The Serotonin Syndrome: From Molecular Mechanisms to Clinical Practice


Abstract
1. Introduction
2. Clinical Context
2.1. Definition and Epidemiology
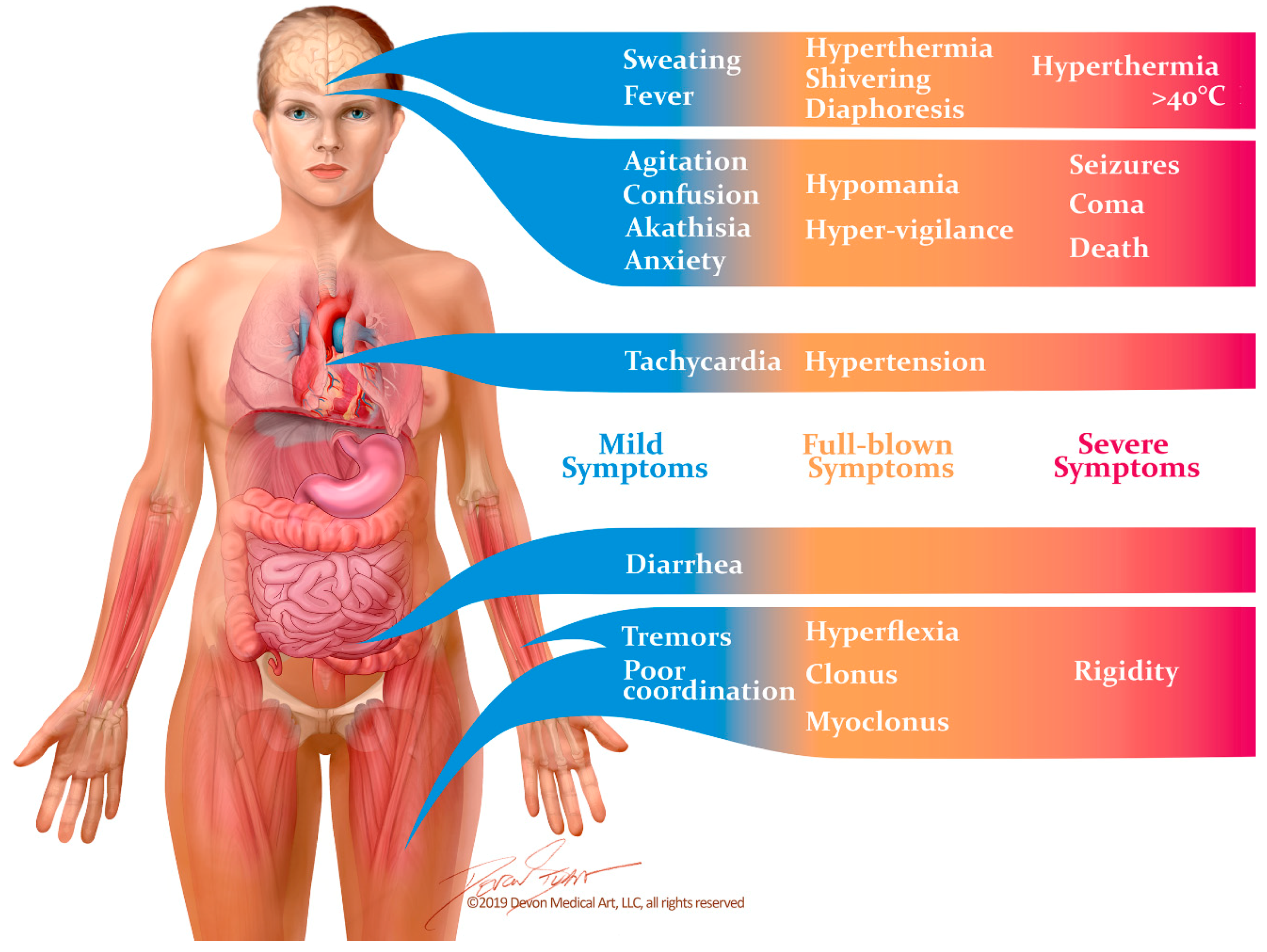
2.2. Manifestations and Diagnosis
2.3. Differential Diagnosis
3. The Molecular Basis for the Serotonin Syndrome
3.1. Animal Models
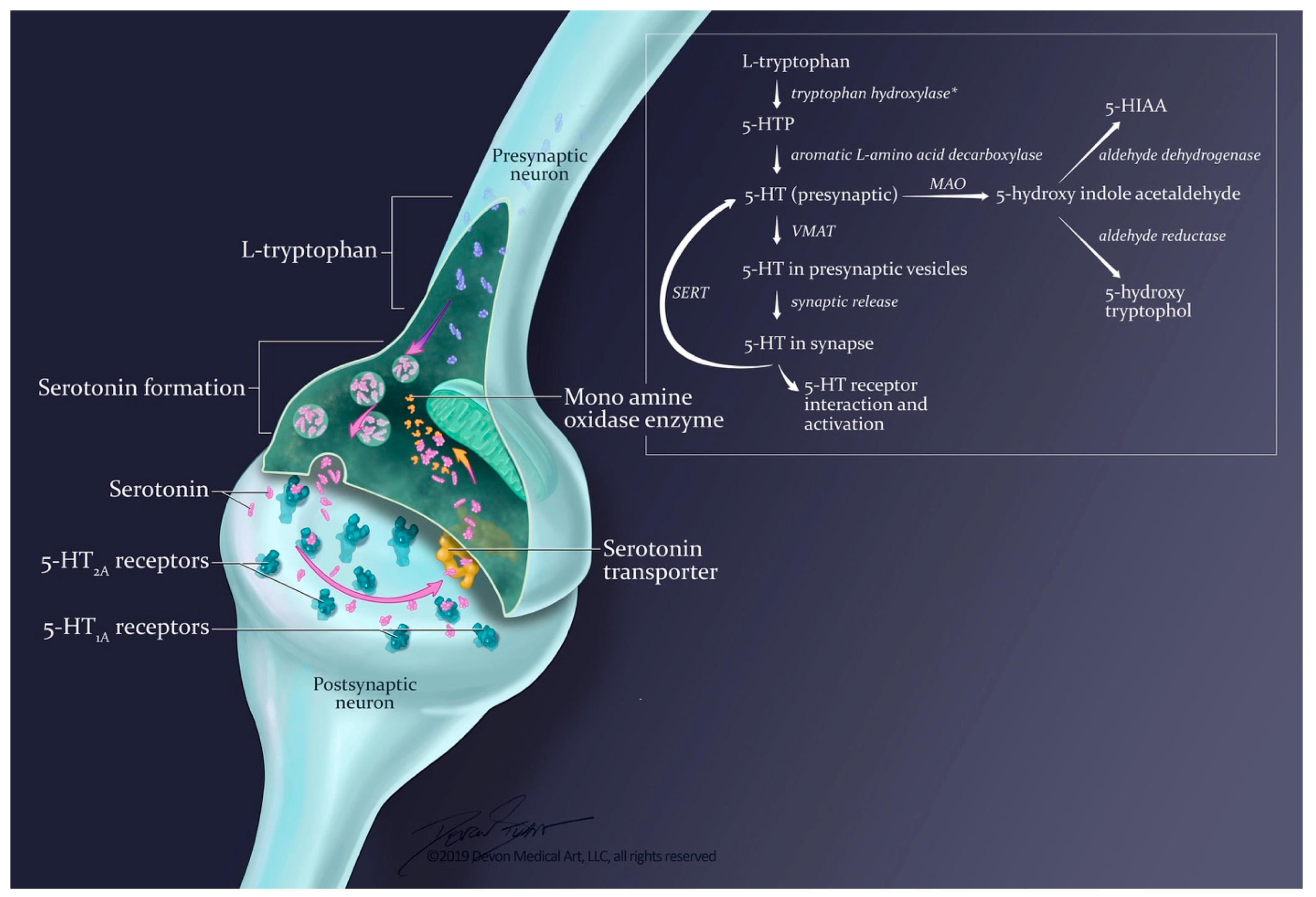
3.2. Molecular Pathways
3.2.1. Synthesis and Release
3.2.2. Termination of Effects: Reuptake and Metabolism
3.2.3. Serotonin Receptors Subtypes
3.3. Medications Triggering the Serotonin Syndrome
3.4. Genetic Polymorphisms
4. Receptor-Targeted Therapy for Serotonin Syndrome
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT | 5-Hydroxytryptamine |
| CYP | Cytochromes p450 |
| MAO-A | Monoamine Oxidase Inhibitor subtype A |
| MAO-B | Monoamine Oxidase Inhibitor subtype B |
| MAOI | Monoamine Oxidase Inhibitor |
| NMDA | N-methyl-D-aspartate |
| SS | Serotonin Syndrome |
| SSRI SNRI | Serotonin Norepinephrine Reuptake Inhibitor |
| TCA | Tricyclic Antidepressant |
References
- Mason, P.J.; Morris, V.A.; Balcezak, T.J. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine 2000, 79, 201–209. [Google Scholar] [CrossRef]
- Mojtabai, R.; Olfson, M. National trends in long-term use of antidepressant medications: Results from the U.S. National health and nutrition examination survey. J. Clin. Psychiatry 2014, 75, 169–177. [Google Scholar] [PubMed]
- Uddin, M.F.; Alweis, R.; Shah, S.R.; Lateef, N.; Shahnawaz, W.; Ochani, R.K.; Dharani, A.M.; Shah, S.A. Controversies in serotonin syndrome diagnosis and management: A review. J. Clin. Diagn. Res. 2017, 11, OE05–OE07. [Google Scholar] [CrossRef]
- Watson, W.A.; Litovitz, T.L.; Klein-Schwartz, W.; Rodgers, G.C., Jr.; Youniss, J.; Reid, N.; Rouse, W.G.; Rembert, R.S.; Borys, D. 2003 annual report of the american association of poison control centers toxic exposure surveillance system. Am. J. Emerg. Med. 2004, 22, 335–404. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.A.; Litovitz, T.L.; Rodgers, G.C., Jr.; Klein-Schwartz, W.; Reid, N.; Youniss, J.; Flanagan, A.; Wruk, K.M. 2004 annual report of the american association of poison control centers toxic exposure surveillance system. Am. J. Emerg. Med. 2005, 23, 589–666. [Google Scholar] [CrossRef]
