Bioinformatic Analysis of Gene Variants from Gastroschisis Recurrence Identifies Multiple Novel Pathogenetic Pathways: Implication for the Closure of the Ventral Body Wall

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Gene Functional Enrichment Analysis
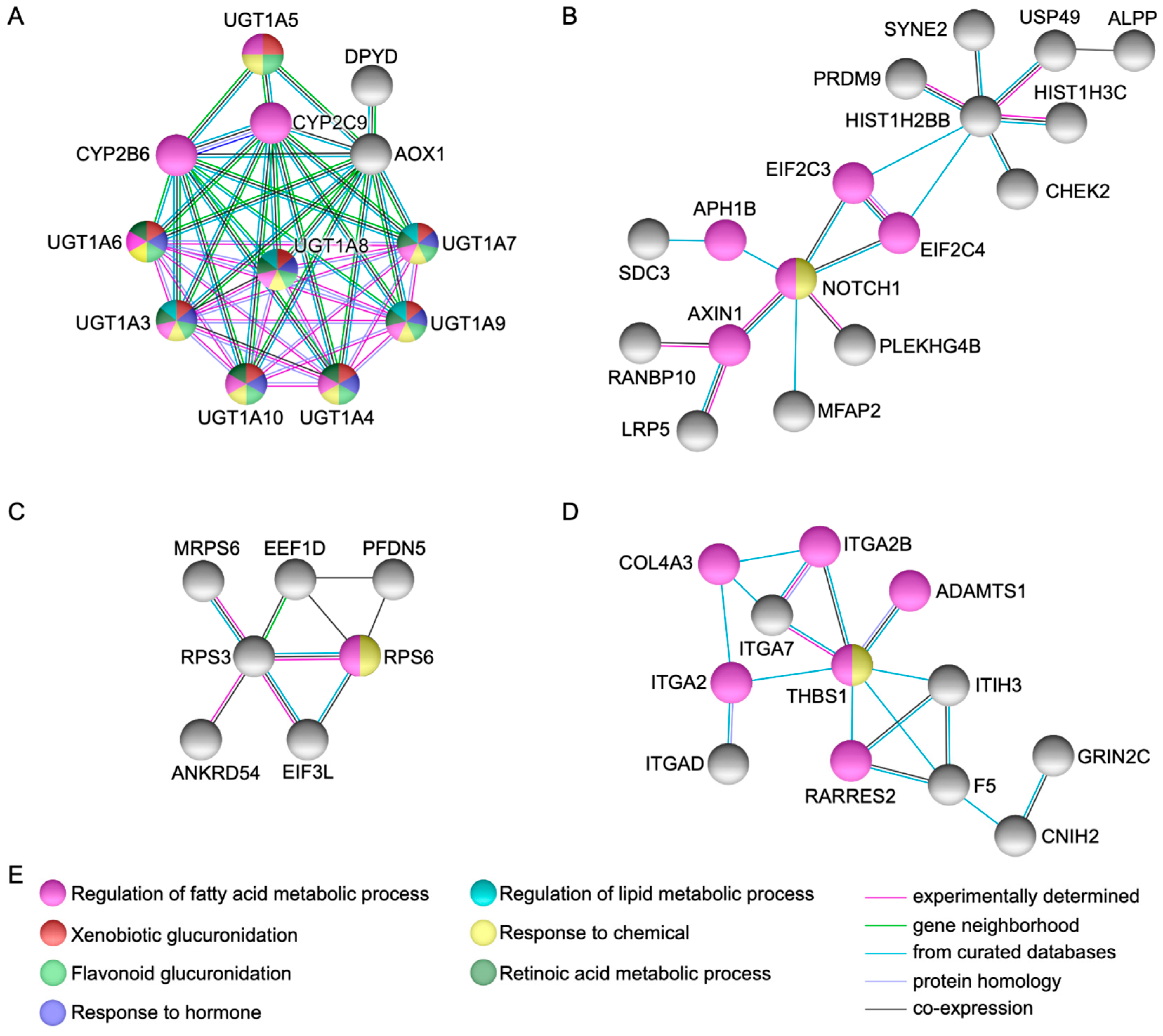
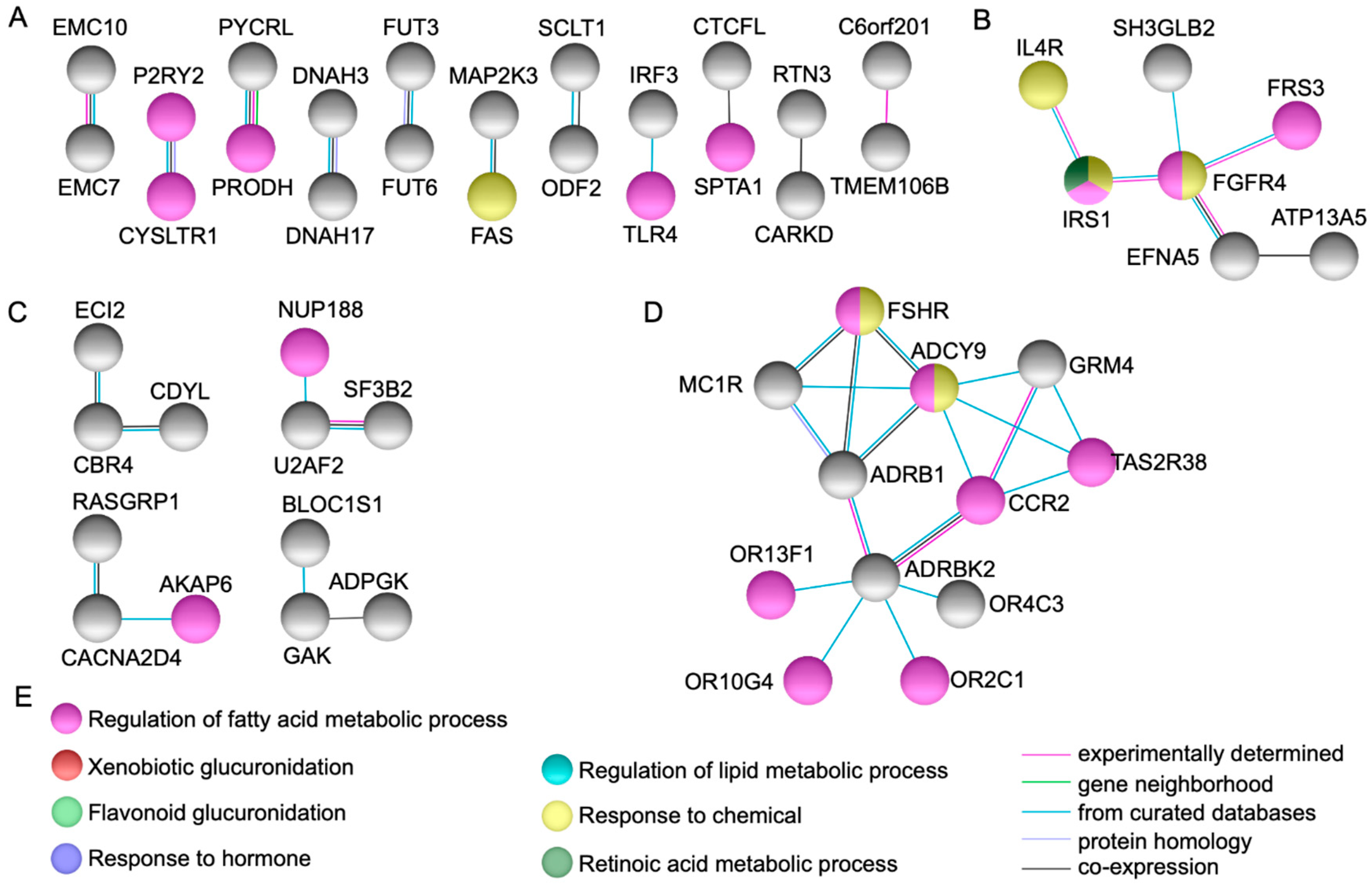
2.2. Protein-Protein Interactions Network Analysis
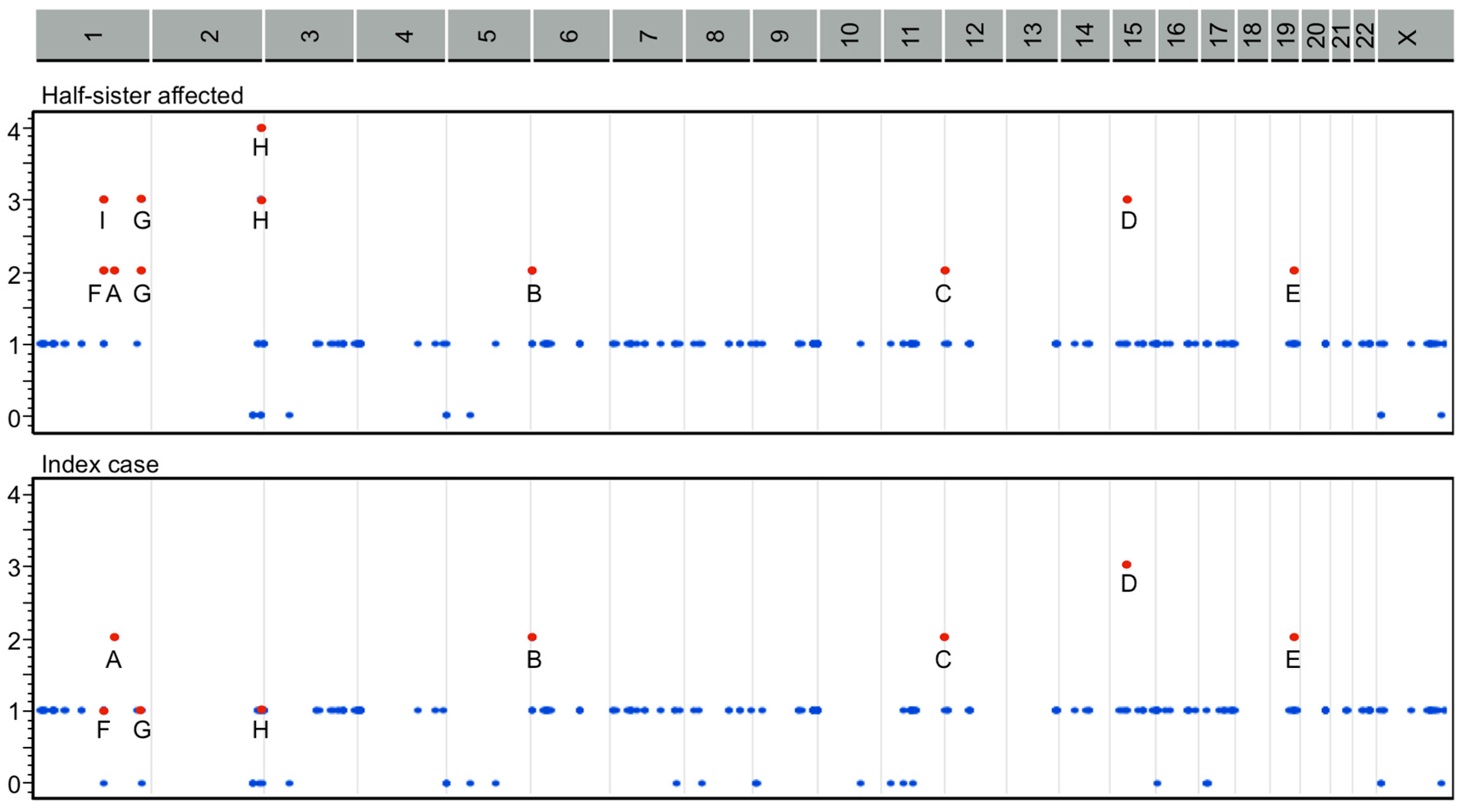
2.3. Classification by Inheritance Pattern
2.4. Pathogenetic Pathways from Selected Genes Co-Segregating with Gastroschisis
3. Discussion
3.1. Novel Candidate Genes and Pathogenetic Pathways
3.2. Theories on the Failure of Body Wall Closure in Human and its Implication for Gastroschisis
3.3. Gastroschisis Clinical Susceptibility
4. Materials and Methods
4.1. Study Participants and Identification of Gene Variants from WES
4.2. Gene Functional Enrichment Analysis
4.3. Gene and Protein Network Analysis
4.4. Classification of Gene Variants by Inheritance Pattern
4.5. Pathogenetic Pathways from GO Functional Categories
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADCY9 | Adenylate cyclase 9 gene |
| ALPP | Alkaline phosphatase placental gene |
| AOX1 | Aldehyde oxidase gene |
| BCAS3 | Microtubule associated cell migration factor gene |
| BP | Biological process |
| CC | Cellular component |
| CDYL | Chromodomain Y like gene |
| CEACAM5 | Carcinoembryonic antigen related cell adhesion molecule gene |
| CFAP65 | Cilia and flagella associated protein 65 gene |
| COL6A3 | Collagen type VI alpha 3 chain gene |
| CROCC | Ciliary rootlet coiled-coil, rootletin gene |
| DPYD | Dihydropyrimidine dehydrogenase gene |
| EVPL | Envoplakin gene |
| F5 | Coagulation factor V gene |
| FDR | False discovery rate |
