25-Hydroxyvitamin D Inhibits Hepatitis C Virus Production in Hepatocellular Carcinoma Cell Line by a Vitamin D Receptor-Independent Mechanism

Abstract
1. Introduction
2. Results
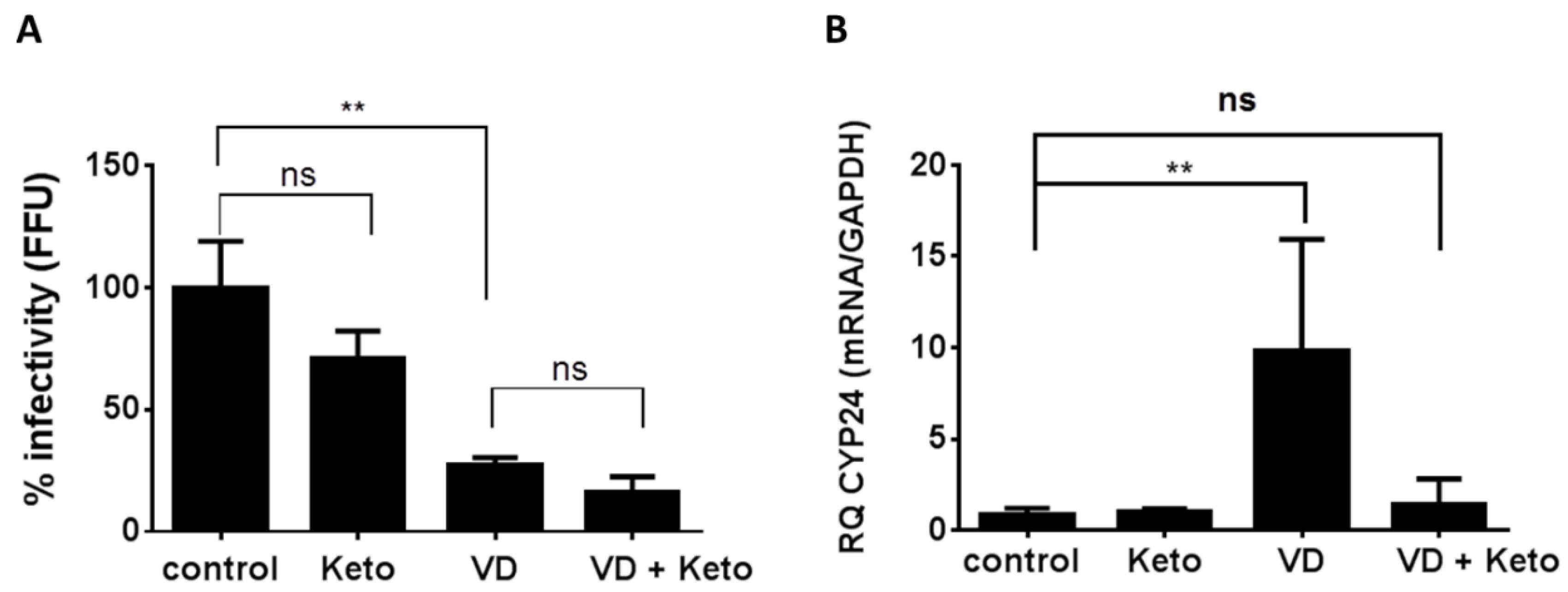
2.1. Involvement of Calcitriol in the Anti-HCV Activity of Vitamin D3
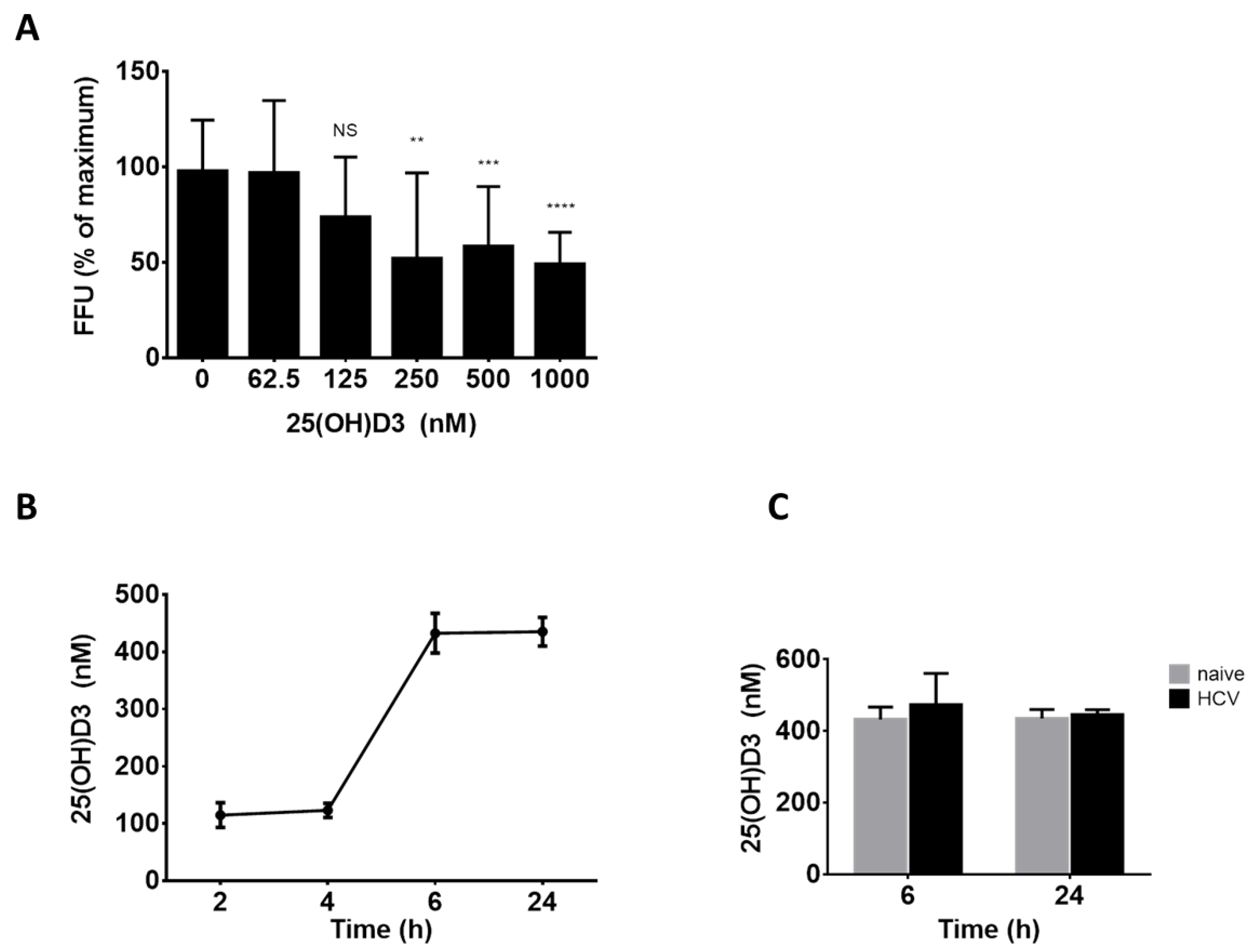
2.2. The Role of 25(OH)D3 as a Direct Mediator of the Antiviral Activity of Vitamin D3
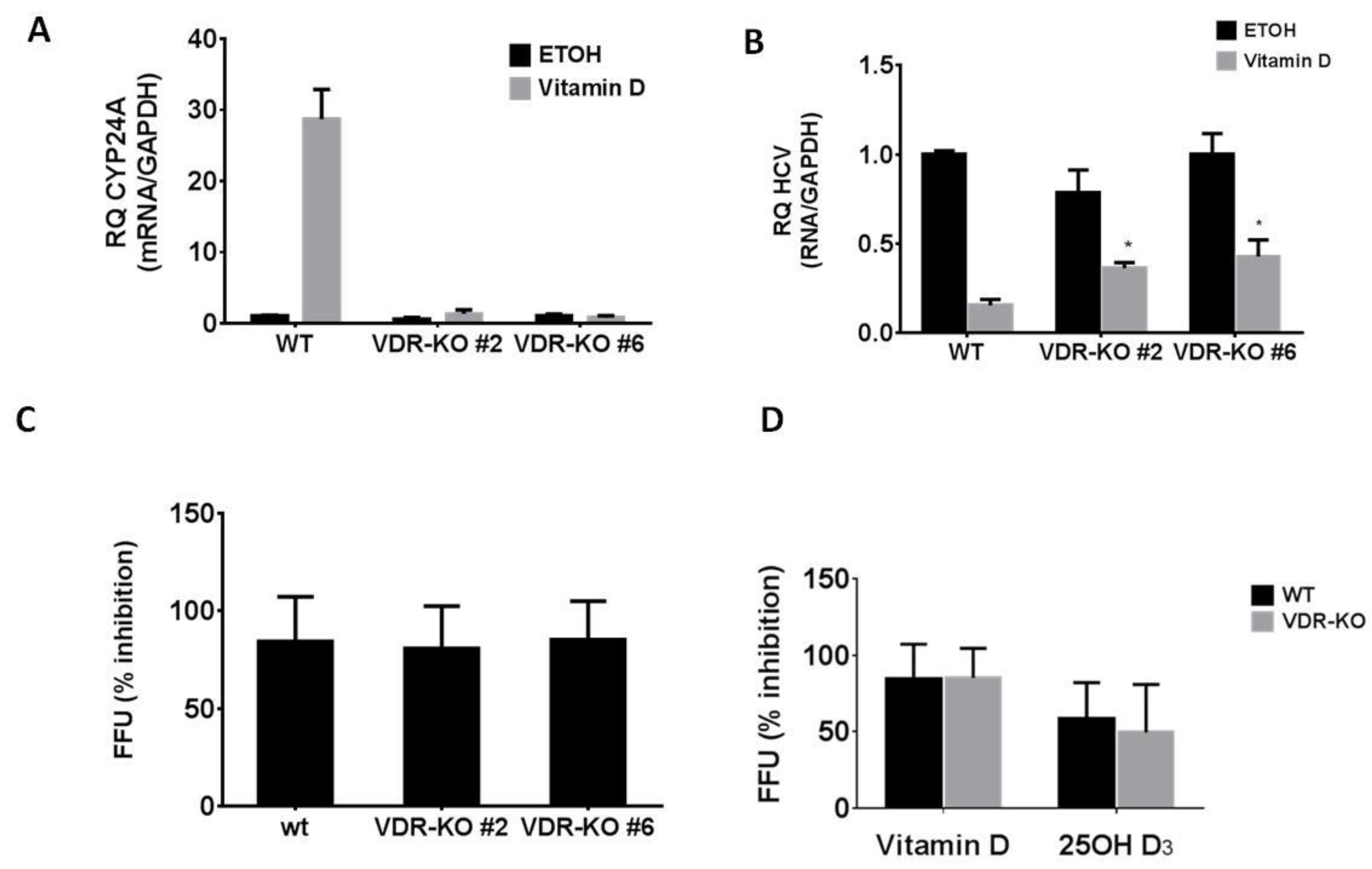
2.3. The Role of VDR in the Anti-HCV Activity of Vitamin D3
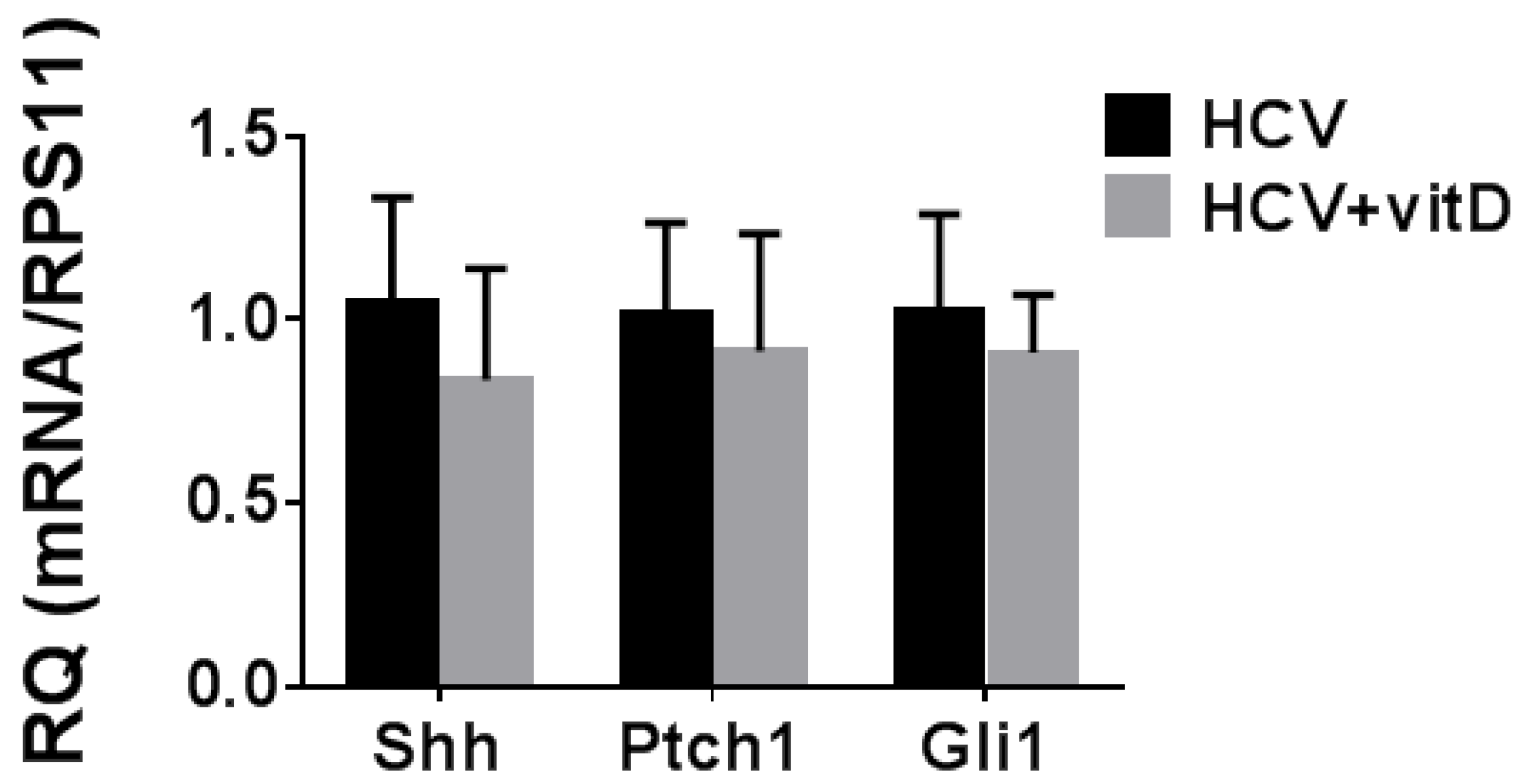
2.4. Involvement of the Hh Pathway in the Anti-HCV Activity of Vitamin D3
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cells
4.3. Inhibition of Infectious Virus Production
4.4. Inhibition of 1,25-Dihydroxyvitamin D Production
4.5. RNA Isolation and cDNA Synthesis
4.6. Quantitative Real-time RT-PCR
4.7. (OH)D3 Determination
4.8. Cell Viability Assay
4.9. VDR Knockout Cells
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lou, Y.R.; Molnár, F.; Peräkylä, M.; Qiao, S.; Kalueff, A.V.; St-Arnaud, R.; Carlberg, C.; Tuohimaa, P. 25-Hydroxyvitamin D3is an agonistic vitamin D receptor ligand. J. Steroid Biochem. Mol. Biol. 2010, 118, 162–170. [Google Scholar] [CrossRef]
- Lou, Y.R.; Laaksi, I.; Syvälä, H.; Bläuer, M.; Tammela, T.L.J.; Ylikomi, T.; Tuohimaa, P. 25-hydroxyvitamin D3 is an active hormone in human primary prostatic stromal cells. FASEB J. 2004, 18, 332–334. [Google Scholar] [CrossRef]