- Watson, W.A.; Litovitz, T.L.; Rodgers, G.C., Jr.; Klein-Schwartz, W.; Youniss, J.; Rose, S.R.; Borys, D.; May, M.E. 2002 annual report of the american association of poison control centers toxic exposure surveillance system. Am. J. Emerg. Med. 2003, 21, 353–421. [Google Scholar] [CrossRef]
- Lai, M.W.; Klein-Schwartz, W.; Rodgers, G.C.; Abrams, J.Y.; Haber, D.A.; Bronstein, A.C.; Wruk, K.M. 2005 annual report of the american association of poison control centers’ national poisoning and exposure database. Clin. Toxicol. 2006, 44, 803–932. [Google Scholar] [CrossRef]
- Sternbach, H. The serotonin syndrome. Am. J. Psychiatry 1991, 148, 705–713. [Google Scholar] [PubMed]
- Martin, T.G. Serotonin syndrome. Ann. Emerg. Med. 1996, 28, 520–526. [Google Scholar] [CrossRef]
- Boyer, E.W.; Shannon, M. The serotonin syndrome. New Engl. J. Med. 2005, 352, 1112–1120. [Google Scholar] [CrossRef]
- Dunkley, E.J.; Isbister, G.K.; Sibbritt, D.; Dawson, A.H.; Whyte, I.M. The hunter serotonin toxicity criteria: Simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003, 96, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Xie, L.; Alley, S.; McCarron, R.M.; Baser, O.; Wang, Z. Epidemiology and economic burden of serotonin syndrome with concomitant use of serotonergic agents: A retrospective study utilizing two large us claims databases. Prim. Care Companion CNS Disord. 2019, 19. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.J.; Dunn, N.R.; Mann, R.D. Antidepressants and the serotonin syndrome in general practice. Br. J. Gen. Pr. 1999, 49, 871–874. [Google Scholar]
- Radomski, J.W.; Dursun, S.M.; Reveley, M.A.; Kutcher, S.P. An exploratory approach to the serotonin syndrome: An update of clinical phenomenology and revised diagnostic criteria. Med. Hypotheses 2000, 55, 218–224. [Google Scholar] [CrossRef]
