Knowledge Generation with Rule Induction in Cancer Omics


 , ,
, ,  and
and 
Abstract
1. Introduction
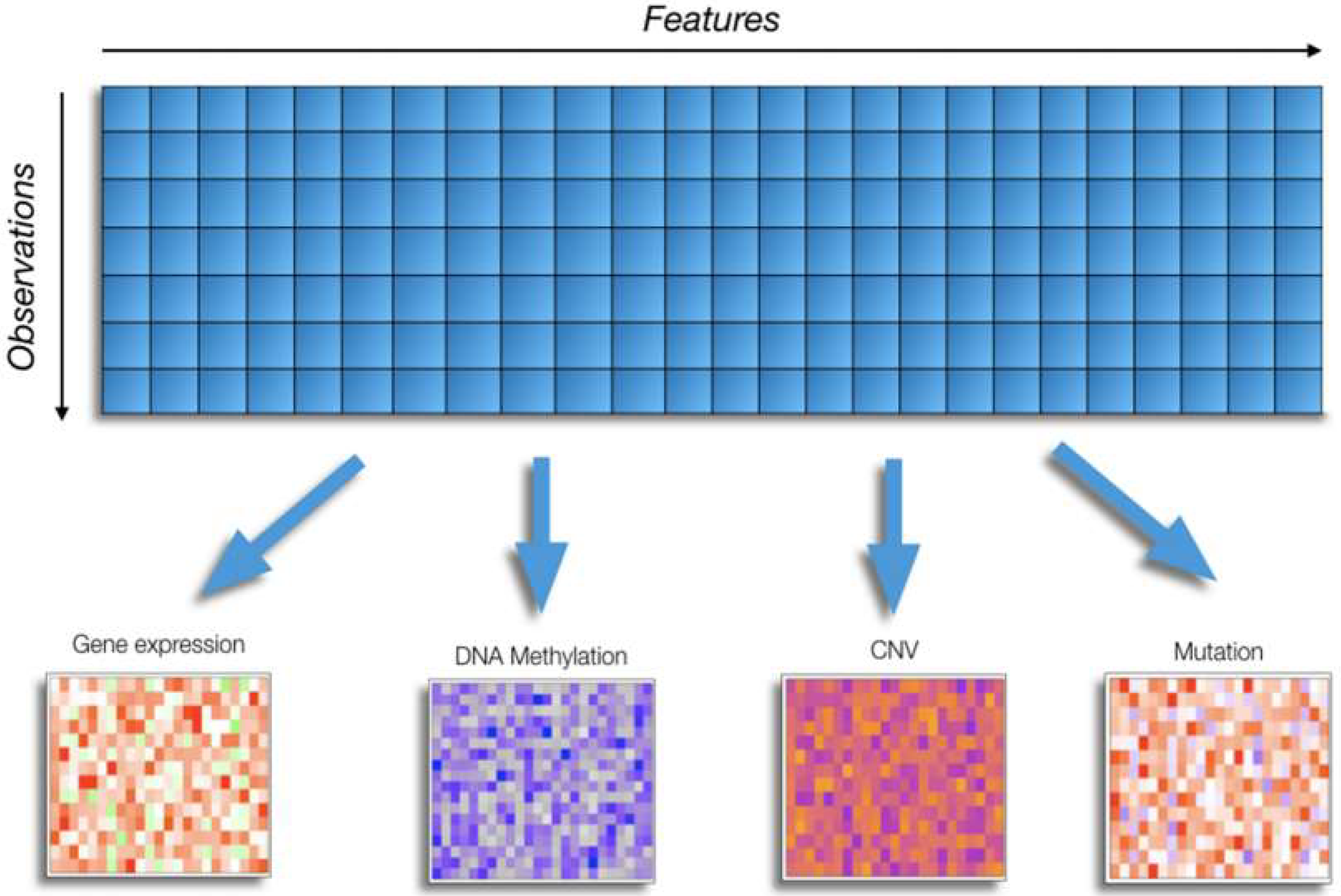
2. Challenges of Omics Data Analysis in Cancer Research
3. Rule Induction
4. Useful Structure of Rules for Cancer Omics Data
- It is important for the algorithm to be stable over the generated rule sets. In particular, one would expect to obtain the same (or at least highly similar) rule sets when running the algorithm on different versions of the learning substrate. This also guarantees that the system is capable of capturing as many as possible of the representable relationships in the chosen formalism. When generating knowledge in cancer studies, it is desirable to generate the complete set only once and to guarantee that it is not dependent on the employed training set.