| FGFR4 | Fibroblast growth factor receptor 4 gene |
| FGFRL1 | Fibroblast growth factor receptor like 1 gene |
| GO | Gene ontology |
| HHIP | Hedgehog interacting protein gene |
| HIST1H2BB | Histone cluster 1 H2B family member B gene |
| IFT140 | Intraflagellar transport 140 gene |
| ITGA2 | Integrin subunit alpha 2 gene |
| ITIH3 | Inter-alpha-trypsin inhibitor heavy chain 3 gene |
| KDM5A | Lysine demethylase 5A gene |
| KP | Kegg pathway |
| KLK14 | Kallikrein related peptidase 14 gene |
| MAP2K3 | Mitogen-activated protein kinase kinase 3 gene |
| MF | Molecular function |
| MYBPC2 | Myosin binding protein C, fast type gene |
| NOTCH1 | Notch 1 gene |
| OBSCN | Obscurin gene |
| OR2C1 | Olfactory receptor family 2 subfamily C member 1 gene |
| OR4C3 | Olfactory receptor family 4 subfamily C member 3 gene |
| OR10G4 | Olfactory receptor family 10 subfamily G member 4 gene |
| OR13F1 | Olfactory receptor family 13 subfamily F member 1 gene |
| PDE4DIP | Phosphodiesterase 4D interacting protein gene |
| PER2 | Period circadian regulator 2 gene |
| PKD1 | Polycystin 1 gene |
| PLEKHG4B | Pleckstrin homology and RhoGEF domain containing G4B gene |
| PLOD1 | Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 gene |
| PTPRD | Protein tyrosine phosphatase receptor type D gene |
| RASGRP1 | RAS Guanyl releasing protein 1 gene |
| RAPGEF1 | Rap guanine nucleotide exchange factor 1 gene |
| RPS3 | Ribosomal protein S3 gene |
| SGCD | Sarcoglycan delta gene |
| SLC9A3 | Solute carrier family 9 member A3 gene |
| SPATA17 | Spermatogenesis associated 17 gene |
| SVS | SNP and variation suite |
| THBS1 | Thrombospondin 1 gene |
| TLR8 | Toll like receptor 8 gene |
| UBE2NL | Ubiquitin conjugating enzyme E2 N like gene |
| UGT1A3 | UDP Glucuronosyltransferase family 1 member A3 gene |