- Jones, G.; Prosser, D.E.; Kaufmann, M. Cytochrome P450-mediated metabolism of vitamin D. J. Lipid Res. 2014, 55, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D.; Patzek, S.; Yang, W. Physiologic and pathophysiologic roles of extra renal CYP27b1: Case report and review. Bone Rep. 2018, 8, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Robin, N.C.; Agoston, Z.; Biechele, T.L.; James, R.G.; Berndt, J.D.; Moon, R.T. Accumulation of the Vitamin D Precursor Cholecalciferol Antagonizes Hedgehog Signaling to Impair Hemogenic Endothelium Formation Mauricio. Stem Cell Rep. 2014, 2, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Spek, C.A.; Zivkovic, D.; van de Water, S.; Rezaee, F.; Peppelenbosch, M.P. Repression of smoothened by patched-dependent (pro-)vitamin D3 secretion. PLoS Biol. 2006, 4, 1397–1410. [Google Scholar] [CrossRef]
- Tang, J.Y.; Xiao, T.Z.; Oda, Y.; Chang, K.S.; Shpall, E.; Wu, A.; So, P.L.; Hebert, J.; Bikle, D.; Epstein, E.H. Vitamin D3 inhibits hedgehog signaling and proliferation in murine basal cell carcinomas. Cancer Prev. Res. 2011, 4, 744–751. [Google Scholar] [CrossRef]
- Banerjee, U.; Ghosh, M.; Kyle Hadden, M. Evaluation of vitamin D3 A-ring analogues as Hedgehog pathway inhibitors. Bioorganic Med. Chem. Lett. 2012, 22, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Hadden, M.K. Hedgehog and Vitamin D Signaling Pathways in Development and Disease, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Vol. 100, ISBN 9780128048245. [Google Scholar]