- The generated rule set, should contain all and only the most relevant relationships, thus keeping the number of rules as lower as possible. This can be obtained by removing the redundant rules and/or the rules covering only a few specific cases of a class.
- The generated rules should be based on all and only the features that are directly related with the predicted variable. In cancer research, the major interest is the identification of key molecular actors useful for (1) the comprehension of the mechanisms leading to the malignant transformation and (2) the identification of therapeutic targets.
- The ideal rule-based model should exhibit highly accurate levels, thus guaranteeing that the set of rules is powerful enough to capture putative relationships between the features and the classes.
- The rule set contains no redundant rules. That means that each association rule of the set must contain information not deducible from other rules
- The obtained rule set should contain all the rules involving relevant features for the phenomenon. Cancer omics data analysis often shows that few features (e.g., expression status of a few genes) are sufficient to classify the disease with appreciable accuracy. Therefore, only one model (e.g., the one with higher accuracy) is usually chosen. In rule induction for knowledge extraction, the aim is not only limited to the classification performances of the model. In this setting, indeed, it is necessary for a rule base to contain as many as possible of the rules involving relevant features, even if this leads to a redundant classification system. Such an approach enables to capture many putative relationships linking causative factors to the phenomenon under study. This approach has been successfully implemented in CAMUR [42] where the rule induction algorithm has been specifically designed to learn alternative and equivalent solutions instead of a single rule set containing few but highly discriminant rules.
5. Feature Selection and Representation
- Technical feature selection strategies,
- knowledge-driven feature selection strategies, and
- hybrid feature selection strategies.
6. Rule Base Evaluation
- Coverage of cases (classification oriented): namely the fraction of observations from the training set whose features’ values satisfies at least one rule. This metric summarizes the fractions of different learnt relationships driving each class from the input features. The extremely high value of this indicator should also be treated with caution since they can be an index of overfitting.
- Coverage of features (knowledge discovery oriented): namely the fraction of relevant features that appear in at least one rule. This metric is particularly important in cancer rule induction, since the main interest in this activity is to explore as many relationships as possible that link the relevant molecular features with the phenotype of interest.
7. Knowledge Representation
8. Integrating Multi-Omics and Non-Omics Features
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TCGA | The Cancer Genome Atlas |
| ICGC | International Cancer Genome Consortium |
| ML | Machine learning |
| SAMGSR | Gene-set analysis-based reduction algorithm |
| RRFE | Reweighted Recursive Feature Elimination |
| GELnet | Generalized Elastic Net |
| ESTs | Expressed Sequence Tags |
| ICA | Independent Component Analysis |
| PCA | Principal Component Analysis |
| NMF | Non-negative Matrix Factorization |
| HTS | High-Throughput Sequencing |
| SVM | Support Vector Machine |
| RF | Random Forest |
| ANFIS | Adaptive Neuro-Fuzzy Inference Systems |
| RIPPER | Repeated Incremental Pruning to Produce Error Reduction |