| UGT1A4 | UDP Glucuronosyltransferase family 1 member A4 gene |
| UGT1A5 | UDP Glucuronosyltransferase family 1 member A5 gene |
| UGT1A6 | UDP Glucuronosyltransferase family 1 member A6 gene |
| UGT1A7 | UDP Glucuronosyltransferase family 1 member A7 gene |
| UGT1A8 | UDP Glucuronosyltransferase family 1 member A8 gene |
| UGT1A9 | UDP Glucuronosyltransferase family 1 member A9 gene |
| UGT1A10 | UDP Glucuronosyltransferase family 1 member A10 gene |
| WD | Week of development |
| WES | Whole exome sequence |
| ZFHX3 | Zinc finger homeobox 3 gene |
| ZNF717 | Zinc finger protein 717 gene |
References
- Salinas-Torres, V.M.; Salinas-Torres, R.A.; Cerda-Flores, R.M.; Martínez-de-Villarreal, L.E. Prevalence, mortality, and spatial distribution of gastroschisis in Mexico. J. Pediatr. Adolesc. Gynecol. 2018, 31, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Torres, V.M.; Salinas-Torres, R.A.; Cerda-Flores, R.M.; Martínez-de-Villarreal, L.E. Genetic variants conferring susceptibility to gastroschisis: A phenomenon restricted to the interaction with the environment? Pediatr. Surg. Int. 2018, 34, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Torres, V.M.; Salinas-Torres, R.A.; Cerda-Flores, R.M.; Gallardo-Blanco, H.L.; Martínez-de-Villarreal, L.E. A clinical-pathogenetic approach on associated anomalies and chromosomal defects supports novel candidate critical regions and genes for gastroschisis. Pediatr. Surg. Int. 2018, 34, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Torres, V.M.; Gallardo-Blanco, H.L.; Salinas-Torres, R.A.; Martínez de Villarreal, L.E. Dataset for Genes and Gene Variants from Familial Gastroschisis. Available online: https://doi.org/10.5281/zenodo.2673050 (accessed on 7 May 2019).
