L.; Gao, Q.; Ou, Z.; Zhu, D.; Zhang, Y. PPARbeta/delta Agonist Provides Neuroprotection by Suppression of IRE1alpha-Caspase-12-Mediated Endoplasmic Reticulum Stress Pathway in the Rotenone Rat Model of Parkinson’s Disease. Mol. Neurobiol. 2016, 53, 3822–3831. [Google Scholar] [CrossRef]
- Swanson, C.; Emborg, M. Expression of peroxisome proliferator-activated receptor-gamma in the substantia nigra of hemiparkinsonian nonhuman primates. Neurol. Res. 2014, 36, 634–646. [Google Scholar] [CrossRef]
- Kapadia, R.; Yi, J.H.; Vemuganti, R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front. Biosci 2008, 13, 1813–1826. [Google Scholar] [CrossRef]
- Breidert, T.; Callebert, J.; Heneka, M.T.; Landreth, G.; Launay, J.M.; Hirsch, E.C. Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson’s disease. J. Neurochem. 2002, 82, 615–624. [Google Scholar] [CrossRef]
- Garrido-Gil, P.; Joglar, B.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Involvement of PPAR-gamma in the neuroprotective and anti-inflammatory effects of angiotensin type 1 receptor inhibition: Effects of the receptor antagonist telmisartan and receptor deletion in a mouse MPTP model of Parkinson’s disease. J. Neuroinflamm. 2012, 9, 38. [Google Scholar] [CrossRef]
- Sadeghian, M.; Marinova-Mutafchieva, L.; Broom, L.; Davis, J.B.; Virley, D.; Medhurst, A.D.; Dexter, D.T. Full and partial peroxisome proliferation-activated receptor-gamma agonists, but not delta agonist, rescue of dopaminergic neurons in the 6-OHDA parkinsonian model is associated with inhibition of microglial activation and MMP expression. J. Neuroimmunol. 2012, 246, 69–77. [Google Scholar] [CrossRef]
- Swanson, C.R.; Joers, V.; Bondarenko, V.; Brunner, K.; Simmons, H.A.; Ziegler, T.E.; Kemnitz, J.W.; Johnson, J.A.; Emborg, M.E. The PPAR-gamma agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J. Neuroinflamm. 2011, 8, 91. [Google Scholar] [CrossRef]
- Hunter, R.L.; Choi, D.Y.; Ross, S.A.; Bing, G. Protective properties afforded by pioglitazone against intrastriatal LPS in Sprague-Dawley rats. Neurosci. Lett. 2008, 432, 198–201. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kao, Y.-C.; Wei, W.-Y.; Tsai, K.-J.; Wang, L.-C. High Fat Diet Suppresses Peroxisome Proliferator-Activated Receptors and Reduces Dopaminergic Neurons in the Substantia Nigra. Int. J. Mol. Sci. 2020, 21, 207. https://doi.org/10.3390/ijms21010207
Kao Y-C, Wei W-Y, Tsai K-J, Wang L-C. High Fat Diet Suppresses Peroxisome Proliferator-Activated Receptors and Reduces Dopaminergic Neurons in the Substantia Nigra. International Journal of Molecular Sciences. 2020; 21(1):207. https://doi.org/10.3390/ijms21010207
Chicago/Turabian StyleKao, Yu-Chia, Wei-Yen Wei, Kuen-Jer Tsai, and Liang-Chao Wang. 2020. "High Fat Diet Suppresses Peroxisome Proliferator-Activated Receptors and Reduces Dopaminergic Neurons in the Substantia Nigra" International Journal of Molecular Sciences 21, no. 1: 207. https://doi.org/10.3390/ijms21010207
APA StyleKao, Y.-C., Wei, W.-Y., Tsai, K.-J., & Wang, L.-C. (2020). High Fat Diet Suppresses Peroxisome Proliferator-Activated Receptors and Reduces Dopaminergic Neurons in the Substantia Nigra. International Journal of Molecular Sciences, 21(1), 207. https://doi.org/10.3390/ijms21010207