- Isbister, G.K.; Buckley, N.A.; Whyte, I.M. Serotonin toxicity: A practical approach to diagnosis and treatment. Med. J. Aust. 2007, 187, 361–365. [Google Scholar]
- Sellers, E.M. Alcohol, barbiturate and benzodiazepine withdrawal syndromes: Clinical management. CMAJ 1988, 139, 113–120. [Google Scholar] [PubMed]
- Wilson, E.; Lader, M. A review of the management of antidepressant discontinuation symptoms. Adv. Psychopharmacol. 2015, 5, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Warner, C.H.; Bobo, W.; Warner, C.; Reid, S.; Rachal, J. Antidepressant discontinuation syndrome. Am. Fam. Physician. 2006, 74, 449–456. [Google Scholar]
- Haddad, P. Newer antidepressants and the discontinuation syndrome. J. Clin. Psychiatry 1997, 58, 17–21. [Google Scholar] [PubMed]
- Buckley, N.A.; Dawson, A.H.; Isbister, G.K. Serotonin syndrome. BMJ 2014, 348, g1626. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.C. Serotonin syndrome. A clinical update. Crit. Care Clin. 1997, 13, 763–783. [Google Scholar] [CrossRef]
- Isbister, G.K.; Buckley, N.A. The pathophysiology of serotonin toxicity in animals and humans: Implications for diagnosis and treatment. Clin. Neuropharmacol. 2005, 28, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Nisijima, K.; Yoshino, T.; Ishiguro, T. Risperidone counteracts lethality in an animal model of the serotonin syndrome. Psychopharmacology 2000, 150, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Haberzettl, R.; Bert, B.; Fink, H.; Fox, M.A. Animal models of the serotonin syndrome: A systematic review. Behav. Brain Res. 2013, 256, 328–345. [Google Scholar] [CrossRef]
- Kalueff, A.V.; Fox, M.A.; Gallagher, P.S.; Murphy, D.L. Hypolocomotion, anxiety and serotonin syndrome-like behavior contribute to the complex phenotype of serotonin transporter knockout mice. Genes. Brain Behav. 2007, 6, 389–400. [Google Scholar] [CrossRef]
- Ener, R.A.; Meglathery, S.B.; Van Decker, W.A.; Gallagher, R.M. Serotonin syndrome and other serotonergic disorders. Pain Med. 2003, 4, 63–74. [Google Scholar] [CrossRef]
- Darmani, N.A.; Ahmad, B. Long-term sequential determination of behavioral ontogeny of 5-ht1a and 5-ht2 receptor functions in the rat. J. Pharm. Exp. 1999, 288, 247–253. [Google Scholar]