| PART | Partial Decision Trees |
| CAMUR | Classifier with Alternative and MUltiple Rule-based models |
| FURIA | Fuzzy Unordered Rule Induction Algorithm |
| MLRules | Maximum Likelihood Rule Ensembles |
| LERS | Learning from Examples based on Rough Sets |
| TSP | Top Scoring Pairs |
| k-TSP | k- Top Scoring Pairs |
| BIOHEL | Bioinformatics-oriented Hierarchical Evolutionary Learning |
| CN2-SD | Clark & Niblet–Subgroup Discovery |
| SDEFSR | Subgroup Discovery with Evolutionary Fuzzy Systems |
| VarSelRF | Variable Selection using Random Forests |
| SVM-RFE | Support Vector Machines–Recursive Feature Elimination |
| FPRF | fuzzy pattern–random forest |
| CART | Classification And Regression Tree |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| Ridge | Tikhonov regularization |
References
- Weinstein, I.B.; Case, K. The history of Cancer Research: Introducing an AACR Centennial series. Cancer Res. 2008, 68, 6861–6862. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Researchers suggest that universal ‘law’ governs tumor growth. J. Natl. Cancer Inst. 2003, 95, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Sogn, J.A.; Anton-Culver, H.; Singer, D.S. Meeting report: NCI think tanks in cancer biology. Cancer Res. 2005, 65, 9117–9120. [Google Scholar] [CrossRef] [PubMed]
- Sompairac, N.; Nazarov, P.V.; Czerwinska, U.; Cantini, L.; Biton, A.; Molkenov, A.; Zhumadilov, Z.; Barillot, E.; Radvanyi, F.; Gorban, A.; et al. Independent Component Analysis for Unraveling the Complexity of Cancer Omics Datasets. Int. J. Mol. Sci. 2019, 20, 4414. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, M.; Ma, S. Integrative Analysis of Cancer Omics Data for Prognosis Modeling. Genes 2019, 10, 604. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.H.; Beam, A.L.; Kohane, I.S. Artificial intelligence in healthcare. Nat. Biomed. Eng. 2018, 2, 719–731. [Google Scholar] [CrossRef]
- Richter, A.N.; Khoshgoftaar, T.M. A review of statistical and machine learning methods for modeling cancer risk using structured clinical data. Artif. Intell. Med. 2018, 90, 1–14. [Google Scholar] [CrossRef]
- Huang, C.; Clayton, E.A.; Matyunina, L.V.; McDonald, L.D.; Benigno, B.B.; Vannberg, F.; McDonald, J.F. Machine learning predicts individual cancer patient responses to therapeutic drugs with high accuracy. Sci. Rep. 2018, 8, 16444. [Google Scholar] [CrossRef]
- Huang, C.; Mezencev, R.; McDonald, J.F.; Vannberg, F. Open source machine-learning algorithms for the prediction of optimal cancer drug therapies. PLoS ONE 2017, 12, e0186906. [Google Scholar] [CrossRef]
- Ali, M.; Aittokallio, T. Machine learning and feature selection for drug response prediction in precision oncology applications. Biophys. Rev. 2019, 11, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Paskov, I.; Paskov, H.; González, A.J.; Leslie, C.S. Multitask learning improves prediction of cancer drug sensitivity. Sci. Rep. 2016, 6, 31619. [Google Scholar] [CrossRef] [PubMed]
- Dorman, S.N.; Baranova, K.; Knoll, J.H.; Urquhart, B.L.; Mariani, G.; Carcangiu, M.L.; Rogan, P.K. Genomic signatures for paclitaxel and gemcitabine resistance in breast cancer derived by machine learning. Mol. Oncol. 2016, 10, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Song, N.; Shen, R.; Arora, A.; Machiela, M.J.; Song, L.; Landi, M.T.; Ghosh, D.; Chatterjee, N.; Baladandayuthapani, V.; et al. Integrating Clinical and Multiple Omics Data for Prognostic Assessment across Human Cancers. Sci. Rep. 2017, 7, 16954. [Google Scholar] [CrossRef] [PubMed]
