Interaction of Mycotoxin Alternariol with Serum Albumin


 , ,
, , 
Abstract
1. Introduction
2. Results
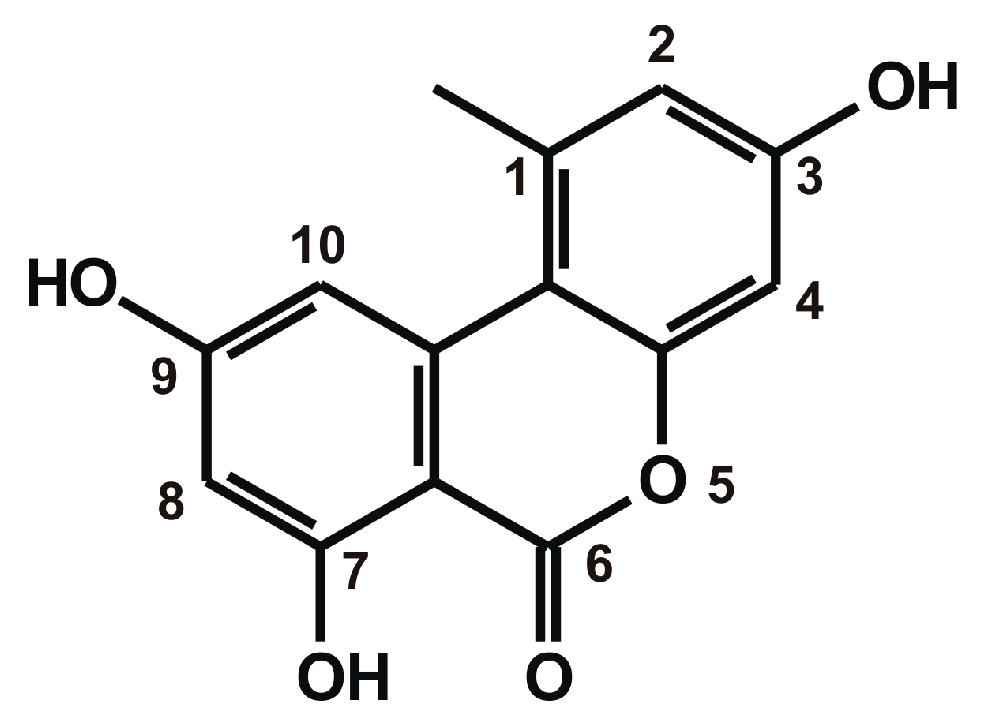
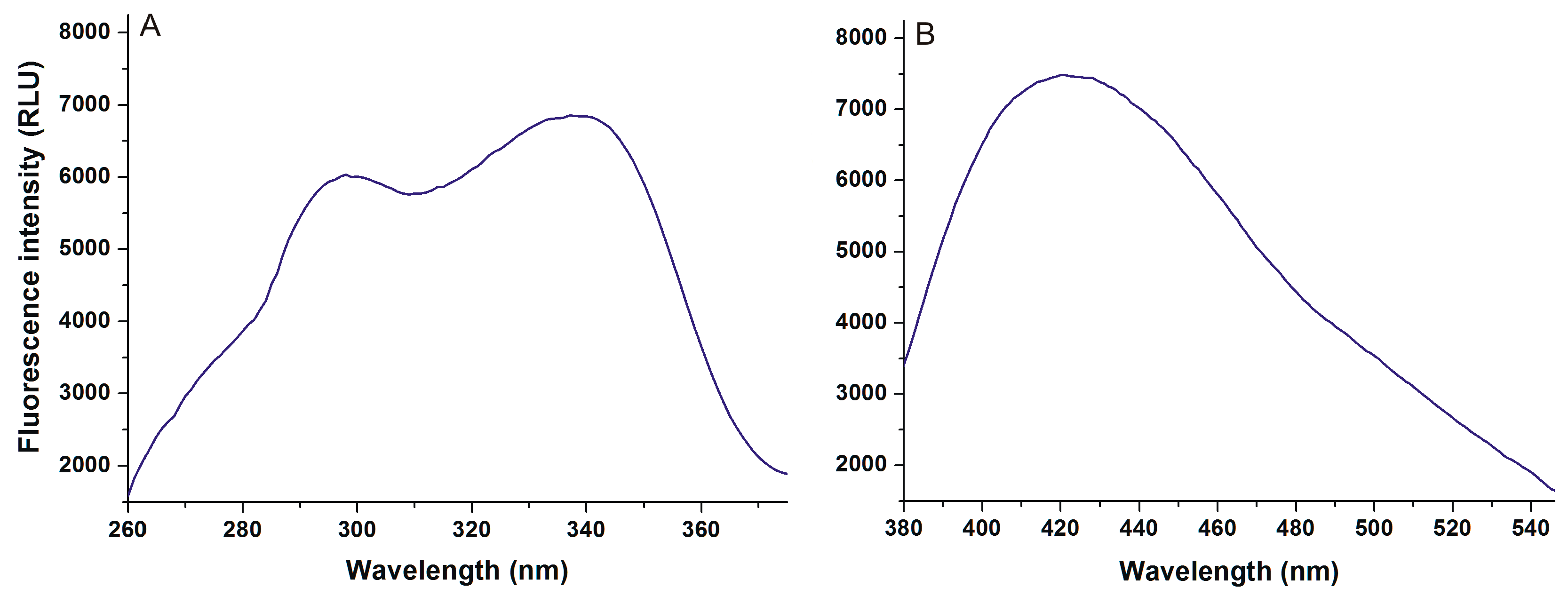
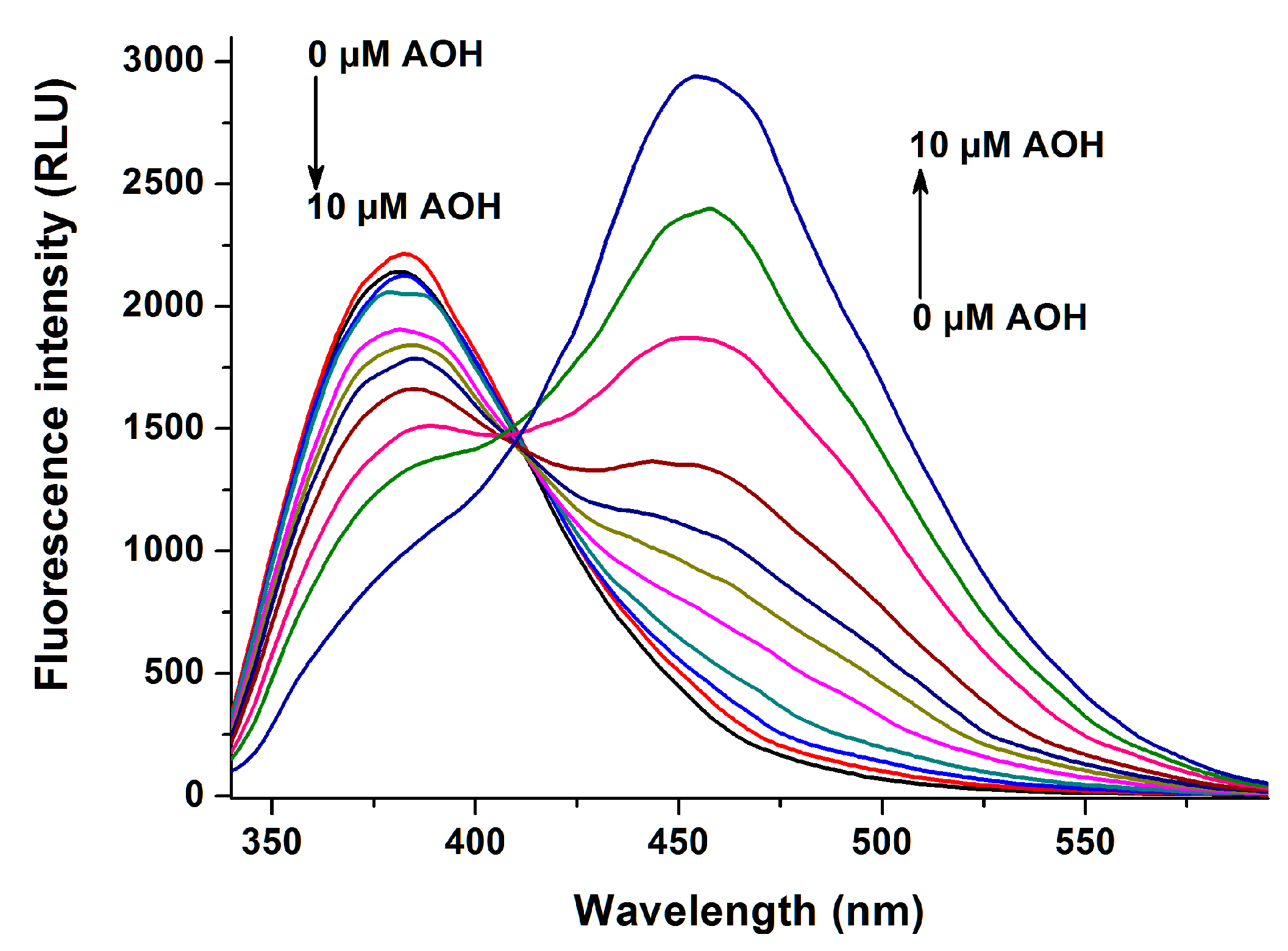
2.1. Fluorescence Spectra of AOH in the Absence and Presence of HSA
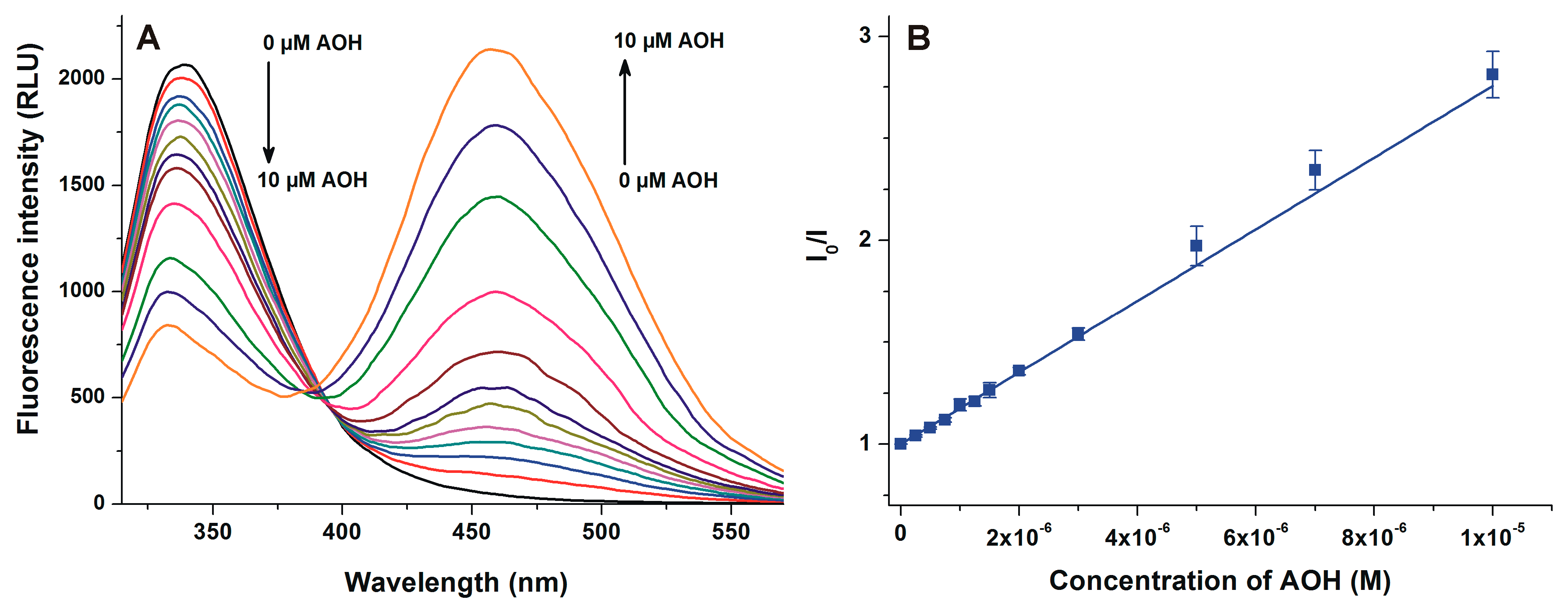
2.2. Fluorescence Quenching Experiments
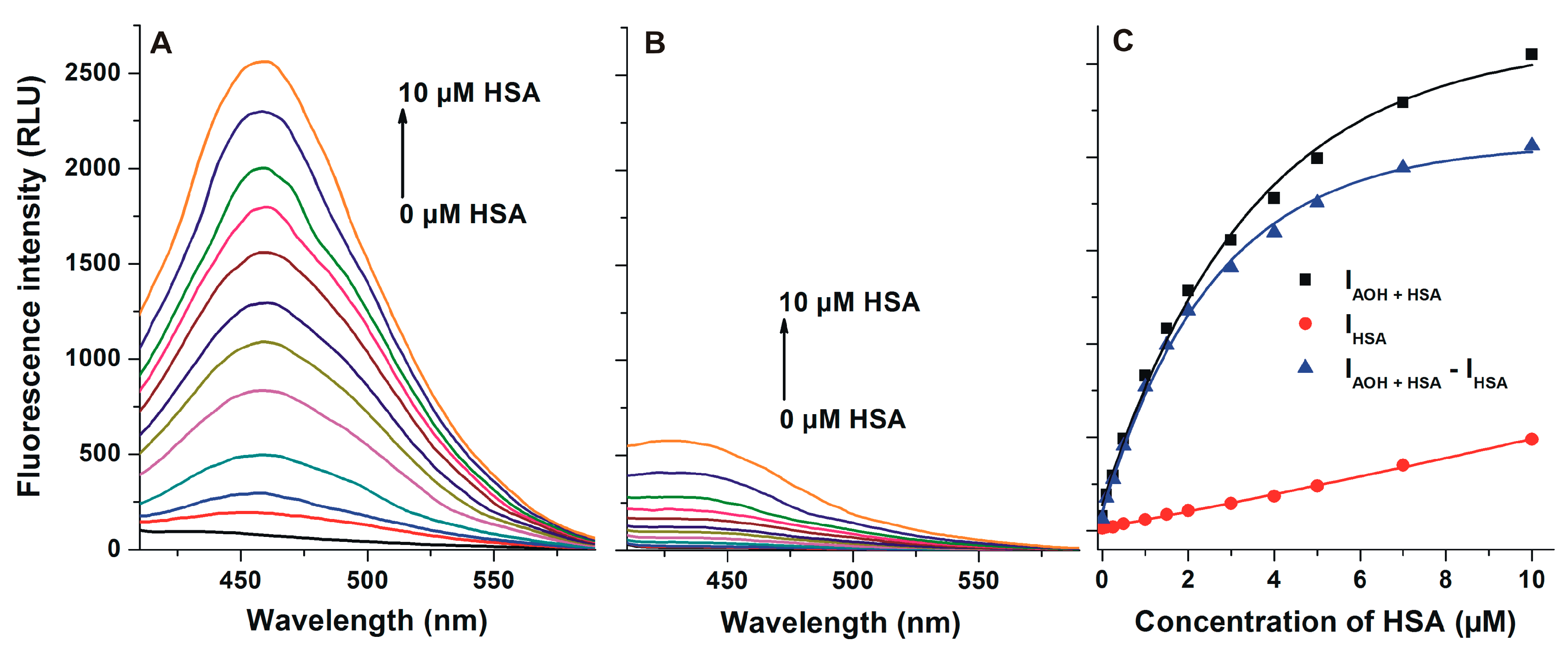
2.3. Interaction of AOH with HSA Based on Energy Transfer
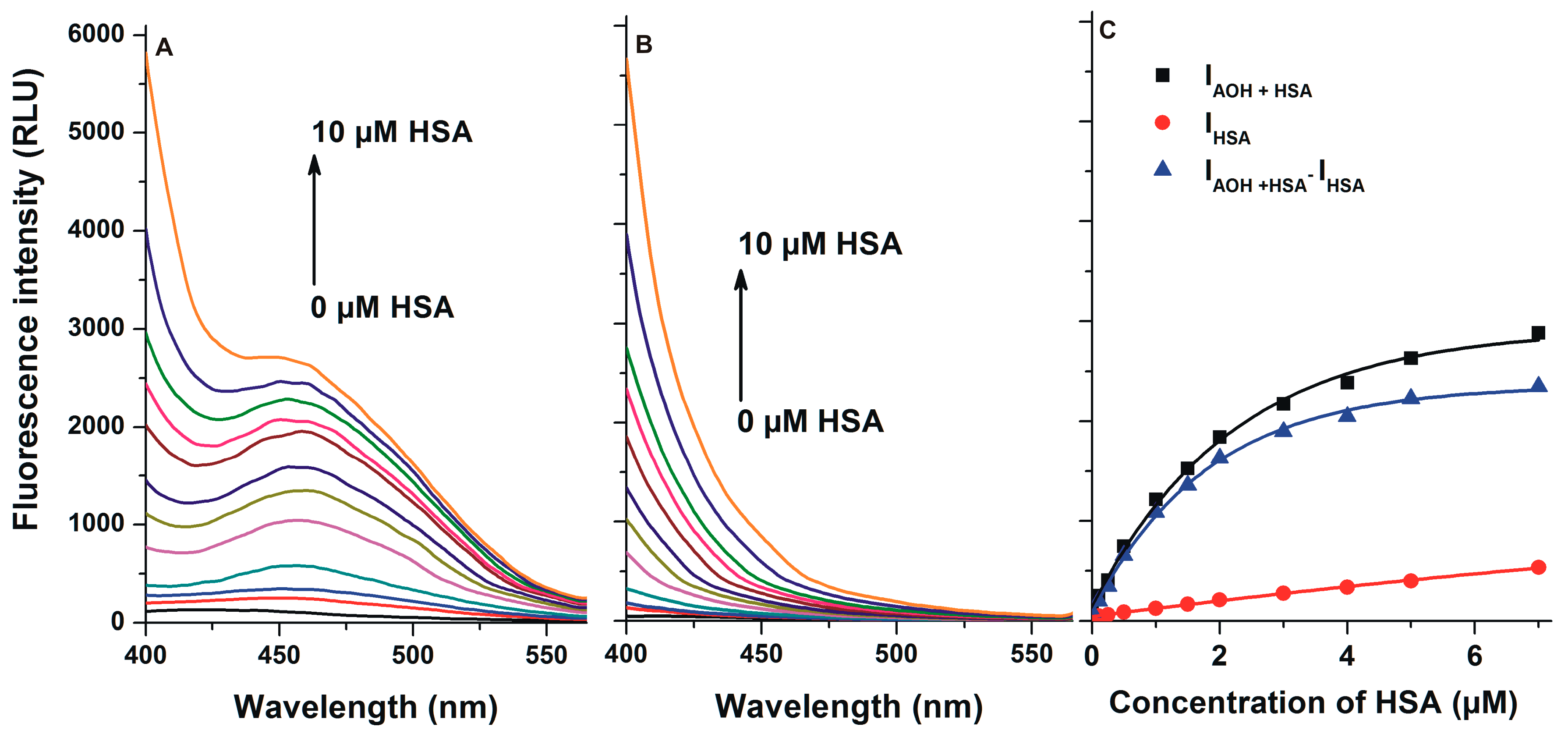
2.4. Stability of AOH-HSA Complex Based on Fluorescence Spectroscopic Measurements
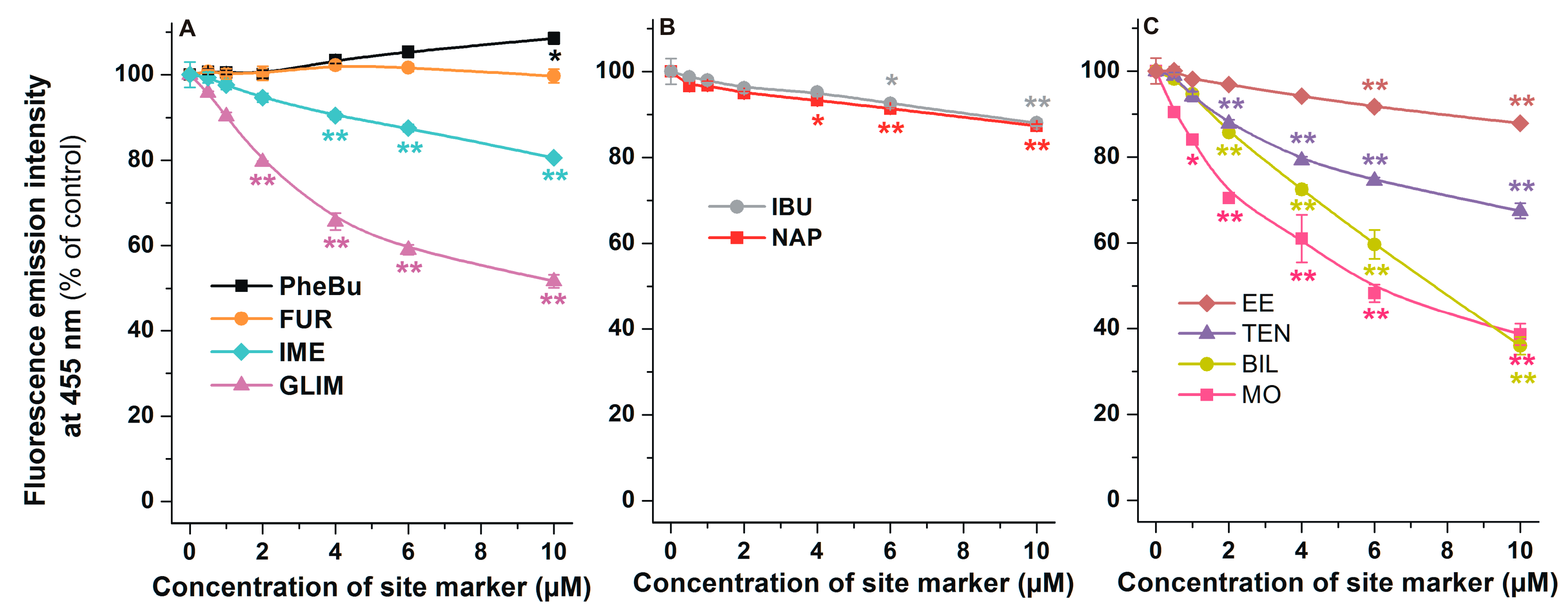
2.5. Effects of Site Markers on the Fluorescence Signal of AOH-HSA Complex
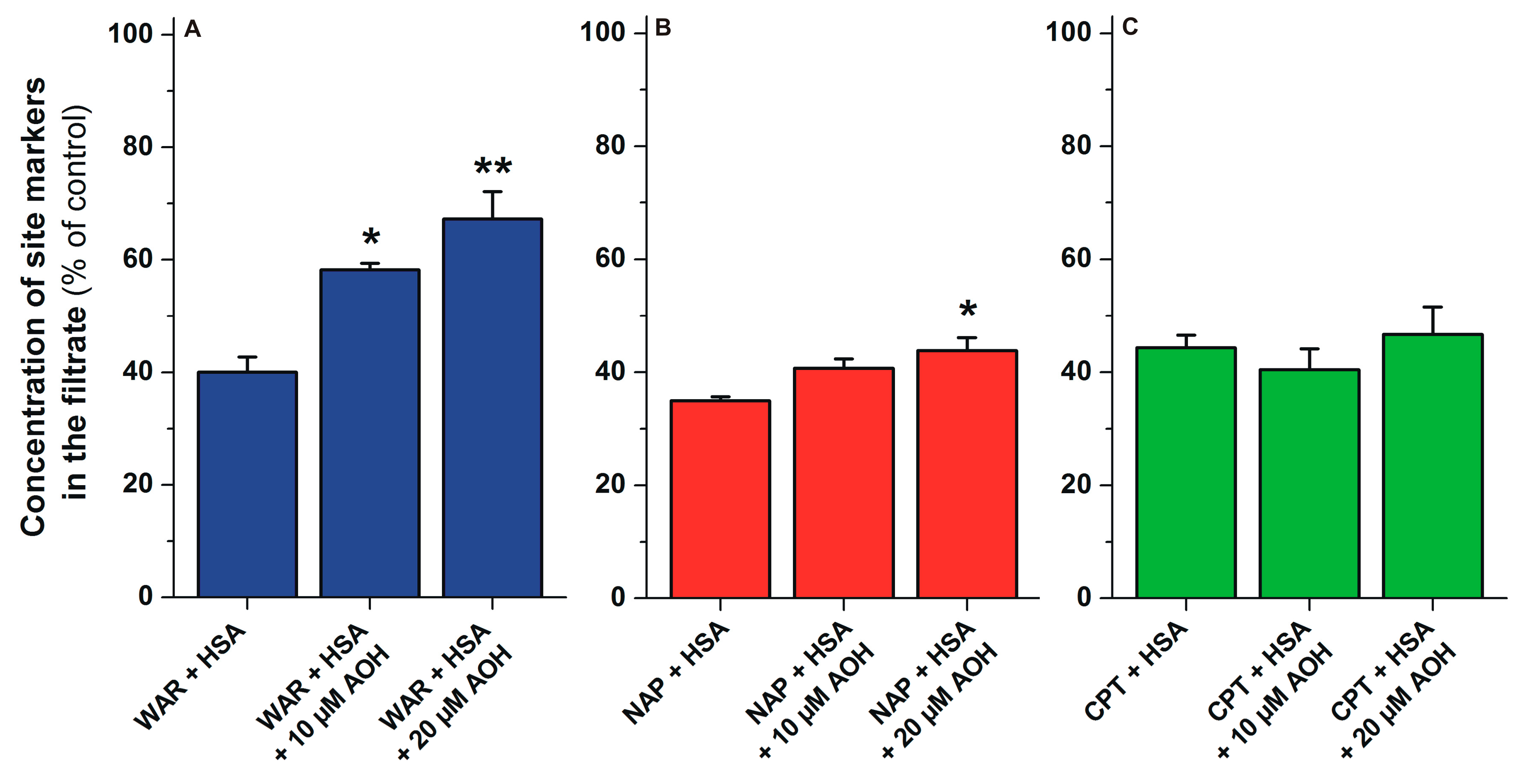
2.6. Displacement of Site Markers from HSA by AOH Based on Ultrafiltration Studies
2.7. Testing the Involvement of SSI and Heme Sites Based on Spectroscopic Studies
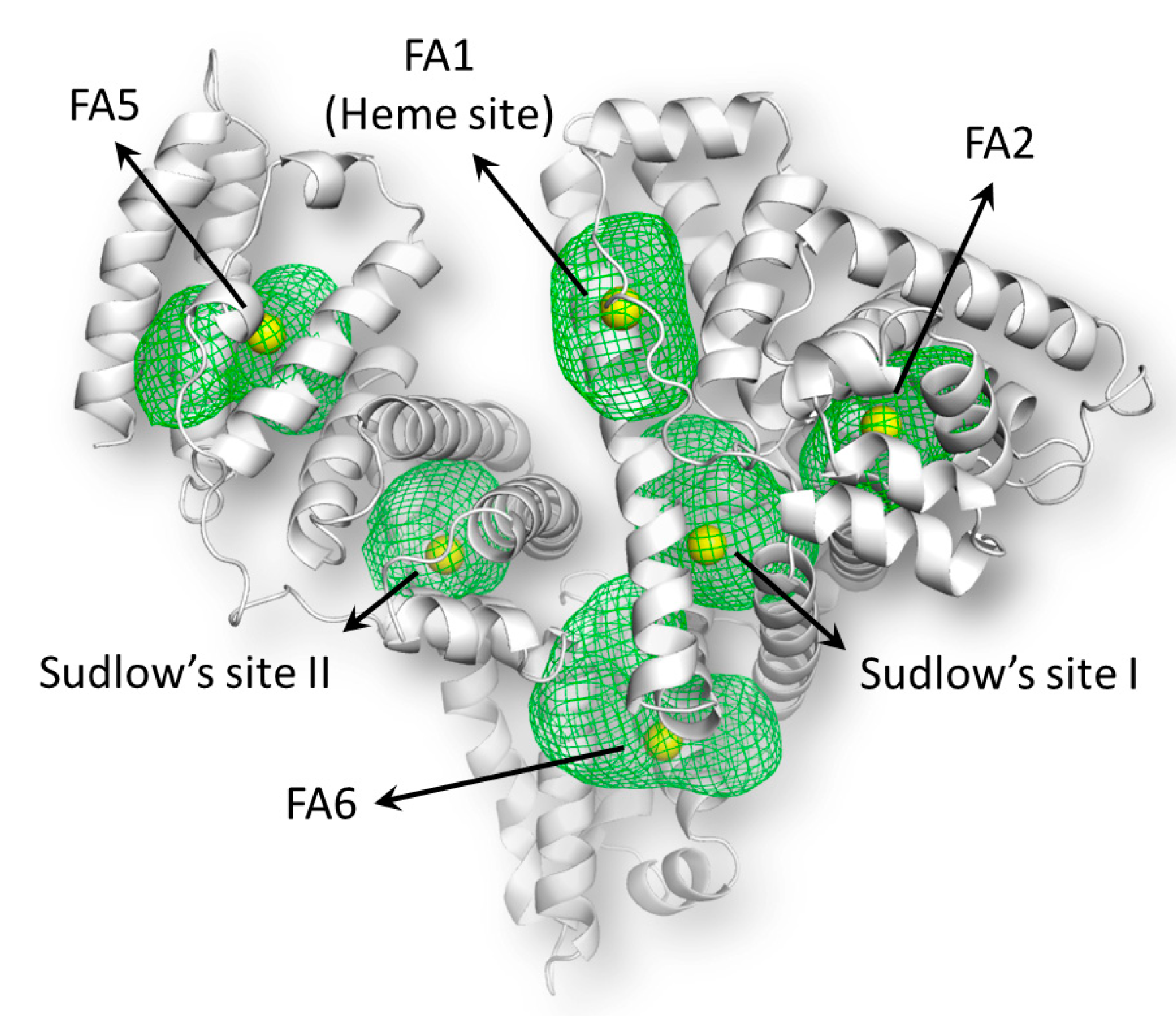
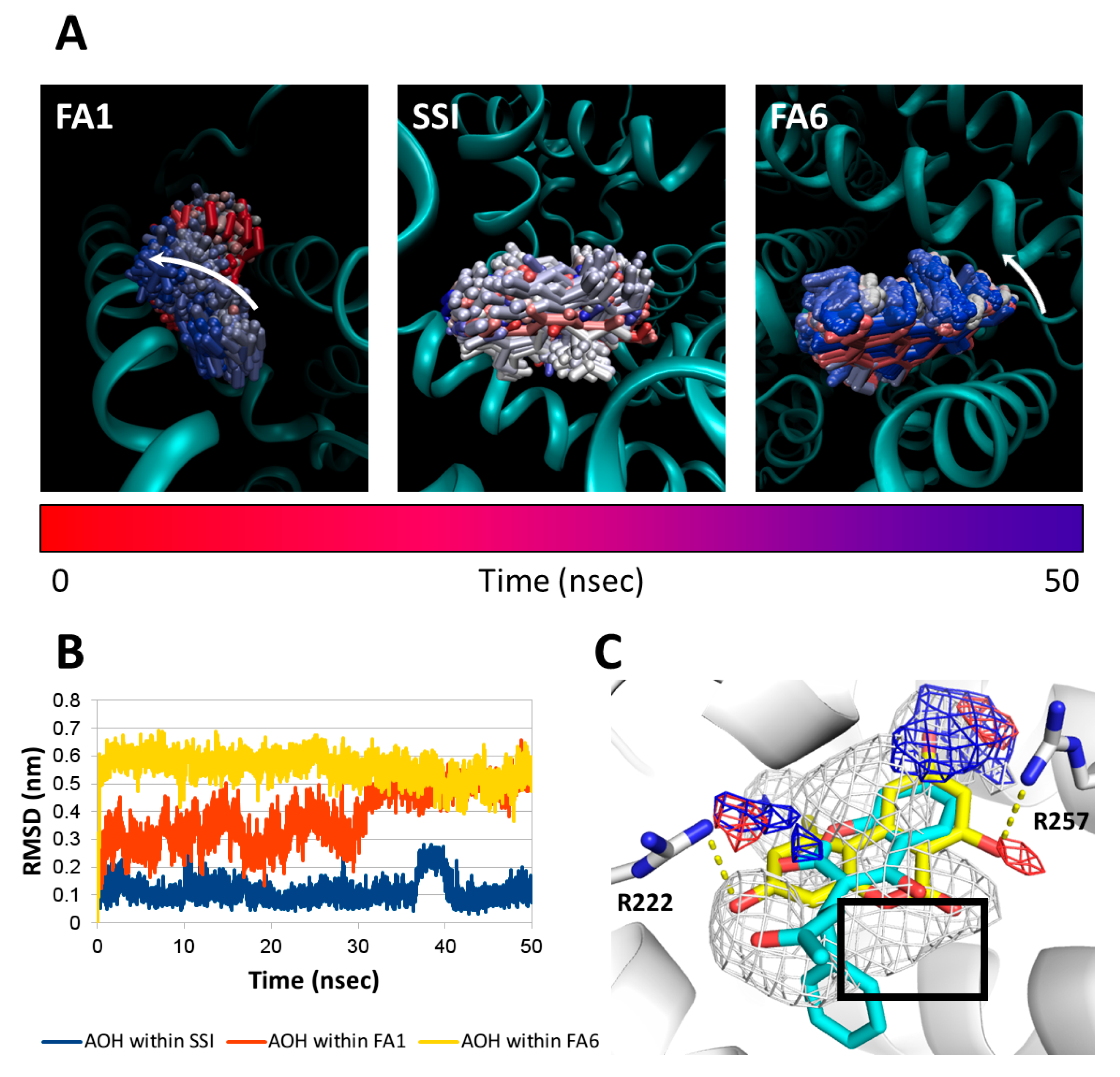
2.8. Molecular Docking Studies
2.9. Species Dependence of AOH-Albumin Interaction
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Spectroscopic Studies
4.3. Ultrafiltration
4.4. HPLC Analyses
4.5. Molecular Docking Studies
4.5.1. Model Preparation
4.5.2. Binding Sites Mapping and Pharmacophoric Modeling
4.5.3. Docking Study and Rescoring Procedure
4.5.4. Molecular Dynamic Simulations
4.6. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AOH | Alternariol |
| BSA | Bovine Serum Albumin |
| FLD | Fluorescence Detector |
| HINT | Hydropathic INTeractions |
| HPLC | High-Performance Liquid Chromatography |
| HSA | Human Serum Albumin |
| MD | Molecular dynamic |
| PDB | Protein Data Bank |
| PSA | Porcine Serum Albumin |
| RMSD | Root-Mean-Square Analysis |
| RSA | Rat Serum Albumin |
| SSI | Sudlow’s Site I |
| SSII | Sudlow’s Site II |
References
- Hussein, H.S.; Brasel, J.M. Toxicity, metabolism, and impact of mycotoxins on humans and animals. Toxicology 2001, 167, 101–134. [Google Scholar] [CrossRef]