- Susa, T.; Iizuka, M.; Okinaga, H.; Tamamori-Adachi, M.; Okazaki, T. Without 1α-hydroxylation, the gene expression profile of 25(OH)D3treatment overlaps deeply with that of 1,25(OH)2D3in prostate cancer cells. Sci. Rep. 2018, 8, 2–11. [Google Scholar] [CrossRef]
- Beloso, C.; Souto, J.; Fábregat, M.; Romanelli, G.; Javiel, G.M.A. Vitamin D deficiency and hepatitis viruses-associated liver diseases: A literature review. World J. Diabetes 2018, 9, 157–164. [Google Scholar] [CrossRef]
- Nimer, A.; Mouch, A. Vitamin D improves viral response in hepatitis C genotype 2-3 naïve patients. World J. Gastroenterol. 2012, 18, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Bitetto, D.; Fabris, C.; Fornasiere, E.; Pipan, C.; Fumolo, E.; Cussigh, A.; Bignulin, S.; Cmet, S.; Fontanini, E.; Falleti, E.; et al. Vitamin D supplementation improves response to antiviral treatment for recurrent hepatitis C. Transpl. Int. 2011, 24, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.M.; Del Campo, J.A.; Ranchal, I.; Lampe, E.; Romero-Gomez, M. Association between vitamin D and hepatitis C virus infection: A meta-analysis. World J. Gastroenterol. 2013, 19, 5917–5924. [Google Scholar] [CrossRef] [PubMed]
- Gal-Tanamy, M.; Bachmetov, L.; Ravid, A.; Koren, R.; Erman, A.; Tur-Kaspa, R.; Zemel, R. Vitamin D: An innate antiviral agent suppressing hepatitis C virus in human hepatocytes. Hepatology 2011, 54, 1570–1579. [Google Scholar] [CrossRef]