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- SNP & Variation Suite ™ (Version 8.x); Golden Helix, Inc.: Bozeman, MT, USA; Available online: http://www.goldenhelix.com (accessed on 31 August 2018).
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [PubMed]
- Rowbotham, S.E.; Illingworth, N.A.; Daly, A.K.; Veal, G.J.; Boddy, A.V. Role of UDP-glucuronosyltransferase isoforms in 13-cis retinoic acid metabolism in humans. Drug Metab. Dispos. 2010, 38, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, B.M.; Rudolph, K.L.; Karagianni, P.; Fonseca, N.A.; White, R.J.; Talianidis, I.; Odom, D.T.; Marioni, J.C.; Kutter, C. High-resolution mapping of transcriptional dynamics across tissue development reveals a stable mRNA-tRNA interface. Genome Res. 2014, 24, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Cormier, S.; Vandormael-Pournin, S.; Babinet, C.; Cohen-Tannoudji, M. Developmental expression of the Notch signaling pathway genes during mouse preimplantation development. Gene Expr. Patterns 2004, 4, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Huss, M.; Tong, G.Q.; Wang, C.; Li Sun, L.; Clarke, N.D.; Robson, P. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev. Cell 2010, 18, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Nordman, E.; Bao, S.; Lee, C.; Wang, X.; Tuch, B.B.; Heard, E.; Lao, K.; Surani, M.A. Deterministic and stochastic allele specific gene expression in single mouse blastomeres. PLoS ONE 2011, 6, e21208. [Google Scholar] [CrossRef]
- Redmond, L.C.; Dumur, C.I.; Archer, K.J.; Haar, J.L.; Lloyd, J.A. Identification of erythroid-enriched gene expression in the mouse embryonic yolk sac using microdissected cells. Dev. Dyn. 2008, 237, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Diez-Roux, G.; Banfi, S.; Sultan, M.; Geffers, L.; Anand, S.; Rozado, D.; Magen, A.; Canidio, E.; Pagani, M.; Peluso, I.; et al. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011, 9, e1000582. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, A.E.; Calarco, P.G.; Damsky, C.H. Developmental regulation of integrin expression at the time of implantation in the mouse embryo. Development 1993, 119, 1175–1186. [Google Scholar] [PubMed]
- Wu, J.E.; Santoro, S.A. Complex patterns of expression suggest extensive roles for the alpha 2 beta 1 integrin in murine development. Dev. Dyn. 1994, 199, 292–314. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.; Horsfall, W.; Conway, E.M.; Schuh, A.C. Early embryonic expression of murine coagulation system components. Thromb. Haemost. 2000, 84, 1023–1030. [Google Scholar] [CrossRef]
- Tamplin, O.J.; Cox, B.J.; Rossant, J. Integrated microarray and ChIP analysis identifies multiple Foxa2 dependent target genes in the notochord. Dev. Biol. 2011, 360, 415–425. [Google Scholar] [CrossRef]
- Visel, A.; Thaller, C.; Eichele, G. GenePaint.org: An atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004, 32, D552–D556. [Google Scholar] [CrossRef]
- Thomas, T.; Voss, A.K.; Petrou, P.; Gruss, P. The murine gene, Traube, is essential for the growth of preimplantation embryos. Dev. Biol. 2000, 227, 324–342. [Google Scholar] [CrossRef]
- Wright, T.J.; Hatch, E.P.; Karabagli, H.; Karabagli, P.; Schoenwolf, G.C.; Mansour, S.L. Expression of mouse fibroblast growth factor and fibroblast growth factor receptor genes during early inner ear development. Dev. Dyn. 2003, 228, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.L.; McMahon, J.A.; McMahon, A.P. FGFR-4, a new member of the fibroblast growth factor receptor family, expressed in the definitive endoderm and skeletal muscle lineages of the mouse. Development 1991, 113, 641–651. [Google Scholar] [PubMed]
- Magdaleno, S.; Jensen, P.; Brumwell, C.L.; Seal, A.; Lehman, K.; Asbury, A.; Cheung, T.; Cornelius, T.; Batten, D.M.; Eden, C.; et al. BGEM: An in situ hybridization database of gene expression in the embryonic and adult mouse nervous system. PLoS Biol. 2006, 4, e86. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.A.; Fu, H.; Luo, P.; Zhao, Q.; Yu, J.; Ferrari, A.; Tenzen, T.; Yuk, D.I.; Tsung, E.F.; Cai, Z.; et al. Mouse brain organization revealed through direct genome-scale TF expression analysis. Science 2004, 306, 2255–2257. [Google Scholar] [CrossRef] [PubMed]
