AKIRIN1: A Potential New Reference Gene in Human Natural Killer Cells and Granulocytes in Sepsis


Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
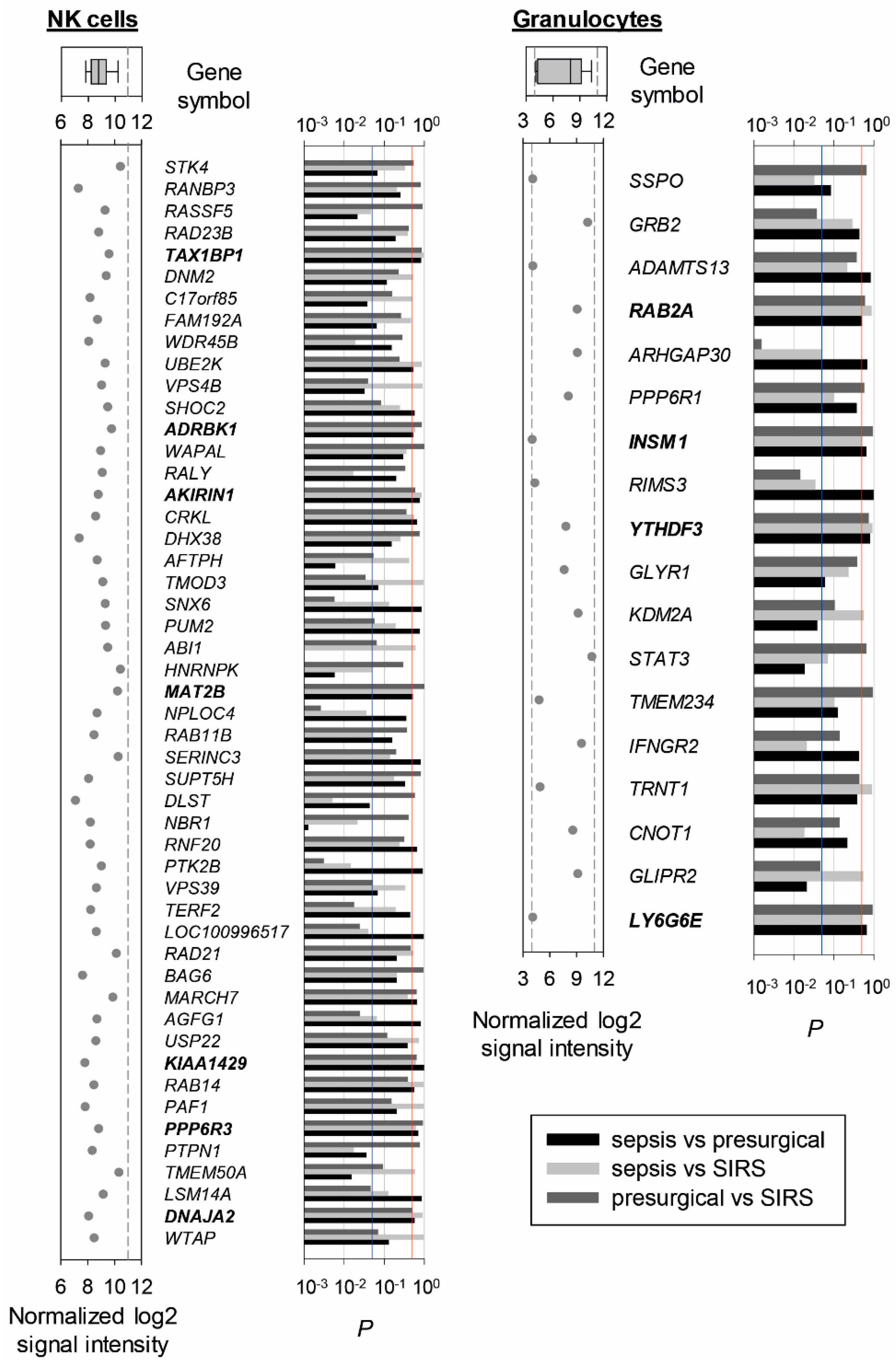
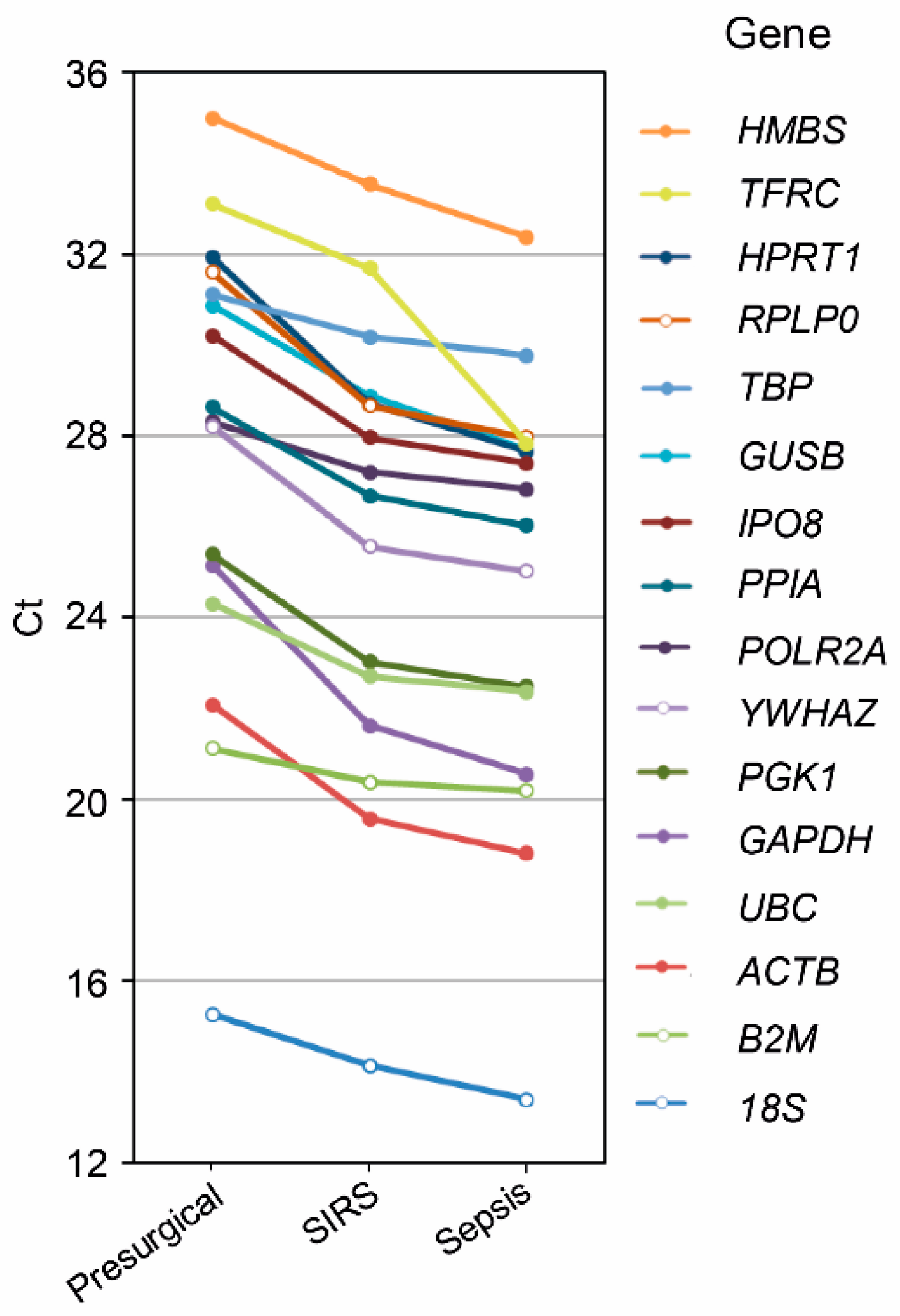
2.2. Reference Gene Discovery in Presurgical, SIRS, and Sepsis Patients
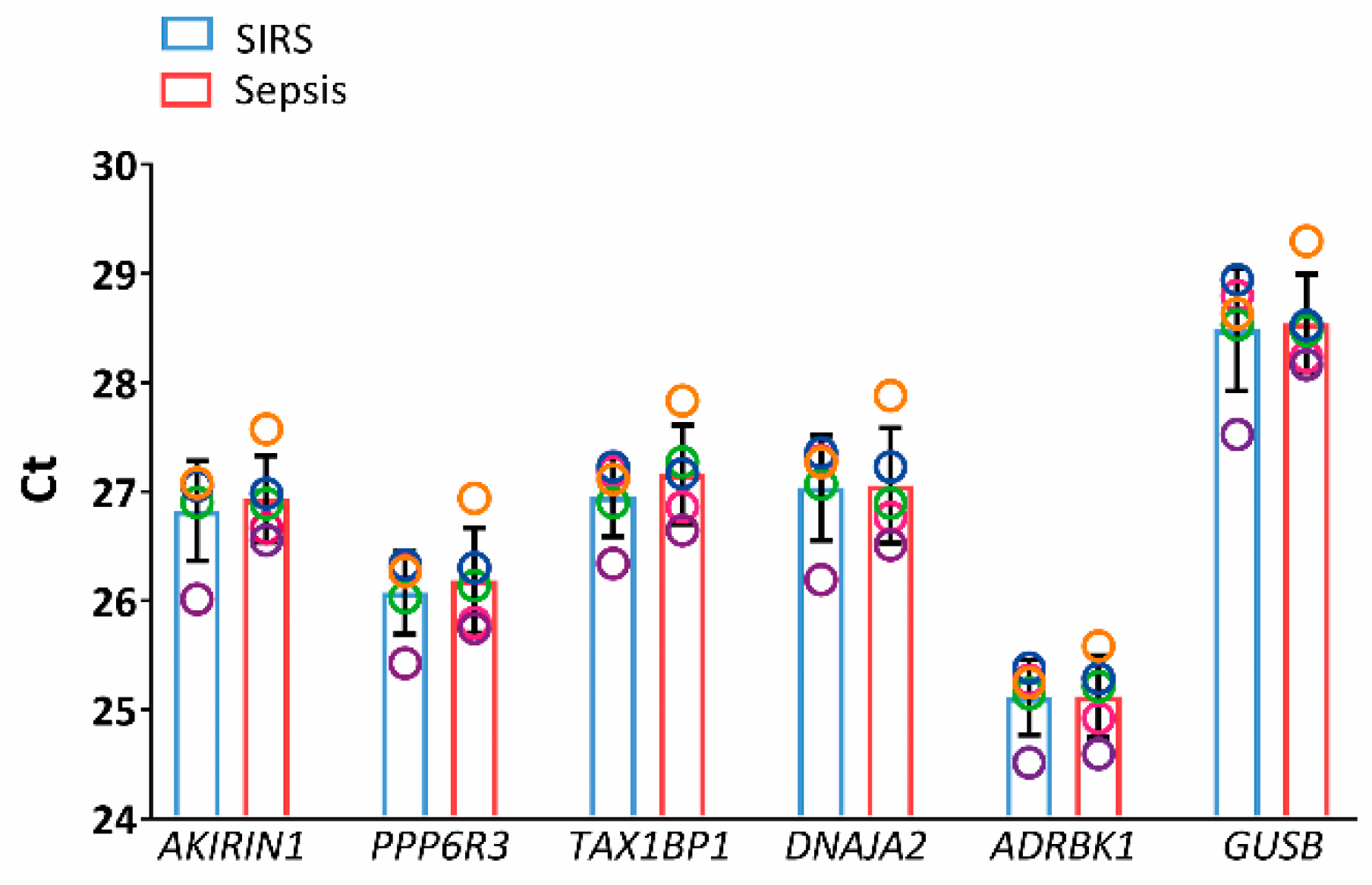
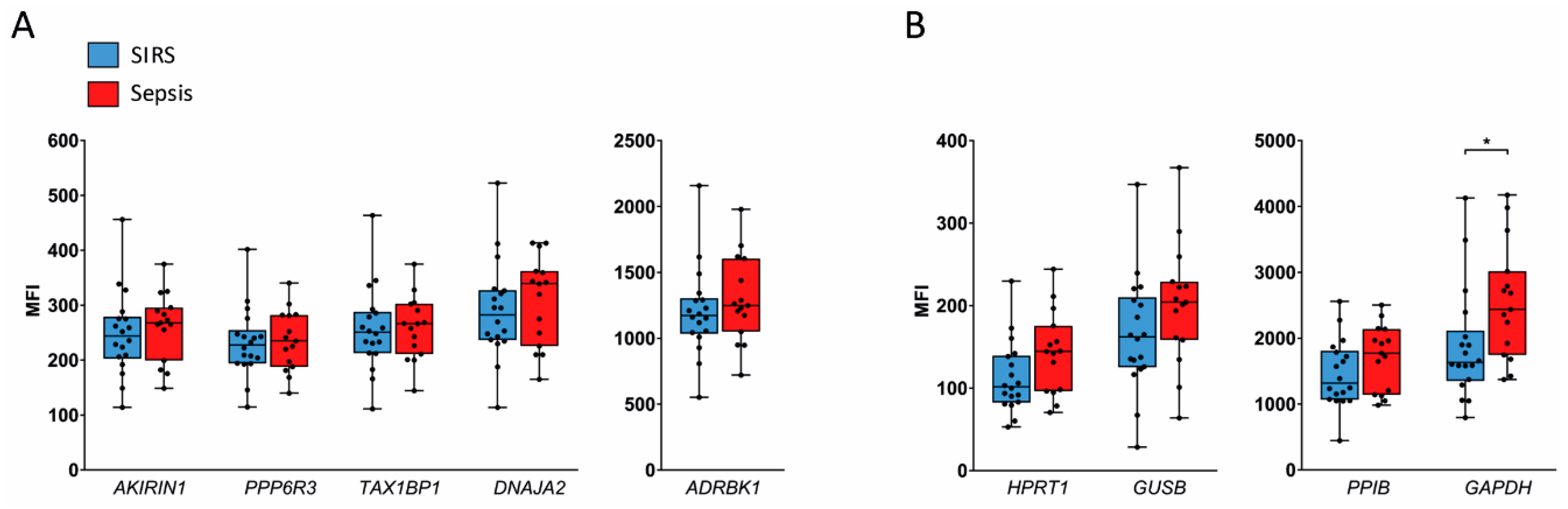
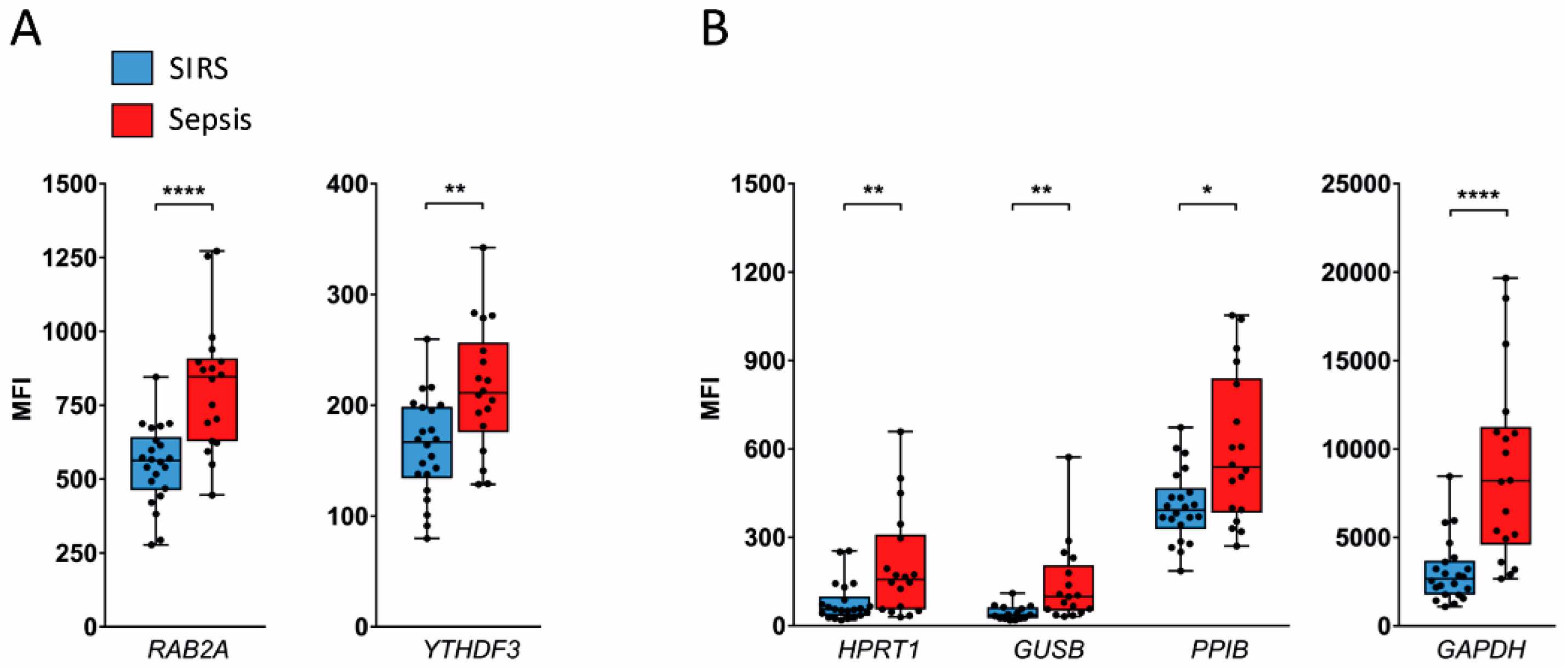
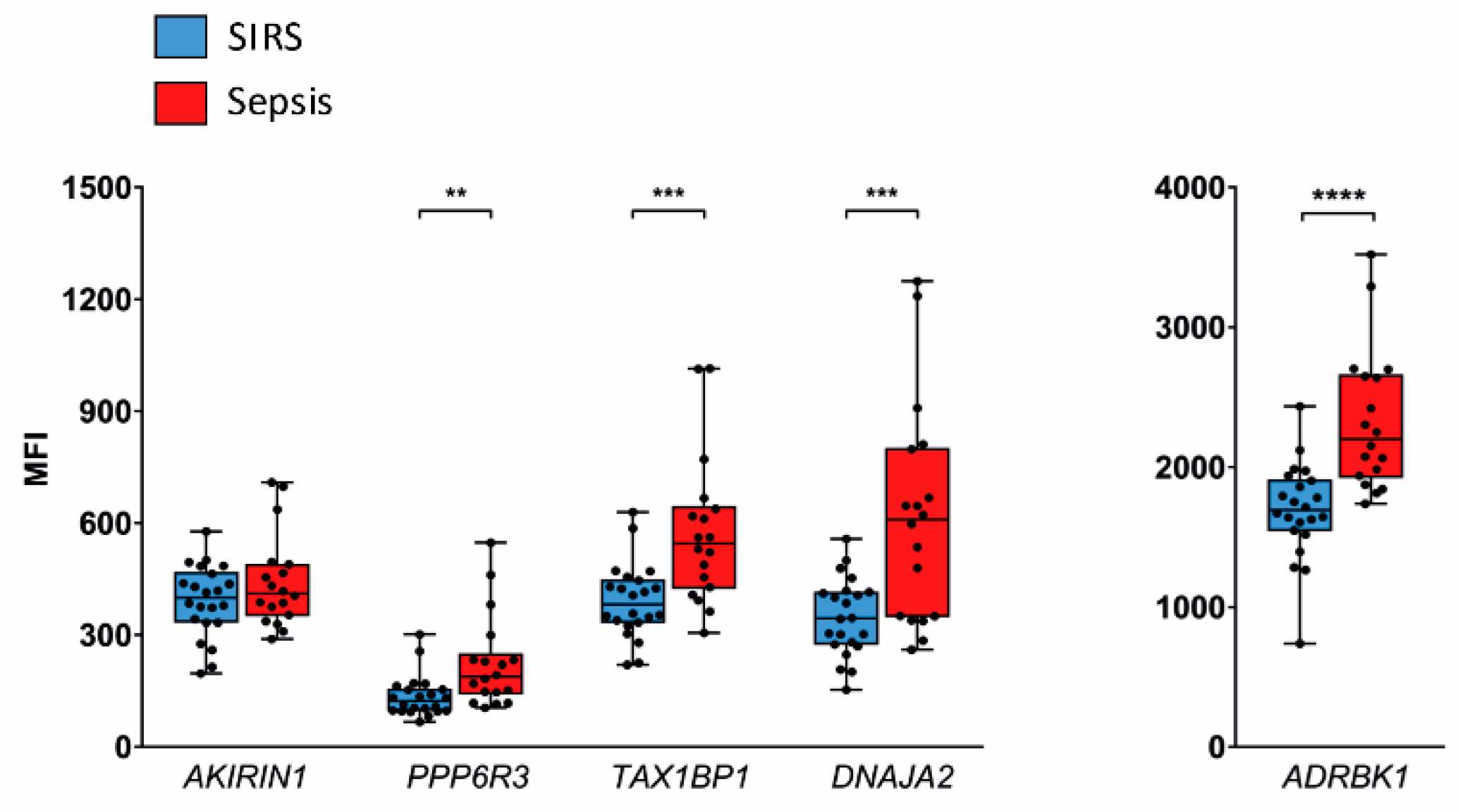
2.3. Validation in SIRS and Sepsis Patients
2.4. AKIRIN1 Expression in External Neutrophil Transcriptome Datasets
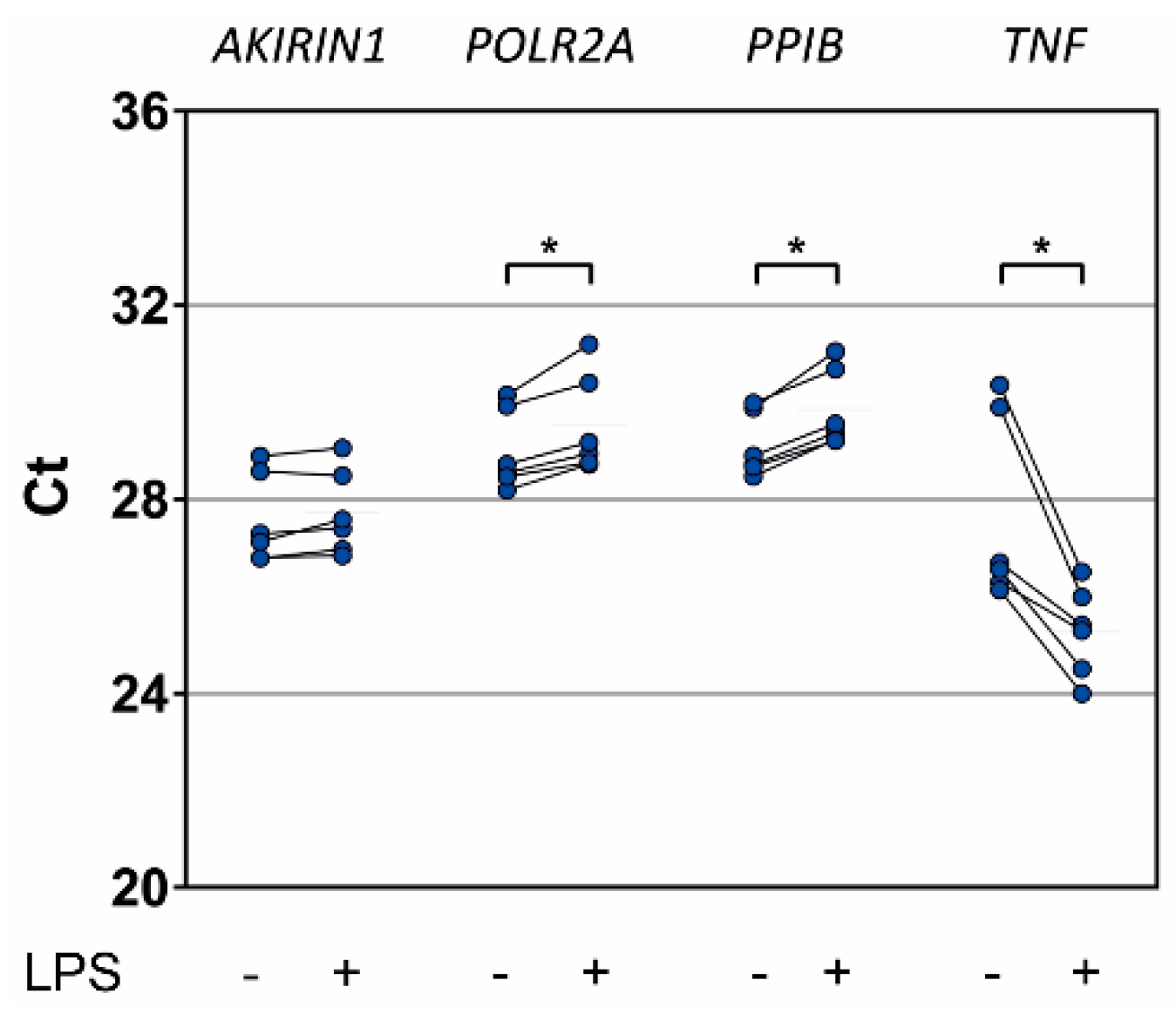
2.5. In Vitro Proinflammatory Stimulation of Healthy Donor Granulocytes
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Cell Isolation and Stimulation
4.3. Flow Cytometry
4.4. Total RNA
4.5. Microarray and Selection of Candidate Endogenous Reference Genes
4.6. RT-PCR
4.7. Multiplex Gene Expression Assay
4.8. External Transcriptome Dataset Retrieval
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADRBK1 | G protein-coupled receptor kinase 2 |
| ALI | Acute lung injury |
| CRP | C-reactive protein |
| Ct | Threshold cycle |
| DNAJA2 | DnaJ heat shock protein family (Hsp40) member A2 |
| FBS | Fetal bovine serum |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GUSB | Glucuronidase beta |
| HMGB1 | High mobility group box 1 protein |
| HPRT1 | Hypoxanthine phosphoribosyltransferase 1 |
| ICU | Intensive care unit |
| IL-15 | Interleukin-15 |
| INSM1 | INSM transcriptional repressor 1 |
| LPS | Lipopolysaccharide |
| LY6G6E NK | Lymphocyte antigen 6 family member G6E Natural killer |
| PBMCs | Peripheral blood mononuclear cells |
| PBS | Phosphate buffered saline |
| POLR2A | RNA polymerase II subunit A |
| PPIB | Peptidylprolyl isomerase B |
| PPP6R3 | Protein phosphatase 6 regulatory subunit 3 |
| P4HA1 | Prolyl 4-hydroxylase subunit alpha 1 |
| QGP | QuantiGene Plex |
| RAB2A | RAB2A, member RAS oncogene family |
| sd | Standard deviation |
| SIRS | Systemic inflammatory response syndrome |
| SOFA | Sequential organ failure assessment score |
| TAX1BP1 | Tax1 binding protein 1 |
| TNF | Tumor necrosis factor |
| YTHDF3 | YTH N6-methyladenosine RNA binding protein 3 |
| WBCs | White blood cell counts |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Full Name | Gene Symbol | Chromosomal Location | Taqman Assay ID | GenBank Accession Number 1 |
|---|---|---|---|---|
| Candidate reference genes identified from microarray for NK cells | ||||
| G protein-coupled receptor kinase 2 2 | ADRBK1 | 11q13.2 | Hs00176395_m1 | NM_001619 |
| akirin1 | AKIRIN1 | 1p34.3 | Hs01047800_g1 | NM_024595 |
| protein phosphatase 6 regulatory subunit 3 | PPP6R3 | 11q13.2 | Hs00217759_m1 | NM_018312 |
| Tax1 binding protein 1 | TAX1BP1 | 7p15.2 | Hs00195718_m1 | NM_006024 |
| DnaJ heat shock protein family (Hsp40) member A2 | DNAJA2 | 16q11.2 | Hs00195365_m1 | NM_005880 |
| Candidate reference genes identified from microarray for granulocytes | ||||
| RAB2A, member RAS oncogene family | RAB2A | 8q12.1-q12.2 | na 3 | NM_002865 |
| INSM transcriptional repressor 1 | INSM1 | 20p11.23 | na | NM_002196 |