- Bland, R.; Walker, E.A.; Hughes, S.V.; Stewart, P.M.; Hewison, M. Constitutive expression. of 25-hydroxyvitamin D3-1alpha-hydroxylase in a transformed human proximal tubule cell line: Evidence for direct regulation of vitamin D metabolism by calcium. Endocrinology. 1999, 140, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Kongsbak, M.; Von Essen, M.R.; Boding, L.; Levring, T.B.; Schjerling, P.; Lauritsen, J.P.H.; Woetmann, A.; Ødum, N.; Bonefeld, C.M.; Geisler, C. Vitamin D up-regulates the vitamin D receptor by protecting it from proteasomal degradation in human CD4+T cells. PLoS ONE 2014, 9, e96695. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.P.; Hollis, B.W.; Patel, S.B.; Patrick, K.S.; Bell, N.H. CYP3A4 is a Human Microsomal Vitamin D 25-Hydroxylase. J. Bone Miner. Res. 2003, 19, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Mason, A. Upregulation of Hedgehog Pathway is Associated with Cellular Permissiveness for Hepatitis C Virus Replication. Hepatology 2009, 19, 389–399. [Google Scholar]
- Huang, S.; He, J.; Zhang, X.; Bian, Y.; Yang, L.; Xie, G.; Zhang, K.; Tang, W.; Stelter, A.A.; Wang, Q.; et al. Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis 2006, 27, 1334–1340. [Google Scholar] [CrossRef]
- Matsumura, T.; Kato, T.; Sugiyama, N.; Tasaka-Fujita, M.; Murayama, A.; Masaki, T.; Wakita, T.; Imawari, M. 25-hydroxyvitamin D 3 suppresses hepatitis C virus production. Hepatology 2012, 56, 1231–1239. [Google Scholar] [CrossRef]