- Siyanov, V.; Baltz, J.M. NHE1 is the sodium-hydrogen exchanger active in acute intracellular pH regulation in preimplantation mouse embryos. Biol. Reprod. 2013, 88, 157. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barr, K.J.; Garrill, A.; Jones, D.H.; Orlowski, J.; Kidder, G.M. Contributions of Na+/H+ exchanger isoforms to preimplantation development of the mouse. Mol. Reprod. Dev. 1998, 50, 146–153. [Google Scholar] [CrossRef]
- Nakamura, K.; Stokes, J.B.; McCray, P.B., Jr. Endogenous and exogenous glucocorticoid regulation of ENaC mRNA expression in developing kidney and lung. Am. J. Physiol. Cell Physiol. 2002, 283, C762–C772. [Google Scholar] [CrossRef][Green Version]
- Schaapveld, R.Q.; Schepens, J.T.; Bächner, D.; Attema, J.; Wieringa, B.; Jap, P.H.; Hendriks, W.J. Developmental expression of the cell adhesion molecule-like protein tyrosine phosphatases LAR, RPTPdelta and RPTPsigma in the mouse. Mech. Dev. 1998, 77, 59–62. [Google Scholar] [CrossRef]
- Kurasawa, M.; Sato, N.; Matsuda, A.; Koshida, S.; Totsuka, T.; Obinata, T. Differential expression of C-protein isoforms in developing and degenerating mouse striated muscles. Muscle Nerve. 1999, 22, 196–207. [Google Scholar] [CrossRef]
- Giudice, J.; Xia, Z.; Wang, E.T.; Scavuzzo, M.A.; Ward, A.J.; Kalsotra, A.; Wang, W.; Wehrens, X.H.; Burge, C.B.; Li, W.; et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat. Commun. 2014, 5, 3603. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.C.; Stokes, G.E. Gastroschisis; report of two cases treated by a modification of the gross operation for omphalocele. Surgery 1953, 33, 112–120. [Google Scholar] [PubMed]
- Duhamel, B. Embryology of exomphalos and allied malformations. Arch. Dis. Child. 1963, 38, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Feldkamp, M.L.; Carey, J.C.; Sadler, T.W. Development of gastroschisis: Review of hypotheses, a novel hypothesis, and implications for research. Am. J. Med. Genet. A 2007, 143A, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A. The myth of gastroschisis. J. Pediatr. Surg. 1975, 10, 235–244. [Google Scholar] [CrossRef]
- Thomas, D.F.; Atwell, J.D. The embryology and surgical management of gastroschisis. Br. J. Surg. 1976, 63, 893–897. [Google Scholar] [CrossRef] [PubMed]
- deVries, P.A. The pathogenesis of gastroschisis and omphalocele. J. Pediatr. Surg. 1980, 15, 245–251. [Google Scholar] [CrossRef]
- Hoyme, H.E.; Higginbottom, M.C.; Jones, K.L. The vascular pathogenesis of gastroschisis: Intrauterine interruption of the omphalomesenteric artery. J. Pediatr. 1981, 98, 228–231. [Google Scholar] [CrossRef]
- Vermeij-Keers, C.; Hartwig, N.G.; van der Werff, J.F. Embryonic development of the ventral body wall and its congenital malformations. Semin. Pediatr. Surg. 1996, 5, 82–89. [Google Scholar] [PubMed]
- Stevenson, R.E.; Rogers, R.C.; Chandler, J.C.; Gauderer, M.W.; Hunter, A.G. Escape of the yolk sac: A hypothesis to explain the embryogenesis of gastroschisis. Clin. Genet. 2009, 75, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Lubinsky, M. A vascular and thrombotic model of gastroschisis. Am. J. Med. Genet. A 2014, 164A, 915–917. [Google Scholar] [CrossRef]
- Salinas-Torres, V.M.; Salinas-Torres, R.A.; Cerda-Flores, R.M.; Martínez-de-Villarreal, L.E. Evaluation of familial factors in a Mexican population-based setting with gastroschisis: Further evidence for an underlying genetic susceptibility. J. Pediatr. Surg. 2018, 53, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Torres, V.M.; Salinas-Torres, R.A.; Cerda-Flores, R.M.; Martínez-de-Villarreal, L.E. Familial occurrence of gastroschisis: A population-based overview on recurrence risk, sex-dependent influence, and geographical distribution. Pediatr. Surg. Int. 2018, 34, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; van Leeuwen, J.; Andrews, B.J.; Boone, C. Genetic network complexity shapes background-dependent phenotypic expression. Trends Genet. 2018, 34, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An expanded view of complex traits: From polygenic to omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Hegde, M. Inferring the effect of genomic variation in the new era of genomics. Hum. Mutat. 2018, 39, 756–773. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, N.G.; Vermeij-Keers, C.; De Vries, H.E.; Kagie, M.; Kragt, H. Limb body wall malformation complex: An embryologic etiology? Hum. Pathol. 1989, 20, 1071–1077. [Google Scholar] [CrossRef]