| YTH N6-methyladenosine RNA binding protein 3 | YTHDF3 | 8q12.3 | na | NM_152758 |
| lymphocyte antigen 6 family member G6E | LY6G6E | 6p21.33 | na | NR_003673 |
| Known endogenous reference genes | ||||
| glyceraldehyde-3-phosphate dehydrogenase | GAPDH | 12p13.31 | na | NM_002046 |
| glucuronidase beta | GUSB | 7q11.21 | Hs00939627_m1 | NM_000181 |
| hypoxanthine phosphoribosyltransferase 1 | HPRT1 | Xq26.2-q26.3 | na | NM_000194 |
| RNA polymerase II subunit A | POLR2A | 17p13.1 | Hs00172187_m1 | na |
| peptidylprolyl isomerase B | PPIB | 15q22.31 | Hs00168719_m1 | NM_000942 |
| Control genes for in vitro cell stimulation | ||||
| prolyl 4-hydroxylase subunit alpha 1 | P4HA1 | 10q22.1 | Hs00914594_m1 | na |
| tumor necrosis factor | TNF | 6p21.33 | Hs00174128_m1 | na |
References
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992, 101, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.M.; Fink, M.P.; Marshall, J.C.; Abraham, E.; Angus, D.; Cook, D.; Cohen, J.; Opal, S.M.; Vincent, J.L.; Ramsay, G.; et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit. Care Med. 2003, 31, 1250–1256. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.M.; Ward, N.S. Introduction. In Sepsis; Ward, N.S., Levy, M.M., Eds.; Humana Press: Providence, RI, USA, 2017; pp. 3–5. ISBN 978-3-319-48470-9. [Google Scholar]
- Engel, C.; Brunkhorst, F.M.; Bone, H.G.; Brunkhorst, R.; Gerlach, H.; Grond, S.; Gruendling, M.; Huhle, G.; Jaschinski, U.; John, S.; et al. Epidemiology of sepsis in Germany: Results from a national prospective multicenter study. Intensive Care Med. 2007, 33, 606–618. [Google Scholar] [CrossRef]
- Lindner, H.A.; Balaban, U.; Sturm, T.; Weiss, C.; Thiel, M.; Schneider-Lindner, V. An Algorithm for Systemic Inflammatory Response Syndrome Criteria-Based Prediction of Sepsis in a Polytrauma Cohort. Crit. Care Med. 2016, 44, 2199–2207. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Jupe, J.; Mack, H.; Coleman, T.P.; Lawrence, S.M.; Fraley, S.I. Emerging Technologies for Molecular Diagnosis of Sepsis. Clin. Microbiol. Rev. 2018, 31, e00089-17. [Google Scholar] [CrossRef]
- Van Engelen, T.S.R.; Wiersinga, W.J.; Scicluna, B.P.; van der Poll, T. Biomarkers in Sepsis. Crit. Care Clin. 2018, 34, 139–152. [Google Scholar] [CrossRef]
- Miller, R.R., 3rd; Lopansri, B.K.; Burke, J.P.; Levy, M.; Opal, S.; Rothman, R.E.; D’Alessio, F.R.; Sidhaye, V.K.; Aggarwal, N.R.; Balk, R.; et al. Validation of a Host Response Assay, SeptiCyte LAB, for Discriminating Sepsis from Systemic Inflammatory Response Syndrome in the ICU. Am. J. Respir. Crit. Care Med. 2018, 198, 903–913. [Google Scholar] [CrossRef]
- Scicluna, B.P.; Klein Klouwenberg, P.M.; van Vught, L.A.; Wiewel, M.A.; Ong, D.S.; Zwinderman, A.H.; Franitza, M.; Toliat, M.R.; Nurnberg, P.; Hoogendijk, A.J.; et al. A molecular biomarker to diagnose community-acquired pneumonia on intensive care unit admission. Am. J. Respir. Crit. Care Med. 2015, 192, 826–835. [Google Scholar] [CrossRef]
- Sweeney, T.E.; Shidham, A.; Wong, H.R.; Khatri, P. A comprehensive time-course-based multicohort analysis of sepsis and sterile inflammation reveals a robust diagnostic gene set. Sci. Transl. Med. 2015, 7, 287ra271. [Google Scholar] [CrossRef]