- Julio, A.; Gutierrez, K.A.; Jones, R.F.; Singhania, A.; Christopher, H.; Woelk, R.T.S.; AWyles, D.L. Vitamin D Metabolites Inhibit Hepatitis C Virus and Modulate Cellular Gene Expression. J. Virol. Antiviral Res. 2014, 3, 3. [Google Scholar]
- Lin, Y.M.; Sun, H.Y.; Chiu, W.T.; Su, H.C.; Chien, Y.C.; Chong, L.W.; Chang, H.C.; Bai, C.H.; Young, K.C.; Tsao, C.W. Calcitriol inhibits HCV infection via blockade of activation of PPAR and interference with endoplasmic reticulum-associated degradation. Viruses 2018, 10, E57. [Google Scholar] [CrossRef]
- Alaei, M.; Negro, F. Hepatitis C virus and glucose and lipid metabolism. Diabetes Metab. 2008, 34, 692–700. [Google Scholar] [CrossRef]
- Asano, L.; Watanabe, M.; Ryoden, Y.; Usuda, K.; Yamaguchi, T.; Khambu, B.; Takashima, M.; Sato, S.-I.; Sakai, J.; Nagasawa, K.; et al. Vitamin D Metabolite, 25-Hydroxyvitamin D, Regulates Lipid Metabolism by Inducing Degradation of SREBP/SCAP. Cell Chem. Biol. 2017, 24, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Näär, A.M. SREBPs: Regulators of cholesterol/lipids as therapeutic targets in metabolic disorders, cancers and viral diseases. Clin. Lipidol. 2012, 7, 27–36. [Google Scholar] [CrossRef]