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef]
- Fente, C.A.; Jaimez, J.; Vázquez, B.I.; Franco, C.M.; Cepeda, A. Determination of alternariol in tomato paste using solid phase extraction and high-performance liquid chromatography with fluorescence detection. Analyst 1998, 123, 2277–2280. [Google Scholar] [CrossRef]
- Broggi, L.; Reynoso, C.; Resnik, S.; Martinez, F.; Drunday, V.; Bernal, Á.R. Occurrence of alternariol and alternariol monomethyl ether in beverages from the Entre Rios Province market, Argentina. Mycotoxin Res. 2012, 29, 17–22. [Google Scholar] [CrossRef]
- Dall’Asta, C.; Cirlini, M.; Falavigna, C. Mycotoxins from Alternaria. Adv. Mol. Toxicol. 2014, 107–121. [Google Scholar] [CrossRef]
- Paterson, R.R.M.; Venâncio, A.; Lima, N.; Guilloux-Bénatier, M.; Rousseaux, S. Predominant mycotoxins, mycotoxigenic fungi and climate change related to wine. Food. Res. Int. 2018, 103, 478–491. [Google Scholar] [CrossRef]
- Scott, P.M. Mycotoxins in Alcoholic Beverages. In Food Contaminants; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2008; pp. 170–191. [Google Scholar]
- Rodríguez-Carrasco, Y.; Mañes, J.; Berrada, H.; Juan, C. Development and Validation of a LC-ESI-MS/MS Method for the Determination of Alternaria Toxins Alternariol, Alternariol Methyl-Ether and Tentoxin in Tomato and Tomato-Based Products. Toxins 2016, 8, 328–340. [Google Scholar] [CrossRef]
- Rico-Yuste, A.; Walravens, J.; Urraca, J.L.; Abou-Hany, R.A.G.; Descalzo, A.B.; Orellana, G.; Rychlik, M.; Saeger, S.D.; Moreno-Bondi, M.C. Analysis of alternariol and alternariol monomethyl ether in foodstuffs by molecularly imprinted solid-phase extraction and ultra-high-performance liquid chromatography tandem mass spectrometry. Food Chem. 2018, 243, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Stypuła-Trębas, S.; Minta, M.; Radko, L.; Jedziniak, P.; Posyniak, A. Nonsteroidal mycotoxin alternariol is a full androgen agonist in the yeast androgen riporter bioassay. Environ. Toxicol. Pharmacol. 2017, 55, 208–211. [Google Scholar] [CrossRef]