- Hartwig, N.G.; Steffelaar, J.W.; Van de Kaa, C.; Schueler, J.A.; Vermeij-Keers, C. Abdominal wall defect associated with persistent cloaca. The embryologic clues in autopsy. Am. J. Clin. Pathol. 1991, 96, 640–647. [Google Scholar] [CrossRef]
- Brewer, S.; Williams, T. Finally, a sense of closure? Animal models of human ventral body wall defects. Bioessays 2004, 26, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- O’Rahilly, R.; Müller, F. Human Embryology and Teratology, 3rd ed.; Wiley-Liss: New York, NY, USA, 2001. [Google Scholar]
- Rittler, M.; Vauthay, L.; Mazzitelli, N. Gastroschisis is a defect of the umbilical ring: Evidence from morphological evaluation of stillborn fetuses. Birth Defects Res. A Clin. Mol. Teratol. 2013, 97, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Smits-van Prooije, A.E.; Vermeij-Keers, C.; Poelmann, R.E.; Mentink, M.M.; Dubbeldam, J.A. The formation of mesoderm and mesectoderm in 5- to 41-somite rat embryos cultured in vitro, using WGA-Au as a marker. Anat. Embryol. (Berl.) 1988, 177, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Brewer, S.; Williams, T. Loss of AP-2alpha impacts multiple aspects of ventral body wall development and closure. Dev. Biol. 2004, 267, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Mekonen, H.K.; Hikspoors, J.P.; Mommen, G.; Köhler, S.E.; Lamers, W.H. Development of the ventral body wall in the human embryo. J. Anat. 2015, 227, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Gasser, R.F. Evidence that some events of mammalian embryogenesis can result from differential growth, making migration unnecessary. Anat. Rec. B New. Anat. 2006, 289, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Hussain, F.; Decuzzi, P. Role of differential adhesion in cell cluster evolution: From vasculogenesis to cancer metastasis. Comput. Methods Biomech. Biomed. Eng. 2015, 18, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Goody, M.F.; Sher, R.B.; Henry, C.A. Hanging on for the ride: Adhesion to the extracellular matrix mediates cellular responses in skeletal muscle morphogenesis and disease. Dev. Biol. 2015, 401, 75–91. [Google Scholar] [CrossRef]
- Bargy, F.; Beaudoin, S. Comprehensive developmental mechanisms in gastroschisis. Fetal Diagn. Ther. 2014, 36, 223–330. [Google Scholar] [CrossRef] [PubMed]
- Shearman, R.M.; Burke, A.C. The lateral somitic frontier in ontogeny and phylogeny. J. Exp. Zool. B. Mol. Dev. Evol. 2009, 312, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Streeter, G.L. Focal deficiencies in fetal tissues and their relation to intra-uterine amputation. Contrib. Embryol. Carnegie Inst. 1930, 22, 1–44. [Google Scholar]
- Bamforth, J.S. Amniotic band sequence: Streeter’s hypothesis reexamined. Am. J. Med. Genet. 1992, 44, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Torres, V.M.; De La O-Espinoza, E.A.; Salinas-Torres, R.A. Severe intrauterine amputations in one dichorionic twin with pentalogy of Cantrell: Further evidence and consideration for mechanical teratogenesis. Pediatr. Dev. Pathol. 2017, 20, 440–443. [Google Scholar] [CrossRef]
- Salinas-Torres, V.M. Fetus with Casamassima-Morton-Nance syndrome and limb-body wall defect: Presentation of a novel association and review of the phenotype. Pediatr. Dev. Pathol. 2016, 19, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Folkerth, R.D.; Habbe, D.M.; Boyd, T.K.; McMillan, K.; Gromer, J.; Sens, M.A.; Elliott, A.J. Prenatal Alcohol, SIDS and Stillbirth (PASS) Research Network. Gastroschisis, destructive brain lesions, and placental infarction in the second trimester suggest a vascular pathogenesis. Pediatr. Dev. Pathol. 2013, 16, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.D.; Chen, B.H.; Kalla, K.; Jernigan, L.; Jones, K.L. Novel risk factor in gastroschisis: Change of paternity. Am. J. Med. Genet. A 2007, 143A, 653–659. [Google Scholar] [CrossRef]
- Frascoli, M.; Jeanty, C.; Fleck, S.; Moradi, P.W.; Keating, S.; Mattis, A.N.; Tang, Q.; MacKenzie, T.C. Heightened immune activation in fetuses with gastroschisis may be blocked by targeting IL-5. J. Immunol. 2016, 196, 4957–4966. [Google Scholar] [CrossRef]
- Kalache, K.D.; Bierlich, A.; Hammer, H.; Bollmann, R. Is unexplained third trimester intrauterine death of fetuses with gastroschisis caused by umbilical cord compression due to acute extra-abdominal bowel dilatation? Prenat. Diagn. 2002, 22, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Luton, D.; de Lagausie, P.; Guibourdenche, J.; Oury, J.; Sibony, O.; Vuillard, E.; Boissinot, C.; Aigrain, Y.; Beaufils, F.; Navarro, J.; et al. Effect of amnioinfusion on the outcome of prenatally diagnosed gastroschisis. Fetal Diagn. Ther. 1999, 14, 152–155. [Google Scholar] [CrossRef] [PubMed]