- Yi, M.; Ma, Y.; Yates, J.; Lemon, S.M. Compensatory Mutations in E1, p7, NS2, and NS3 Enhance Yields of Cell Culture-Infectious Intergenotypic Chimeric Hepatitis C Virus. J. Virol. 2007, 81, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lim, K.; Kim, J.S.; Bae, S. Cas-analyzer: An online tool for assessing genome editing results using NGS data. Bioinformatics 2017, 33, 286–288. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number | Primer Name | Sequence (5′→3′) | Product Size (bp) |
|---|---|---|---|
| NM_001128915.1 | Cyp24A2-S | ACCCAAAGGAATTGTCCGCA | 111 |
| Cyp24A1-AS | CAAAACGCGATGGGGAGTTC | ||
| NM_024514.4 | Cyp2r1-S | TGGAGGCATATCAACTGTGGT | 133 |
| Cyp2r1-AS | GAGTAAGCCTCCCATTTTTGTCA | ||
| NM_000775.4 | Cyp2J2-S | TGGACCCCACCAAACTCTCT | 153 |
| Cyp2J2-AS | GGATTGCCTGTGTGCTTT | ||
| NM_000784.4 | Cyp27A1-S | GTTCACCACGGAAGGACACC | 163 |
| Cyp27A1-AS | GTTCCCCGAAGCACTCTCTG | ||
| NM_017460.6 | Cyp3A4-S | TGTGGGGCTTTTATGATGGT | 117 |
| Cyp3A4-AS | GACCAAAAGGCCTCCGGTTT | ||
| NM_000785.4 | Cyp27B1-S | GTGCTAAGACTGTACCCTGTGG | 150 |
| Cyp27B1-AS | ATTTGGCTCTGGGAACTGG | ||
| ENSG00000111424 | VDR-S | AGGGCGAATCATGTATGAGG | 396 |
| VDR-AS | TGCTTCTTCTCCCTCCCTTT | ||
| NM_000193.3 | SHH-S | GAAACTCCGAGCGATTTAAGGA | 228 |
| SHH-AS | GGCCCTCGTAGTGCAGAGA | ||
| NM_001083603.2 | PTCH1-S | TCTTGGTGTTGGTGTGGATG | 145 |
| PTCH1-AS | ATTGCTGATGGACGTGAGG | ||
| NM_005269.2 | Gli1-S | CATCAGGGAGGAAAGCAGAC | 146 |
| Gli1-AS | CATTGCCAGTCATTTCCACAC | ||
| NM_001015.5 | RPS11-S | GCCCTCAATAGCCTCCTTGG | 149 |
| RPS11-AS | TTCAGACTGAGCGTGCCTAC | ||
| NM_002046.7 | GAPDH-S | GAAGGTGAAGGTCGGAGTC | 226 |