- EFSA, European Food Safety Authority: Scientific Opinion on the risks for animal and public health related to the presence of Alternaria toxins in feed and food. EFSA J. 2011, 9, 2407. [CrossRef]
- Solhaug, A.; Eriksen, G.S.; Holme, J.A. Mechanisms of Action and Toxicity of the Mycotoxin Alternariol: A Review. Basic Clin. Pharmacol. Toxicol. 2016, 119, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-T.; Qian, Y.-Z.; Zhang, P.; Dong, W.-H.; Qi, Y.-M.; Guo, H.-T. Etiological role of Alternaria alternata in human esophageal cancer. Chin. Med. J. 1992, 105, 394–400. [Google Scholar] [PubMed]
- Fleck, S.C.; Sauter, F.; Pfeiffer, E.; Metzler, M.; Hartwig, A.; Köberle, B. DNA damage and repair kinetics of the Alternaria mycotoxins alternariol, altertoxin II and stemphyltoxin III in cultured cells. Mutat. Res. 2016, 798–799, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ostry, V. Alternaria mycotoxins: an overview of chemical characterization, producers, toxicity, analysis and occurrence in foodstuffs. World Mycotoxin J. 2008, 1, 175–188. [Google Scholar] [CrossRef]
- Tiessen, C.; Fehr, M.; Schwarz, C.; Baechler, S.; Domnanich, K.; Böttler, U.; Pahlke, G.; Marko, D. Modulation of the cellular redox status by the Alternaria toxins alternariol and alternariol monomethyl ether. Toxicol. Lett. 2013, 216, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, L.; Wagner, J.; Metzler, M. Estrogenic and clastogenic potential of the mycotoxin alternariol in cultured mammalian cells. Food Chem. Toxicol. 2006, 44, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Demaegdt, H.; Daminet, B.; Evrard, A.; Scippo, M.-L.; Muller, M.; Pussemier, L.; Callebaut, A.; Vandermeiren, K. Endocrine activity of mycotoxins and mycotoxin mixtures. Food Chem. Toxicol. 2016, 96, 107–116. [Google Scholar] [CrossRef]