- Esteva, A.; Kuprel, B.; Novoa, R.A.; Ko, J.; Swetter, S.M.; Blau, H.M.; Thrun, S. Dermatologist-level classification of skin cancer with deep neural networks. Nature 2017, 542, 115–118. [Google Scholar] [CrossRef]
- Grapov, D.; Fahrmann, J.; Wanichthanarak, K.; Khoomrung, S. Rise of Deep Learning for Genomic, Proteomic, and Metabolomic Data Integration in Precision Medicine. J. Integr. Biol. 2018, 22, 630–636. [Google Scholar] [CrossRef]
- Kaufman, S.; Rosset, S.; Perlich, C.; Stitelman, O. Leakage in data mining: Formulation, detection, and avoidance. ACM Trans. Knowl. Discov. Data 2012, 6, 4. [Google Scholar] [CrossRef]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15, 20170387. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef]
- Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome Profiling in Human Diseases: New Advances and Perspectives. Int. J. Mol. Sci. 2017, 18, 1652. [Google Scholar] [CrossRef] [PubMed]
- Vitali, F.; Li, Q.; Schissler, A.G.; Berghout, J.; Kenost, C.; Lussier, Y.A. Developing a ‘personalome’ for precision medicine: Emerging methods that compute interpretable effect sizes from single-subject transcriptomes. Brief. Bioinform. 2019, 20, 789–805. [Google Scholar] [CrossRef] [PubMed]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J.K. Review of applications of high-throughput sequencing in personalized medicine: Barriers and facilitators of future progress in research and clinical application. Brief. Bioinform. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Altman, N.; Krzywinski, M. The curse(s) of dimensionality. Nat. Methods 2018, 15, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.W.B.; Wang, W.; Wong, L. Why Batch Effects Matter in Omics Data, and How to Avoid Them. Trends Biotechnol. 2017, 35, 498–507. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Tamborero, D.; Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Kandoth, C.; Reimand, J.; Lawrence, M.S.; Getz, G.; Bader, G.D.; Ding, L.; et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013, 3, 2650. [Google Scholar] [CrossRef]
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Dubitzky, W.; Granzow, M.; Berrar, D. Comparing symbolic and subsymbolic machine learning approaches to classification of cancer and gene identification. In Methods of Microarray Data Analysis; Springer: Boston, MA, USA, 2002; pp. 151–165. [Google Scholar]
- Zilke, J.R.; Loza Mencía, E.; Janssen, F. DeepRED—Rule Extraction from Deep Neural Networks. In Discovery Science. Lecture Notes in Computer Science; Calders, T., Ceci, M., Malerba, D., Eds.; Springer: Cham, Switzerland, 2016; p. 9956. [Google Scholar]
- Bologna, G. A Simple Convolutional Neural Network with Rule Extraction. Appl. Sci. 2019, 9, 2411. [Google Scholar] [CrossRef]
- Friedman, J.H.; Popescu, B.E. Predictive learning via rule ensembles. Ann. Appl. Stat. 2008, 2, 916–954. [Google Scholar] [CrossRef]
- Mashayekhi, M.; Gras, R. Rule Extraction from Random Forest: The RF+HC Methods. In Advances in Artificial Intelligence. Lecture Notes in Computer Science; Barbosa, D., Milios, E., Eds.; Springer: Cham, Switzerland, 2015; p. 9091. [Google Scholar]
- Barakat, N.H.; Bradley, A.P. Rule Extraction from Support Vector Machines: A Sequential Covering Approach. IEEE Trans. Knowl. Data Eng. 2007, 19, 729–741. [Google Scholar] [CrossRef]
- Sharma, M.; Mukharjee, S. Artificial Neural Network Fuzzy Inference System (ANFIS) for brain tumor detection. arXiv 2012, arXiv:1212.0059. [Google Scholar]
- Paper, C.; Adib, M.; Sarker, M.H.; Ahmed, S.; Ariwa, E. Networked Digital Technologies; Communications in Computer and Information Science; Springer: Berlin, Germany, 2014; p. 293. [Google Scholar]