- O’Connell, G.C.; Treadway, M.B.; Tennant, C.S.; Lucke-Wold, N.; Chantler, P.D.; Barr, T.L. Shifts in Leukocyte Counts Drive the Differential Expression of Transcriptional Stroke Biomarkers in Whole Blood. Transl. Stroke Res. 2019, 10, 26–35. [Google Scholar]
- O’Connell, G.C.; Treadway, M.B.; Petrone, A.B.; Tennant, C.S.; Lucke-Wold, N.; Chantler, P.D.; Barr, T.L. Leukocyte Dynamics Influence Reference Gene Stability in Whole Blood: Data-Driven qRT-PCR Normalization Is a Robust Alternative for Measurement of Transcriptional Biomarkers. Lab. Med. 2017, 48, 346–356. [Google Scholar] [CrossRef]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef]
- Cabrera, C.P.; Manson, J.; Shepherd, J.M.; Torrance, H.D.; Watson, D.; Longhi, M.P.; Hoti, M.; Patel, M.B.; O’Dwyer, M.; Nourshargh, S.; et al. Signatures of inflammation and impending multiple organ dysfunction in the hyperacute phase of trauma: A prospective cohort study. PLoS Med. 2017, 14, e1002352. [Google Scholar] [CrossRef]
- Tak, T.; van Groenendael, R.; Pickkers, P.; Koenderman, L. Monocyte Subsets Are Differentially Lost from the Circulation during Acute Inflammation Induced by Human Experimental Endotoxemia. J. Innate Immun. 2017, 9, 464–474. [Google Scholar] [CrossRef]
- Shen, X.F.; Cao, K.; Jiang, J.P.; Guan, W.X.; Du, J.F. Neutrophil dysregulation during sepsis: An overview and update. J. Cell Mol. Med. 2017, 21, 1687–1697. [Google Scholar] [CrossRef]
- Zonneveld, R.; Molema, G.; Plotz, F.B. Analyzing Neutrophil Morphology, Mechanics, and Motility in Sepsis: Options and Challenges for Novel Bedside Technologies. Crit. Care Med. 2016, 44, 218–228. [Google Scholar] [CrossRef]
- Guo, Y.; Patil, N.K.; Luan, L.; Bohannon, J.K.; Sherwood, E.R. The biology of natural killer cells during sepsis. Immunology 2018, 153, 190–202. [Google Scholar] [CrossRef]
- Sonego, F.; Castanheira, F.V.; Ferreira, R.G.; Kanashiro, A.; Leite, C.A.; Nascimento, D.C.; Colon, D.F.; Borges Vde, F.; Alves-Filho, J.C.; Cunha, F.Q. Paradoxical Roles of the Neutrophil in Sepsis: Protective and Deleterious. Front Immunol. 2016, 7, 155. [Google Scholar] [CrossRef]
- Souza-Fonseca-Guimaraes, F.; Adib-Conquy, M.; Cavaillon, J.M. Natural killer (NK) cells in antibacterial innate immunity: Angels or devils? Mol. Med. 2012, 18, 270–285. [Google Scholar] [CrossRef]
- McHugh, L.; Seldon, T.A.; Brandon, R.A.; Kirk, J.T.; Rapisarda, A.; Sutherland, A.J.; Presneill, J.J.; Venter, D.J.; Lipman, J.; Thomas, M.R.; et al. A Molecular Host Response Assay to Discriminate Between Sepsis and Infection-Negative Systemic Inflammation in Critically Ill Patients: Discovery and Validation in Independent Cohorts. PLoS Med. 2015, 12, e1001916. [Google Scholar] [CrossRef]
- Sanders, R.; Mason, D.J.; Foy, C.A.; Huggett, J.F. Considerations for accurate gene expression measurement by reverse transcription quantitative PCR when analysing clinical samples. Anal. Bioanal. Chem. 2014, 406, 6471–6483. [Google Scholar] [CrossRef]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6, 279–284. [Google Scholar] [CrossRef]
- Mane, V.P.; Heuer, M.A.; Hillyer, P.; Navarro, M.B.; Rabin, R.L. Systematic method for determining an ideal housekeeping gene for real-time PCR analysis. J. Biomol. Tech. 2008, 19, 342–347. [Google Scholar]