- Fanali, G.; Masi, A.; Trezza, V.; Marino, M.; Fazano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Levitt, D.G.; Levitt, M.D. Human serum albumin homeostasis: a new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int. J. Gen. Med. 2016, 9, 229–255. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Notari, S.; Fanali, G.; Fesce, R.; Fasano, M. Allosteric Modulation of Drug Binding to Human Serum Albumin. Mini Rev. Med. Chem. 2006, 6, 483–489. [Google Scholar] [CrossRef]
- Yamasaki, K.; Chuang, V.T.G.; Maruyama, T.; Otagiri, M. Albumin–drug interaction and its clinical implication. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 5435–5443. [Google Scholar] [CrossRef]
- Lee, P.; Wu, X. Review: Modifications of Human Serum Albumin and Their Binding Effect. Curr. Pharm. Des. 2015, 21, 1862–1865. [Google Scholar] [CrossRef]
- Wang, Z.; Ho, J.X.; Ruble, J.R.; Rose, J.; Rüker, F.; Ellenburg, M.; Murphy, R.; Click, J.; Soistman, E.; Wilkerson, L.; et al. Structural studies of several clinically important oncology drugs in complex with human serum albumin. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 5356–5374. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Spectroscopic techniques in the study of protein binding. A fluorescence technique for the evaluation of the albumin binding and displacement of warfarin and warfarin-alcohol. Clin. Exp. Pharmacol. Physiol. 1975, 2, 129–140. [Google Scholar] [CrossRef]
- Zsila, F. Subdomain IB Is the Third Major Drug Binding Region of Human Serum Albumin: Toward the Three-Sites Model. Mol. Pharm. 2013, 10, 1668–1682. [Google Scholar] [CrossRef]
- Perry, J.L.; Il’ichev, Y.V.; Kempf, V.R.; McClendon, J.; Park, G.; Manderville, R.A.; Rüker, F.; Dockal, M.; Simon, J.D. Binding of ochratoxin A derivatives to human serum albumin. J. Phys. Chem. B 2003, 107, 6644–6647. [Google Scholar] [CrossRef]
- Kőszegi, T.; Poór, M. Ochratoxin A: Ochratoxin A: Molecular Interactions, Mechanisms of Toxicity and Prevention at the Molecular Level. Toxins 2016, 8, 111. [Google Scholar] [CrossRef]