- Nascimento, A.C.A.; Prudêncio, R.B.C.; de Souto, M.C.P.; Costa, I.G. Mining Rules for the Automatic Selection Process of Clustering Methods Applied to Cancer Gene Expression Data. In Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2009; p. 5769. [Google Scholar]
- Geman, D.; d’Avignon, C.; Naiman, D.; Winslow, R. Classifying Gene Expression Profiles from Pairwise mRNA Comparisons. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Naiman, D.; Xu, L.; Winslow, R.; Geman, D. Simple decision rules for classifying human cancers from gene expression profiles. Bioinformatics 2005, 21, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Regev, A.; Elidan, G. Expression Profiles. Bioinformatics 2001, 17, 3896–3904. [Google Scholar]
- Cestarelli, V.; Fiscon, G.; Felici, G.; Bertolazzi, P.; Weitschek, E. CAMUR: Knowledge extraction from RNA-seq cancer data through equivalent classification rules. Bioinformatics 2016, 32, 697–704. [Google Scholar] [CrossRef]
- Weitschek, E.; Lauro, S.D.; Cappelli, E.; Bertolazzi, P.; Felici, G. CamurWeb: A classification software and a large knowledge base for gene expression data of cancer. BMC Bioinform. 2018, 19, 354. [Google Scholar] [CrossRef]
- Celli, F.; Cumbo, F.; Weitschek, E. Classification of Large DNA Methylation Datasets for Identifying Cancer Drivers. Big Data Res. 2018, 13, 21–28. [Google Scholar] [CrossRef]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef]
- Quinlan, J.R. C4.5: Programs for Machine Learning; Morgan Kaufmann Publishers: Burlington, MA, USA, 1993. [Google Scholar]
- Frank, E.; Hall, M.A.; Witten, I.H. The WEKA Workbench. In Online Appendix for "Data Mining: Practical Machine Learning Tools and Techniques", 4th ed.; Morgan Kaufmann Publishers: Burlington, MA, USA, 2016. [Google Scholar]
- Cohen, W.W. Fast effective rule induction. In Proceedings of the Twelfth International Conference of Machine learning, Tahoe City, CA, USA, 9–12 July 1995; pp. 115–123. [Google Scholar]
- Eibe, F.; Witten, I.H. Generating Accurate Rule Sets Without Global Optimization. In Proceedings of the Fifteenth International Conference on Machine Learning, Madison, WI, USA, 24–27 July 1998; pp. 144–151. [Google Scholar]
- Gasparovica, M.; Aleksejeva, L. Using Fuzzy Unordered Rule Induction Algorithm for cancer data classification. Breast Cancer 2011, 13, 1229. [Google Scholar]
- Dembczyński, K.; Kotłowski, W.; Słowiński, R. Maximum likelihood rule ensembles. In Proceedings of the 25th International Conference on Machine Learning (ICML 2008), Helsinki, Finland, 5–9 July 2008. [Google Scholar]
- Grzymala-Busse, J.W. A local version of the MLEM2 algorithm for rule induction. Fundam. Inform. 2010, 100, 1–18. [Google Scholar] [CrossRef]
- Bacardit, J.; Krasnogor, N. Biohel: Bioinformatics-oriented hierarchical evolutionary learning. In Nottingham eprints; University of Nottingham: Nottingham, UK, 2006. [Google Scholar]
- Lavrač, N.; Kavšek, B.; Flach, P.; Todorovsky, L. Subgroup Discovery with CN2-SD. J. Mach. Learn. Res. 2004, 5, 153–188. [Google Scholar]
- García, Á.; Charte, F.; González, P.; Carmona, C.; Jesus, M. Subgroup Discovery with Evolutionary Fuzzy Systems in R: The SDEFSR Package. R J. 2016, 8, 307. [Google Scholar] [CrossRef]
- Kira, K.; Rendell, L. The Feature Selection Problem: Traditional Methods and a New Algorithm. In Proceedings of the AAAI-92 Tenth National Conference on Artificial Intelligence, San Jose, CA, USA, 12–16 July 1992; pp. 129–134. [Google Scholar]
- Vergara, J.R.; Estévez, P.A. A Review of Feature Selection Methods Based on Mutual Information. Neural Comput. Appl. 2014, 24, 175–186. [Google Scholar] [CrossRef]
- Kursa, M.B.; Jankowski, A.; Witold, R.R. Boruta-A system for feature selection. Fundam. Inform. 2010, 101, 271–285. [Google Scholar] [CrossRef]