- Zhang, Y.; Li, D.; Sun, B. Do Housekeeping Genes Exist? PLoS ONE 2015, 10, e0123691. [Google Scholar] [CrossRef]
- Kozera, B.; Rapacz, M. Reference genes in real-time PCR. J. Appl. Genet. 2013, 54, 391–406. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef]
- Ecker, S.; Chen, L.; Pancaldi, V.; Bagger, F.O.; Fernandez, J.M.; Carrillo de Santa Pau, E.; Juan, D.; Mann, A.L.; Watt, S.; Casale, F.P.; et al. Genome-wide analysis of differential transcriptional and epigenetic variability across human immune cell types. Genome Biol. 2017, 18, 18. [Google Scholar] [CrossRef]
- Sturm, T.; Leiblein, J.; Schneider-Lindner, V.; Kirschning, T.; Thiel, M. Association of Microcirculation, Macrocirculation, and Severity of Illness in Septic Shock: A Prospective Observational Study to Identify Microcirculatory Targets Potentially Suitable for Guidance of Hemodynamic Therapy. J. Intensive Care Med. 2018, 33, 256–266. [Google Scholar] [CrossRef]
- Tang, B.M.P.; McLean, A.S.; Dawes, I.W.; Huang, S.J.; Lin, R.C.Y. The Use of Gene-Expression Profiling to Identify Candidate Genes in Human Sepsis. Am J Respir Crit Care Med 2007, 176, 676–684. [Google Scholar] [CrossRef]
- Tang, B.M.; McLean, A.S.; Dawes, I.W.; Huang, S.J.; Cowley, M.J.; Lin, R.C. Gene-expression profiling of gram-positive and gram-negative sepsis in critically ill patients. Crit. Care Med. 2008, 36, 1125–1128. [Google Scholar] [CrossRef]
- Silva, E.; Arcaroli, J.; He, Q.; Svetkauskaite, D.; Coldren, C.; Nick, J.A.; Poch, K.; Park, J.S.; Banerjee, A.; Abraham, E. HMGB1 and LPS induce distinct patterns of gene expression and activation in neutrophils from patients with sepsis-induced acute lung injury. Intensive Care Med. 2007, 33, 1829–1839. [Google Scholar] [CrossRef]
- Coldren, C.D.; Nick, J.A.; Poch, K.R.; Woolum, M.D.; Fouty, B.W.; O’Brien, J.M.; Gruber, M.P.; Zamora, M.R.; Svetkauskaite, D.; Richter, D.A.; et al. Functional and genomic changes induced by alveolar transmigration in human neutrophils. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L1267–L1276. [Google Scholar] [CrossRef]
- Maslove, D.M.; Wong, H.R. Gene expression profiling in sepsis: Timing, tissue, and translational considerations. Trends Mol. Med. 2014, 20, 204–213. [Google Scholar] [CrossRef]
- Maslove, D.M.; Shapira, T.; Tyryshkin, K.; Veldhoen, R.A.; Marshall, J.C.; Muscedere, J. Validation of diagnostic gene sets to identify critically ill patients with sepsis. J. Crit. Care 2019, 49, 92–98. [Google Scholar] [CrossRef]
- Pena, O.M.; Hancock, D.G.; Lyle, N.H.; Linder, A.; Russell, J.A.; Xia, J.; Fjell, C.D.; Boyd, J.H.; Hancock, R.E. An Endotoxin Tolerance Signature Predicts Sepsis and Organ Dysfunction at Initial Clinical Presentation. EBioMedicine 2014, 1, 64–71. [Google Scholar] [CrossRef]
- Kangelaris, K.N.; Prakash, A.; Liu, K.D.; Aouizerat, B.; Woodruff, P.G.; Erle, D.J.; Rogers, A.; Seeley, E.J.; Chu, J.; Liu, T.; et al. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1102–L1113. [Google Scholar] [CrossRef]