- Poór, M.; Lemli, B.; Bálint, M.; Hetényi, C.; Sali, N.; Kőszegi, T.; Kunsági-Máté, S. Interaction of citrinin with human serum albumin. Toxins 2015, 7, 5155–5166. [Google Scholar] [CrossRef]
- Poór, M.; Kunsági-Máté, S.; Bálint, M.; Hetényi, C.; Gerner, Z.; Lemli, B. Interaction of mycotoxin zealarenone with human serum albumin. J. Photochem. Photobiol. B Biol. 2017, 16–24. [Google Scholar] [CrossRef]
- González-Jiménez, J.; Cortijo, M. Resonance energy transfer between tryptophan-214 in human serum albumin and acrylodan, prodan, and promen. Protein J. 2004, 23, 351–355. [Google Scholar] [CrossRef]
- Cozzini, P.; Fornabaio, M.; Marabotti, A.; Abraham, D.J.; Kellogg, G.E.; Mozzarelli, A. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 1. Models without explicit constrained water. J. Med. Chem. 2002, 45, 2469–2483. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Galaverna, G.; Dall’Asta, C. In silico analysis sheds light on the structural basis underlying the ribotoxicity of trichothecenes-A tool for supporting the hazard identification process. Toxicol. Lett. 2017, 270, 80–87. [Google Scholar] [CrossRef]
- Ehrlich, V.A.; Dellafiora, L.; Mollergues, J.; Dall’Asta, C.; Serrant, P.; Marin-Kuan, M.; Lo Piparo, E.; Schilter, B.; Cozzini, P. Hazard assessment through hybrid in vitro/in silico approach: The case of zearalenone. ALTEX 2015, 32, 275–286. [Google Scholar] [CrossRef]
- Petitpas, I.; Bhattacharya, A.A.; Twine, S.; East, M.; Curry, S. Crystal structure analysis of warfarin binding to human serum albumin: Anatomy of drug site I. J. Biol. Chem. 2001, 276, 22804–22809. [Google Scholar] [CrossRef] [PubMed]
- Dobretsov, G.E.; Syrejschikova, T.I.; Smolina, N.V. On Mechanisms of Fluorescence Quenching by Water. Biophysics 2014, 59, 231–237. [Google Scholar] [CrossRef]
- Sytnik, A.; Litvinyuk, I. Energy transfer to a proton-transfer fluorescence probe: Tryptophan to a flavonol in human serum albumin. Proc. Natl. Acad. Sci. USA 1996, 93, 12959–12963. [Google Scholar] [CrossRef]