- Diaz-Uriarte, R. GeneSrF and varSelRF: A web-based tool and R package for gene selection and classification using random forest. BMC Bioinform. 2007, 8, 328. [Google Scholar] [CrossRef]
- Adorada, A.; Permatasari, R.; Wirawan, P.W.; Wibowo, A.; Sujiwo, A. Support Vector Machine - Recursive Feature Elimination (SVM - RFE) for Selection of MicroRNA Expression Features of Breast Cancer. In Proceedings of the 2018 2nd International Conference on Informatics and Computational Sciences (ICICoS), Semarang, Indonesia, 30–31 October 2018; IEEE: Piscataway, NJ, USA, 2018. [Google Scholar]
- Fortino, V.; Kinaret, P.; Fyhrquist, N.; Alenius, H.; Greco, D. A robust and accurate method for feature selection and prioritization from multi-class OMICs data. PLoS ONE 2014, 9, e107801. [Google Scholar] [CrossRef]
- Breiman, L. Classification and Regression Trees; Wadsworth International Group: San Francisco, CA, USA, 1984. [Google Scholar]
- Quinlan, J.R. Induction of Decision Trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef]
- Santosa, F.; Symes, W.W. Linear inversion of band-limited reflection seismograms. SIAM J. Sci. Stat. Comput. 1986, 7, 1307–1330. [Google Scholar] [CrossRef]
- Hoerl, A.E.; Kennard, R.W. Ridge regression: Biased estimation for nonorthogonal problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and Variable Selection via the Elastic Net. J. R. Stat. Soc. 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Dinu, I.; Potter, J.D.; Mueller, T.; Qi, L.; Adeniyi, J.A.; Gian, S.J.; Gunilla, E.; Famulski, K.S.; Halloran, P.; Yasui, Y. Gene-set analysis and reduction. Brief. Bioinform. 2009, 10, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Johannes, M.; Brase, J.C.; Fröhlich, H.; Gade, S.; Gehrmann, M.; Fälth, M.; Sültmann, H.; Beißbarth, T. Integration of pathway knowledge into a reweighted recursive feature elimination approach for risk stratification of cancer patients. Bioinformatics 2010, 26, 2136–2144. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, A.; Carlin, D.E.; Paull, E.O.; Baertsch, R.; Stuart, J.M. Pathway-Based Genomics Prediction using Generalized Elastic Net. PLoS Comput. Biol. 2016, 12, e1004790. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Chen, J.; Yan, K.; Jin, Q.; Xue, Y.; Gao, Z. A hybrid feature selection algorithm for gene expression data classification. Neurocomputing 2017, 256, 56–62. [Google Scholar] [CrossRef]
- Lee, C.P.; Leu, Y. A novel hybrid feature selection method for microarray data analysis. Appl. Soft Comput. 2011, 11, 208–213. [Google Scholar] [CrossRef]
- Zhou, W.; Koudijs, K.K.M.; Böhringer, S. Influence of batch effect correction methods on drug induced differential gene expression profiles. BMC Bioinform. 2019, 20, 437. [Google Scholar] [CrossRef]
- Papiez, A.; Marczyk, M.; Polanska, J.; Polanski, A. BatchI: Batch effect Identification in high-throughput screening data using a dynamic programming algorithm. Bioinformatics 2019, 35, 1885–1892. [Google Scholar] [CrossRef]
- Oytam, Y.; Sobhanmanesh, F.; Duesing, K.; Bowden, J.C.; Osmond-McLeod, M.; Ross, J. Risk-conscious correction of batch effects: Maximising information extraction from high-throughput genomic datasets. BMC Bioinform. 2016, 17, 332. [Google Scholar] [CrossRef]
- Yi, H.; Raman, A.T.; Zhang, H.; Allen, G.I.; Liu, Z. Detecting hidden batch factors through data-adaptive adjustment for biological effects. Bioinformatics 2018, 34, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Mitra, G.; Sundareisan, S.; Sarkar, B.K. A simple data discretizer. arXiv 2017, arXiv:1710.05091. [Google Scholar]