| GAPDH-AS | GAAGATGGTGATGGGATTTC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravid, A.; Rapaport, N.; Issachar, A.; Erman, A.; Bachmetov, L.; Tur-Kaspa, R.; Zemel, R. 25-Hydroxyvitamin D Inhibits Hepatitis C Virus Production in Hepatocellular Carcinoma Cell Line by a Vitamin D Receptor-Independent Mechanism. Int. J. Mol. Sci. 2019, 20, 2367. https://doi.org/10.3390/ijms20092367
Ravid A, Rapaport N, Issachar A, Erman A, Bachmetov L, Tur-Kaspa R, Zemel R. 25-Hydroxyvitamin D Inhibits Hepatitis C Virus Production in Hepatocellular Carcinoma Cell Line by a Vitamin D Receptor-Independent Mechanism. International Journal of Molecular Sciences. 2019; 20(9):2367. https://doi.org/10.3390/ijms20092367
Chicago/Turabian StyleRavid, Amiram, Noa Rapaport, Assaf Issachar, Arie Erman, Larisa Bachmetov, Ran Tur-Kaspa, and Romy Zemel. 2019. "25-Hydroxyvitamin D Inhibits Hepatitis C Virus Production in Hepatocellular Carcinoma Cell Line by a Vitamin D Receptor-Independent Mechanism" International Journal of Molecular Sciences 20, no. 9: 2367. https://doi.org/10.3390/ijms20092367
APA StyleRavid, A., Rapaport, N., Issachar, A., Erman, A., Bachmetov, L., Tur-Kaspa, R., & Zemel, R. (2019). 25-Hydroxyvitamin D Inhibits Hepatitis C Virus Production in Hepatocellular Carcinoma Cell Line by a Vitamin D Receptor-Independent Mechanism. International Journal of Molecular Sciences, 20(9), 2367. https://doi.org/10.3390/ijms20092367