- Xu, L.; Hu, Y.-X.; Li, Y.-C.; Liu, Y.-F.; Zhang, L.; Ai, H.-X.; Liu, H.-S. Study on the interaction of paeoniforin with human serum albumin (HSA) by spectroscopic and molecular docking techniques. Chem. Cent. J. 2017, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- Sueck, F.; Poór, M.; Faisal, Z.; Gertzen, C.G.W.; Cramer, B.; Lemli, B.; Kunsági-Máté, S.; Gohlke, H.; Humpf, H.U. Interaction of Ochratoxin A and Its Thermal Degradation Product 2’R-Ochratoxin A with Human Serum Albumin. Toxins 2018, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Faisal, Z.; Lemli, B.; Szerencsés, D.; Kunsági-Máté, S.; Bálint, M.; Hetényi, C.; Kuzma, M.; Mayer, M.; Poór, M. Interactions of zearalenone and its reduced metabolites α-zearalenol and β-zearalenol with serum albumins: Species differences, binding sites, and thermodynamics. Mycotox. Res. 2018, 34, 269–278. [Google Scholar] [CrossRef]
- Poór, M.; Boda, G.; Mohos, V.; Kuzma, M.; Bálint, M.; Hetényi, C.; Bencsik, T. Pharmacokinetic interaction of diosmetin and silibinin with other drugs: Inhibition of CYP2C9-mediated biotransformation and displacement from serum albumin. Biomed. Pharmacother. 2018, 102, 912–921. [Google Scholar] [CrossRef]
- Zsila, F.; Bikádi, Z.; Simonyi, M. Probing the binding of the flavonoid, quercetin to human serum albumin by circular dichroism, electronic absorption spectroscopy and molecular modelling methods. Biochem. Pharmacol. 2003, 65, 447–456. [Google Scholar] [CrossRef]
- Rimac, H.; Dufour, C.; Debeljak, Ž.; Zorc, B.; Bojić, M. Warfarin and Flavonoids Do Not Share the Same Binding Region in Binding to the IIA Subdomain of Human Serum Albumin. Molecules 2017, 22, 1153. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural Basis of the Drug-binding Specificity of Human Serum Albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Notari, S.; Menegatti, E.; Fasano, M. Heme impairs allosterically drug binding to human serum albumin Sudlow’s site I. Biochem. Biophys. Res. Commun. 2005, 334, 481–486. [Google Scholar] [CrossRef]