- Bauer, M.; Giamarellos-Bourboulis, E.J.; Kortgen, A.; Moller, E.; Felsmann, K.; Cavaillon, J.M.; Guntinas-Lichius, O.; Rutschmann, O.; Ruryk, A.; Kohl, M.; et al. A Transcriptomic Biomarker to Quantify Systemic Inflammation in Sepsis - A Prospective Multicenter Phase II Diagnostic Study. EBioMedicine 2016, 6, 114–125. [Google Scholar] [CrossRef][Green Version]
- Davenport, E.E.; Burnham, K.L.; Radhakrishnan, J.; Humburg, P.; Hutton, P.; Mills, T.C.; Rautanen, A.; Gordon, A.C.; Garrard, C.; Hill, A.V.S.; et al. Genomic landscape of the individual host response and outcomes in sepsis: A prospective cohort study. Lancet Respir. Med. 2016, 4, 259–271. [Google Scholar] [CrossRef]
- Schaack, D.; Siegler, B.H.; Tamulyte, S.; Weigand, M.A.; Uhle, F. The immunosuppressive face of sepsis early on intensive care unit—A large-scale microarray meta-analysis. PLoS ONE 2018, 13, e0198555. [Google Scholar] [CrossRef]
- Sweeney, T.E.; Azad, T.D.; Donato, M.; Haynes, W.A.; Perumal, T.M.; Henao, R.; Bermejo-Martin, J.F.; Almansa, R.; Tamayo, E.; Howrylak, J.A.; et al. Unsupervised Analysis of Transcriptomics in Bacterial Sepsis Across Multiple Datasets Reveals Three Robust Clusters. Crit. Care Med. 2018, 46, 915–925. [Google Scholar] [CrossRef]
- Burnham, K.L.; Davenport, E.E.; Radhakrishnan, J.; Humburg, P.; Gordon, A.C.; Hutton, P.; Svoren-Jabalera, E.; Garrard, C.; Hill, A.V.S.; Hinds, C.J.; et al. Shared and Distinct Aspects of the Sepsis Transcriptomic Response to Fecal Peritonitis and Pneumonia. Am. J. Respir. Crit. Care Med. 2017, 196, 328–339. [Google Scholar] [CrossRef]
- Sweeney, T.E.; Perumal, T.M.; Henao, R.; Nichols, M.; Howrylak, J.A.; Choi, A.M.; Bermejo-Martin, J.F.; Almansa, R.; Tamayo, E.; Davenport, E.E.; et al. A community approach to mortality prediction in sepsis via gene expression analysis. Nat. Commun. 2018, 9, 694. [Google Scholar] [CrossRef]
- Tsalik, E.L.; Langley, R.J.; Dinwiddie, D.L.; Miller, N.A.; Yoo, B.; van Velkinburgh, J.C.; Smith, L.D.; Thiffault, I.; Jaehne, A.K.; Valente, A.M.; et al. An integrated transcriptome and expressed variant analysis of sepsis survival and death. Genome Med. 2014, 6, 111. [Google Scholar] [CrossRef]
- Demaret, J.; Venet, F.; Plassais, J.; Cazalis, M.-A.; Vallin, H.; Friggeri, A.; Lepape, A.; Rimmelé, T.; Textoris, J.; Monneret, G. Identification of CD177 as the most dysregulated parameter in a microarray study of purified neutrophils from septic shock patients. Immunol. Lett. 2016, 178, 122–130. [Google Scholar] [CrossRef]
- Vieira da Silva Pellegrina, D.; Severino, P.; Vieira Barbeiro, H.; Maziero Andreghetto, F.; Tadeu Velasco, I.; Possolo de Souza, H.; Machado, M.C.C.; Reis, E.M.; Pinheiro da Silva, F. Septic Shock in Advanced Age: Transcriptome Analysis Reveals Altered Molecular Signatures in Neutrophil Granulocytes. PLoS ONE 2015, 10, e0128341. [Google Scholar] [CrossRef]
- De Kleijn, S.; Kox, M.; Sama, I.E.; Pillay, J.; van Diepen, A.; Huijnen, M.A.; van der Hoeven, J.G.; Ferwerda, G.; Hermans, P.W.M.; Pickkers, P. Transcriptome Kinetics of Circulating Neutrophils during Human Experimental Endotoxemia. PLoS ONE 2012, 7, e38255. [Google Scholar] [CrossRef]