- McClintock, T.S.; Glasser, C.E.; Bose, S.C.; Bergman, D.A. Tissue expression patterns identify mouse cilia genes. Physiol. Genomics 2008, 32, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Stauber, M.; Weidemann, M.; Dittrich-Breiholz, O.; Lobschat, K.; Alten, L.; Mai, M.; Beckers, A.; Kracht, M.; Gossler, A. Identification of FOXJ1 effectors during ciliogenesis in the foetal respiratory epithelium and embryonic left-right organiser of the mouse. Dev. Biol. 2017, 423, 170–188. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.L.; Hsu, P.P.; Glienke, J.; Rubanyi, G.M.; Brooks, A.R. Hedgehog-interacting protein is highly expressed in endothelial cells but down-regulated during angiogenesis and in several human tumors. BMC Cancer 2004, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, R.; D’Agati, V.; Daoust, M.; Trudel, M. Murine Pkd1 is a developmentally regulated gene from morula to adulthood: Role in tissue condensation and patterning. Dev. Dyn. 1999, 214, 337–348. [Google Scholar] [CrossRef]
- Weinmaster, G.; Roberts, V.J.; Lemke, G. A homolog of Drosophila Notch expressed during mammalian development. Development 1991, 113, 199–205. [Google Scholar] [PubMed]
- Schmidts, M.; Frank, V.; Eisenberger, T.; Al Turki, S.; Bizet, A.A.; Antony, D.; Rix, S.; Decker, C.; Bachmann, N.; Bald, M.; et al. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney Disease. Hum. Mutat. 2013, 34, 714–724. [Google Scholar] [CrossRef]
- Ryan, A.K.; Blumberg, B.; Rodriguez-Esteban, C.; Yonei-Tamura, S.; Tamura, K.; Tsukui, T.; de la Peña, J.; Sabbagh, W.; Greenwald, J.; Choe, S.; et al. Pitx2 determines left-right asymmetry of internal organs in vertebrates. Nature 1998, 394, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Hikspoors, J.P.J.M.; Peeters, M.M.J.P.; Mekonen, H.K.; Kruepunga, N.; Mommen, G.M.C.; Cornillie, P.; Köhler, S.E.; Lamers, W.H. The fate of the vitelline and umbilical veins during the development of the human liver. J. Anat. 2017, 231, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Ramsdell, A.F.; Yost, H.J. Molecular mechanisms of vertebrate left-right development. Trends Genet. 1998, 14, 459–465. [Google Scholar] [CrossRef]
- Benjamin, B.G.; Ethen, M.K.; Van Hook, C.L.; Myers, C.A.; Canfield, M.A. Gastroschisis prevalence in Texas 1999–2003. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 178–185. [Google Scholar] [PubMed]
- Khodr, Z.G.; Lupo, P.J.; Canfield, M.A.; Chan, W.; Cai, Y.; Mitchell, L.E. Hispanic ethnicity and acculturation, maternal age and the risk of gastroschisis in the National Birth Defects Prevention Study. Birth Defects Res. A Clin. Mol. Teratol. 2013, 97, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.M.; Ananth, C.V.; Siddiq, Z.; D’Alton, M.E.; Wright, J.D. Gastroschisis: Epidemiology and mode of delivery, 2005–2013. Am. J. Obstet. Gynecol. 2016, 215, e1–e9. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Category | GO term | Count | P Values (FDR) |
|---|---|---|---|
| BP | Xenobiotic glucuronidation | 8 | 7.99 × 10−11 |
| BP | Negative regulation of glucuronosyltransferase activity | 7 | 1.42 × 10−9 |
| BP | Flavonoid glucuronidation | 8 | 2.26 × 10−7 |
| BP | Response to hormone | 30 | 1.65 × 10−4 |
| BP | Response to chemical | 75 | 3.9 × 10−4 |
| BP | Regulation of fatty acid metabolic process | 9 | 7.61 × 10−4 |
| BP | Retinoic acid metabolic process | 4 | 2.05 × 10−2 |
| CC | Integral component of membrane | 70 | 4.95 × 10−3 |
| CC | Membrane part | 75 | 5.55 × 10−3 |
| CC | Endoplasmic reticulum part | 23 | 8.92 × 10−3 |
| CC | Cytoplasmic part | 102 | 2.18 × 10−2 |
| MF | Glucuronosyltransferase activity | 8 | 1.58 × 10−6 |
| MF | Retinoid binding | 5 | 7.67 × 10−3 |
| MF | Active transmembrane transporter activity | 12 | 1.27 × 10−2 |
| KP | Drug metabolism – other enzymes | 9 | 1.32 × 10−8 |
| KP | Pentose and glucuronate interconversions | 8 | 2.83 × 10−8 |
| KP | Retinol metabolism | 9 | 1.1 × 10−7 |
| KP | Steroid hormone biosynthesis | 8 | 5.85 × 10−7 |
| KP | Metabolism of xenobiotics by cytochrome P450 | 8 | 3.45 × 10−6 |
| Pathogenetic Pathway | GO Terms | Genes Involved |
|---|---|---|