- Poór, M.; Li, Y.; Matisz, G.; Kiss, L.; Kunsági-Máté, S.; Kőszegi, T. Quantitation of species differences in albumin–ligand interactions for bovine, human and rat serum albumins using fluorescence spectroscopy: A test case with some Sudlow’s site I ligands. J. Lumin. 2014, 145, 767–773. [Google Scholar] [CrossRef]
- Hagelberg, S.; Hult, K.; Fuchs, R. Toxicokinetics of ochratoxin A in several species and its plasma-binding properties. J. Appl. Toxicol. 1989, 9, 91–96. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Larsson, T.; Wedborg, M.; Turner, D. Correction of inner-filter effect in fluorescence excitation-emission matrix spectrometry using Raman scatter. Anal. Chim. Acta 2007, 583, 357–363. [Google Scholar] [CrossRef]
- Mohos, V.; Bencsik, T.; Boda, G.; Fliszár-Nyúl, E.; Lemli, B.; Kunsági-Máté, S.; Poór, M. Interactions of casticin, ipriflavone, and resveratrol with serum albumin and their inhibitory effects on CYP2C9 and CYP3A4 enzymes. Biomed. Pharmacother. 2018, 107, 777–784. [Google Scholar] [CrossRef]
- Wen, Y.; Fan, Y.; Zhang, M.; Feng, Y.-Q. Determination of camptothecin and 10-hydroxycamptothecin in human plasma using polymer monolithic in-tube solid phase microextraction combined with high-performance liquid chromatography. Anal. Bioanal. Chem. 2005, 204–210. [Google Scholar] [CrossRef]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Carosati, E.; Sciabola, S.; Cruciani, G. Hydrogen bonding interactions of covalently bonded fluorine atoms: From crystallographic data to a new angular function in the GRID force field. J. Med. Chem. 2004, 47, 5114–5125. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, E.G.; Abraham, D.J. Hydrophobicity: is LogP (o/w) more than the sum of its parts? Eur. J. Med. Chem. 2000, 35, 651–661. [Google Scholar] [CrossRef]