- Cummings, M.; Sarveswaran, J.; Homer-Vanniasinkam, S.; Burke, D.; Orsi, N.M. Glyceraldehyde-3-phosphate dehydrogenase is an inappropriate housekeeping gene for normalising gene expression in sepsis. Inflammation 2014, 37, 1889–1894. [Google Scholar] [CrossRef]
- Piehler, A.P.; Grimholt, R.M.; Ovstebø, R.; Berg, J.P. Gene expression results in lipopolysaccharide-stimulated monocytes depend significantly on the choice of reference genes. BMC Immunol. 2010, 11, 21. [Google Scholar] [CrossRef]
- Chen, X.; Huang, Z.; Wang, H.; Jia, G.; Liu, G.; Guo, X.; Tang, R.; Long, D. Role of akirin in skeletal myogenesis. Int. J. Mol. Sci. 2013, 14, 3817–3823. [Google Scholar] [CrossRef]
- Macqueen, D.J.; Johnston, I.A. Evolution of the multifaceted eukaryotic akirin gene family. BMC Evol. Biol. 2009, 9, 34. [Google Scholar] [CrossRef]
- Goto, A.; Matsushita, K.; Gesellchen, V.; El Chamy, L.; Kuttenkeuler, D.; Takeuchi, O.; Hoffmann, J.A.; Akira, S.; Boutros, M.; Reichhart, J.M. Akirins are highly conserved nuclear proteins required for NF-kappaB-dependent gene expression in drosophila and mice. Nat. Immunol. 2008, 9, 97–104. [Google Scholar] [CrossRef]
- Polanowska, J.; Chen, J.-X.; Soulé, J.; Omi, S.; Belougne, J.; Taffoni, C.; Pujol, N.; Selbach, M.; Zugasti, O.; Ewbank, J.J. Evolutionary plasticity in the innate immune function of Akirin. PLoS Genet. 2018, 14, e1007494. [Google Scholar] [CrossRef]
- Tartey, S.; Takeuchi, O. Chromatin Remodeling and Transcriptional Control in Innate Immunity: Emergence of Akirin2 as a Novel Player. Biomolecules 2015, 5, 1618–1633. [Google Scholar] [CrossRef]
- Vavouri, T.; Semple, J.I.; Lehner, B. Widespread conservation of genetic redundancy during a billion years of eukaryotic evolution. Trends Genet. 2008, 24, 485–488. [Google Scholar] [CrossRef]
- Mar, J.C.; Matigian, N.A.; Mackay-Sim, A.; Mellick, G.D.; Sue, C.M.; Silburn, P.A.; McGrath, J.J.; Quackenbush, J.; Wells, C.A. Variance of gene expression identifies altered network constraints in neurological disease. PLoS Genet. 2011, 7, e1002207. [Google Scholar] [CrossRef]
- Mason, E.A.; Mar, J.C.; Laslett, A.L.; Pera, M.F.; Quackenbush, J.; Wolvetang, E.; Wells, C.A. Gene expression variability as a unifying element of the pluripotency network. Stem Cell Rep. 2014, 3, 365–377. [Google Scholar] [CrossRef]
- Alemu, E.Y.; Carl, J.W., Jr.; Corrada Bravo, H.; Hannenhalli, S. Determinants of expression variability. Nucleic Acids Res. 2014, 42, 3503–3514. [Google Scholar] [CrossRef]
- Vincent, J.L.; Moreno, R.; Takala, J.; Willatts, S.; De Mendonca, A.; Bruining, H.; Reinhart, C.K.; Suter, P.M.; Thijs, L.G. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996, 22, 707–710. [Google Scholar] [CrossRef]
- Figueiredo, A.S.; Killian, D.; Schulte, J.; Sticht, C.; Lindner, H.A. Whole transcriptome data of primary human NK cells under hypoxia and interleukin 15 priming: A 2x2 factorial design experiment. Data Brief 2017, 14, 77–83. [Google Scholar] [CrossRef]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef]
- Kauffmann, A.; Gentleman, R.; Huber, W. arrayQualityMetrics—A bioconductor package for quality assessment of microarray data. Bioinformatics 2009, 25, 415–416. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- de Torrente, L.; Zimmerman, S.; Taylor, D.; Hasegawa, Y.; Wells, C.A.; Mar, J.C. pathVar: A new method for pathway-based interpretation of gene expression variability. PeerJ 2017, 5, e3334. [Google Scholar] [CrossRef][Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]