| Xenobiotic | Xenobiotic and flavonoid glucuronidation, retinol metabolism, negative regulation of fatty acid metabolic process, drug metabolic process, steroid hormone biosynthesis, negative regulation of cellular carbohydrate metabolic process, cellular response to xenobiotic stimulus, chemical carcinogenesis, negative regulation of lipid metabolic process, cellular hormone metabolic process | UGT1A4, UGT1A3, UGT1A10, UGT1A8, UGT1A7, UGT1A6, UGT1A5, AOX1, UGT1A9 |
| Regulation of metabolic processes | Negative regulation of catalytic activity, negative regulation of molecular function, regulation of transferase activity, carboxylic acid metabolic process, response to growth factor, regulation of hydrolase activity, response to endogenous stimulus, detection of stimulus, regulation of protein modification process, folate biosynthesis | UGT1A4, UGT1A3, PLEKHG4B, COL6A3, RASGRP1, HHIP, THBS1, ADCY9, PER2, KDM5A, SLC9A3, BCAS3, OR2C1, OR4C3, RPS3, OR13F1, OBSCN, UGT1A10, UGT1A8, UGT1A7, UGT1A6, PKD1, UGT1A9, RAPGEF1, FGFRL1, ZFHX3, MAP2K3, FGFR4, ITGA2, TLR8, OR10G4, ITIH3, NOTCH1, ALPP, PLOD1 |
| Regulation of cell adhesion | Cell-substrate adhesion, cell adhesion, regulation of cell junction assembly, negative regulation of anoikis, epidermis morphogenesis and development, epithelium development, cell-cell adhesion via plasma-membrane adhesion molecules, focal adhesion, wound healing | COL6A3, RASGRP1, THBS1, CEACAM5, PTPRD, BCAS3, HHIP, PKD1, RAPGEF1, FGFRL1, ITGA2, F5, KLK14, EVPL, FGFR4, PLOD1, NOTCH1, MYBPC2 |
| Regulation of gene expression | Circadian regulation of gene expression, developmental biology, multi-organism reproductive process, negative regulation of nucleic acid-templated transcription | PER2, ZNF717, KDM5A, ZFHX3, COL6A3, RASGRP1, KLK14, HIST1H2BB, CDYL, EVPL, FGFR4, ITGA2, NOTCH1 |
| Inflammatory response | Toll receptor signaling pathway, inflammatory response, regulation of cytokine biosynthetic process | UBE2NL, MAP2K3, TLR8, RASGRP1, THBS1, AOX1, ITGA2, NOTCH1 |
| Regulation of vascular development | Circulatory system development, blood vessel development, hemostasis, blood coagulation | HHIP, RASGRP1, THBS1, CEACAM5, ITGA2, F5, ITIH3, BCAS3, SGCD, PKD1, RAPGEF1, FGFRL1, NOTCH1 |
| Keratinization | Formation of the cornified envelope | KLK14, EVPL |
| Left-right symmetry | Left-right axis specification | NOTCH1 |
| Epigenetic | Histone modification, chromatin organization, DNA methylation | PER2, KDM5A, HIST1H2BB, CDYL |
| Ubiquitination | Protein ubiquitination | UBE2NL, PER2, RPS3 |
| Regulation of protein synthesis | Protein-containing complex assembly | RASGRP1, RPS3, PDE4DIP, FGFRL1, HIST1H2BB, F5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salinas-Torres, V.M.; Gallardo-Blanco, H.L.; Salinas-Torres, R.A.; Cerda-Flores, R.M.; Lugo-Trampe, J.J.; Villarreal-Martínez, D.Z.; Martínez de Villarreal, L.E. Bioinformatic Analysis of Gene Variants from Gastroschisis Recurrence Identifies Multiple Novel Pathogenetic Pathways: Implication for the Closure of the Ventral Body Wall. Int. J. Mol. Sci. 2019, 20, 2295. https://doi.org/10.3390/ijms20092295
Salinas-Torres VM, Gallardo-Blanco HL, Salinas-Torres RA, Cerda-Flores RM, Lugo-Trampe JJ, Villarreal-Martínez DZ, Martínez de Villarreal LE. Bioinformatic Analysis of Gene Variants from Gastroschisis Recurrence Identifies Multiple Novel Pathogenetic Pathways: Implication for the Closure of the Ventral Body Wall. International Journal of Molecular Sciences. 2019; 20(9):2295. https://doi.org/10.3390/ijms20092295
Chicago/Turabian StyleSalinas-Torres, Víctor M., Hugo L. Gallardo-Blanco, Rafael A. Salinas-Torres, Ricardo M. Cerda-Flores, José J. Lugo-Trampe, Daniel Z. Villarreal-Martínez, and Laura E. Martínez de Villarreal. 2019. "Bioinformatic Analysis of Gene Variants from Gastroschisis Recurrence Identifies Multiple Novel Pathogenetic Pathways: Implication for the Closure of the Ventral Body Wall" International Journal of Molecular Sciences 20, no. 9: 2295. https://doi.org/10.3390/ijms20092295
APA StyleSalinas-Torres, V. M., Gallardo-Blanco, H. L., Salinas-Torres, R. A., Cerda-Flores, R. M., Lugo-Trampe, J. J., Villarreal-Martínez, D. Z., & Martínez de Villarreal, L. E. (2019). Bioinformatic Analysis of Gene Variants from Gastroschisis Recurrence Identifies Multiple Novel Pathogenetic Pathways: Implication for the Closure of the Ventral Body Wall. International Journal of Molecular Sciences, 20(9), 2295. https://doi.org/10.3390/ijms20092295