- Dellafiora, L.; Galaverna, G.; Righi, F.; Cozzini, P.; Dall’Asta, C. Assessing the hydrolytic fate of the masked mycotoxin zearalenone-14-glucoside—A warning light for the need to look at the “maskedome”. Food Chem. Toxicol. 2017, 99, 9–16. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| logKSV (SV-plot; Figure 4) λex = 295 nm, λem = 340 nm | logK (Hyperquad; Figure 4) λex = 295 nm, λem = 340 nm | logK (Hyperquad; Figure 5) λex = 295 nm, λem = 455 nm | logK (Hyperquad; Figure 3) λex = 345 nm, λem = 455 nm |
|---|---|---|---|
| 5.31 ± 0.03 | 5.60 ± 0.01 | 5.78 ± 0.02 | 5.64 ± 0.05 |
| Binding Sites | HINT Score | Expected Interaction |
|---|---|---|
| FA1 (Heme site) | 852.44 | Positive |
| FA2 | −127.25 | Negative |
| FA5 | −356.23 | Negative |
| FA6 | 160.34 | Positive |
| Sudlow’s site I (SSI) | 399.35 | Positive |
| Sudlow’s site II (SSII) | −91.69 | Negative |
| Complexes | AOH-HSA | AOH-BSA | AOH-PSA | AOH-RSA |
|---|---|---|---|---|
| logK | 5.64 ± 0.05 | 5.91 ± 0.04 | 5.61 ± 0.04 | 6.53 ± 0.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fliszár-Nyúl, E.; Lemli, B.; Kunsági-Máté, S.; Dellafiora, L.; Dall’Asta, C.; Cruciani, G.; Pethő, G.; Poór, M. Interaction of Mycotoxin Alternariol with Serum Albumin. Int. J. Mol. Sci. 2019, 20, 2352. https://doi.org/10.3390/ijms20092352
Fliszár-Nyúl E, Lemli B, Kunsági-Máté S, Dellafiora L, Dall’Asta C, Cruciani G, Pethő G, Poór M. Interaction of Mycotoxin Alternariol with Serum Albumin. International Journal of Molecular Sciences. 2019; 20(9):2352. https://doi.org/10.3390/ijms20092352
Chicago/Turabian StyleFliszár-Nyúl, Eszter, Beáta Lemli, Sándor Kunsági-Máté, Luca Dellafiora, Chiara Dall’Asta, Gabriele Cruciani, Gábor Pethő, and Miklós Poór. 2019. "Interaction of Mycotoxin Alternariol with Serum Albumin" International Journal of Molecular Sciences 20, no. 9: 2352. https://doi.org/10.3390/ijms20092352
APA StyleFliszár-Nyúl, E., Lemli, B., Kunsági-Máté, S., Dellafiora, L., Dall’Asta, C., Cruciani, G., Pethő, G., & Poór, M. (2019). Interaction of Mycotoxin Alternariol with Serum Albumin. International Journal of Molecular Sciences, 20(9), 2352. https://doi.org/10.3390/ijms20092352