| NK cells | |||
| Patients contributing eligible NK cell preparations/enrolled patients | Presurgical (n = 19/20) | SIRS (n = 16/19) | Sepsis (n = 10/20) |
| Patient characteristics | |||
| Age (years) | 63.6 ± 12.3 | 61.8 ± 17.4 | 61.8 ± 17.6 |
| Sex (Male/Female) | 10/9 | 12/4 | 5/5 |
| SOFA score | na 2 | 5.3 ± 2.3 | 13.1 ± 3.7 **** |
| CRP (mg/L) | na 3 | 80 ± 49 4 | 213 ± 105 *** |
| Lactate (mmol/L) | na 2 | 1.8 ± 1.3 5 | 4.1 ± 2.2 6 ** |
| WBC (cells/nL) | 7.0 ± 1.9 7 | 12.5 ± 4.8 *** | 14.3 ± 9.1 * |
| SIRS etiology | |||
| Abdominal surgery | N.A.8 | n = 9 | N.A. |
| Thoracic surgery | N.A. | n = 3 | N.A. |
| Polytrauma | N.A. | n = 3 | N.A. |
| Lumbar spinal stabilization | N.A. | n = 1 | N.A. |
| Septic focus | |||
| Abdominal | N.A. | N.A. | n = 5 |
| Pulmonal | N.A. | N.A. | n = 5 |
| Granulocytes | |||
| Patients contributing eligible granulocyte preparations/enrolled patients | Presurgical (n = 11/20) | SIRS (n = 16/19) | Sepsis (n = 15/20) |
| Patient characteristics | |||
| Age (years) | 60.6 ± 12.9 | 63.1 ± 17.6 | 62.5 ± 17.6 |
| Sex (Male/Female) | 5/6 | 12/4 | 7/8 |
| SOFA score | na 2 | 5.1 ± 2.4 | 12.7 ± 3.2 **** |
| CRP (mg/L) | na 3 | 75 ± 48 9 | 226 ± 101 **** |
| Lactate (mmol/L) | na 2 | 2.0 ± 1.9 10 | 3.5 ± 2.1 11 ** |
| WBC (cells/nL) | 7.6 ± 2.4 7 | 11.8 ± 4.6 | 16.6 ± 11.6 * |
| SIRS etiology | |||
| Abdominal surgery | N.A. | n = 10 | N.A. |
| Thoracic surgery | N.A. | n = 1 | N.A. |
| Polytrauma | N.A. | n = 4 | N.A. |
| Lumbar spinal stabilization | N.A. | n = 1 | N.A. |
| Septic focus | |||
| Abdominal | N.A. | N.A. | n = 11 |
| Pulmonal | N.A. | N.A. | n = 4 |
| Patients contributing both NK cells and granulocytes/enrolled patients | n = 10/20 | n = 14/19 | n = 9/20 |
| NK cells | ||
| Patients contributing eligible granulocyte preparations/enrolled patients | SIRS (n = 18/22) | Sepsis (n = 15/23) |
| Patient characteristics | ||
| Age (years) | 68.7 ± 15.0 | 71.5 ± 11.8 |
| Sex (Male/Female) | 13/5 | 7/8 |
| SOFA score | 6.3 ± 2.7 | 10.4 ± 2.0 **** |
| CRP (mg/L) | 59 ± 38 2 | 323 ± 111 **** |
| Lactate (mmol/L) | 1.6 ± 1.3 3 | 2.3 ± 1.7 4 |
| WBC (cells/nL) | 16.7 ± 5.9 | 18.6 ± 4.9 |
| SIRS etiology | ||
| Abdominal surgery | n = 7 | N.A. |
| Thoracic surgery | n = 4 | N.A. |
| Vascular surgery | n = 2 | N.A. |
| Polytrauma | n = 4 | N.A. |
| Otolaryngology surgery | n = 1 | N.A. |
| Septic focus | ||
| Abdominal | N.A. | n = 7 |
| Pulmonal | N.A. | n = 5 |
| Soft tissue | N.A. | n = 2 |
| Urogenital | N.A. | n = 1 |
| Granulocytes | ||
| SIRS (n = 22/22) | Sepsis (n = 18/23) | |
| Patient characteristics | ||
| Age (years) | 66.3 ± 14.9 | 69.4 ± 13.3 |
| Sex (Male/Female) | 16/17 | 9/9 |
| SOFA score | 6.5 ± 2.9 | 10.8 ± 2.7 **** |
| CRP (mg/L) | 59 ± 47 2 | 313 ± 123 **** |
| Lactate (mmol/L) | 1.9 ± 2.0 5 | 2.9 ± 2.4 6 * |
| WBC (cells/nL) | 16.7 ± 6.4 | 15.3 ± 5.9 |
| SIRS etiology | ||
| Abdominal surgery | n = 8 | N.A. |
| Thoracic surgery | n = 4 | N.A. |