- Gallo, C.A.; Cecchini, R.L.; Carballido, J.A.; Micheletto, S.; Ponzoni, I. Discretization of gene expression data revised. Brief. Bioinform. 2016, 17, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Huerta, E.B.; Duval, B.; Hao, J.K. Fuzzy logic for elimination of redundant information of microarray data. Genom. Proteom. Bioinform. 2008, 6, 61–73. [Google Scholar] [CrossRef]
- Glez-Peña, D.; Alvarez, R.; Díaz, F.; Fdez-Riverola, F. DFP: A Bioconductor package for fuzzy profile identification and gene reduction of microarray data. BMC Bioinform. 2009, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Mirza, B.; Wang, W.; Wang, J.; Choi, H.; Chung, N.C.; Ping, P. Machine Learning and Integrative Analysis of Biomedical Big Data. Genes 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.H.; Hassen, R.M.K.; Le, N.T. Combinatorial Roles of DNA Methylation and Histone Modifications on Gene Expression; Springer International Publishing: Cham, Switzerland, 2015; pp. 123–135. [Google Scholar]
- Cappelli, E.; Felici, G.; Weitschek, E. Combining DNA methylation and RNA sequencing data of cancer for supervised knowledge extraction. BioData Min. 2018, 11, 22. [Google Scholar] [CrossRef]


{kind=link}
| Tool | Strategy | Output | Implementation | Language |
|---|---|---|---|---|
| C4.5 [46] | Decision tree | Decision trees | WEKA [47]/J48 | Java, R, Python |
| RIPPER (Repeated Incremental Pruning to Produce Error Reduction) [48] | Sequential covering | Rule set | WEKA/JRip | Java, R, Python |
| PART (Partial Decision Trees) [49] | Sequential covering | Rule set | WEKA/PART | Java, R, Python |
| CAMUR (Classifier with Alternative and MUltiple Rule-based models) [42,43] | Sequential covering | Rule set | CAMUR website 1,2 | Java |
| BIGBIOCL [44] | Sequential covering | Rule set | BIGBIOCL github 3 | Java |
| FURIA (Fuzzy Unordered Rule Induction Algorithm) [50] | Sequential covering | Fuzzy rule set | WEKA/FURIA | Java, R, Python |
| MLRules (Maximum Likelihood Rule Ensembles) [51] | Sequential covering and probability estimation | Rule set | MLRules website 4 | Java |
| LERS (Learning from Examples based on Rough Sets) [52] | Rough set theory | Rule set | R/RoughSets package | R |
| TSP (Top Scoring Pairs) [39] | Rank based | Rule set | R/tspair package | R |
| k-TSP (k - Top Scoring Pairs) [40] | Rank based | Rule set | R/switchbox package | R |
| BIOHEL (Bioinformatics-oriented Hierarchical Evolutionary Learning) [53] | Evolutionary rule learning | Rule set | BIOHEL website 5 | C++ |
| CN2-SD (Clark & Niblet – Subgroup Discovery) [54] | Subgroup discovery | Rule set | CN2-SD website 6 | Java |
| SDEFSR (Subgroup Discovery with Evolutionary Fuzzy Systems) [55] | Subgroup discovery | Fuzzy rule set | R/SDEFSR | R |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scala, G.; Federico, A.; Fortino, V.; Greco, D.; Majello, B. Knowledge Generation with Rule Induction in Cancer Omics. Int. J. Mol. Sci. 2020, 21, 18. https://doi.org/10.3390/ijms21010018
Scala G, Federico A, Fortino V, Greco D, Majello B. Knowledge Generation with Rule Induction in Cancer Omics. International Journal of Molecular Sciences. 2020; 21(1):18. https://doi.org/10.3390/ijms21010018
Chicago/Turabian StyleScala, Giovanni, Antonio Federico, Vittorio Fortino, Dario Greco, and Barbara Majello. 2020. "Knowledge Generation with Rule Induction in Cancer Omics" International Journal of Molecular Sciences 21, no. 1: 18. https://doi.org/10.3390/ijms21010018
APA StyleScala, G., Federico, A., Fortino, V., Greco, D., & Majello, B. (2020). Knowledge Generation with Rule Induction in Cancer Omics. International Journal of Molecular Sciences, 21(1), 18. https://doi.org/10.3390/ijms21010018