| Vascular surgery | n = 3 | N.A. |
| Polytrauma | n = 6 | N.A. |
| Otolaryngology surgery | n = 1 | N.A. |
| Septic focus | ||
| Abdominal | N.A. | n = 8 |
| Pulmonal | N.A. | n = 8 |
| Soft tissue | N.A. | n = 1 |
| Urogenital | N.A. | n = 1 |
| Patients contributing both NK cells and granulocytes/enrolled patients | n = 18/22 | n = 15/23 |
| GEO Profile ID 1 | Comparison Groups (n) | Mean Fold Differences 3 | Publication | ||||
|---|---|---|---|---|---|---|---|
| p-Values 2 | |||||||
| 48169967 | Group number | 1 | 2 | 3 | 4 | Tang et al., 2008 [32] | |
| 1 Controls (17) | 1.04 | 0.93 | 0.79 | ||||
| 2 Mixed infection sepsis (12) | 0.76 | 0.89 | 0.76 | ||||
| 3 Gram-negative sepsis (25) | 0.54 | 0.35 | 0.85 | ||||
| 4 Gram-positive sepsis (18) | 0.12 | 0.08 | 0.23 | ||||
| 41143967 | Group number | 1 | 2 | 3 | 4 | Tang et al., 2007 [31] | |
| 1 Controls training set (13) | 0.80 | 0.82 | 0.81 | ||||
| 2 Controls validation set (8) | 0.15 | 1.02 | 1.02 | ||||
| 3 Sepsis training set (26) | 0.09 | 0.86 | 1.00 | ||||
| 4 Sepsis validation set (38) | 0.07 | 0.89 | 0.96 | ||||
| 28190858 | Group number | 1 | 2 | 3 | Silva et al., 2007 [33] | ||
| 1 In vitro controls (8) | 0.91 | 0.99 | |||||
| 2 In vitro LPS treated (8) | 0.39 | 1.09 | |||||
| 3 In vitro HMGB1 treated (8) | 0.94 | 0.51 | |||||
| 29128858 | Group number | 1 | 2 | 3 | 4 | 5 | Coldren et al., 2006 [34] |
| 1 In vitro controls (5) | 0.83 | 0.40 | 0.38 | 0.36 | |||
| 2 In vitro LPS (5) | 0.33 | 0.49 | 0.46 | 0.44 | |||
| 3 Circulating pre-LPS (12) | <10−3 | 0.04 | 0.94 | 0.90 | |||
| 4 Circulating post-LPS (14) | <10−3 | 0.03 | 0.52 | 0.96 | |||
| 5 Alveolar post-LPS (15) | <10−3 | 0.03 | 0.49 | 0.77 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coulibaly, A.; Velásquez, S.Y.; Sticht, C.; Figueiredo, A.S.; Himmelhan, B.S.; Schulte, J.; Sturm, T.; Centner, F.-S.; Schöttler, J.J.; Thiel, M.; et al. AKIRIN1: A Potential New Reference Gene in Human Natural Killer Cells and Granulocytes in Sepsis. Int. J. Mol. Sci. 2019, 20, 2290. https://doi.org/10.3390/ijms20092290
Coulibaly A, Velásquez SY, Sticht C, Figueiredo AS, Himmelhan BS, Schulte J, Sturm T, Centner F-S, Schöttler JJ, Thiel M, et al. AKIRIN1: A Potential New Reference Gene in Human Natural Killer Cells and Granulocytes in Sepsis. International Journal of Molecular Sciences. 2019; 20(9):2290. https://doi.org/10.3390/ijms20092290
Chicago/Turabian StyleCoulibaly, Anna, Sonia Y. Velásquez, Carsten Sticht, Ana Sofia Figueiredo, Bianca S. Himmelhan, Jutta Schulte, Timo Sturm, Franz-Simon Centner, Jochen J. Schöttler, Manfred Thiel, and et al. 2019. "AKIRIN1: A Potential New Reference Gene in Human Natural Killer Cells and Granulocytes in Sepsis" International Journal of Molecular Sciences 20, no. 9: 2290. https://doi.org/10.3390/ijms20092290
APA StyleCoulibaly, A., Velásquez, S. Y., Sticht, C., Figueiredo, A. S., Himmelhan, B. S., Schulte, J., Sturm, T., Centner, F.-S., Schöttler, J. J., Thiel, M., & Lindner, H. A. (2019). AKIRIN1: A Potential New Reference Gene in Human Natural Killer Cells and Granulocytes in Sepsis. International Journal of Molecular Sciences, 20(9), 2290. https://doi.org/10.3390/ijms20092290