- Goodwin, G.M.; Green, A.R. A behavioural and biochemical study in mice and rats of putative selective agonists and antagonists for 5-ht1 and 5-ht2 receptors. Br. J. Pharm. 1985, 84, 743–753. [Google Scholar] [CrossRef]
- Forster, E.A.; Cliffe, I.A.; Bill, D.J.; Dover, G.M.; Jones, D.; Reilly, Y.; Fletcher, A. A pharmacological profile of the selective silent 5-ht1a receptor antagonist, way-100635. Eur. J. Pharm. 1995, 281, 81–88. [Google Scholar] [CrossRef]
- Smith, L.M.; Peroutka, S.J. Differential effects of 5-hydroxytryptamine1a selective drugs on the 5-ht behavioral syndrome. Pharm. Biochem. Behav. 1986, 24, 1513–1519. [Google Scholar] [CrossRef]
- Tricklebank, M.D.; Forler, C.; Fozard, J.R. The involvement of subtypes of the 5-ht1 receptor and of catecholaminergic systems in the behavioural response to 8-hydroxy-2-(di-n-propylamino)tetralin in the rat. Eur J. Pharm. 1984, 106, 271–282. [Google Scholar] [CrossRef]
- Mazzola-Pomietto, P.; Aulakh, C.; Wozniak, K.; Hill, J.; Murphy, D. Evidence that 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (doi)-induced hyperthermia in rats is mediated by stimulation of 5-ht2a receptors | springerlink. Psychopharmacology 2019, 117, 193–199. [Google Scholar] [CrossRef]
- Nisijima, K.; Shioda, K.; Yoshino, T.; Takano, K.; Kato, S. Diazepam and chlormethiazole attenuate the development of hyperthermia in an animal model of the serotonin syndrome. Neurochem. Int. 2003, 43, 155–164. [Google Scholar] [CrossRef]
- Gudelsky, G.A.; Koenig, J.I.; Meltzer, H.Y. Thermoregulatory responses to serotonin (5-ht) receptor stimulation in the rat. Evidence for opposing roles of 5-ht2 and 5-ht1a receptors. Neuropharmacology 1986, 25, 1307–1313. [Google Scholar]
- Nisijima, K.; Yoshino, T.; Yui, K.; Katoh, S. Potent serotonin (5-ht)(2a) receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-ht syndrome. Brain Res. 2001, 890, 23–31. [Google Scholar] [CrossRef]
- Shioda, K.; Nisijima, K.; Yoshino, T.; Kato, S. Extracellular serotonin, dopamine and glutamate levels are elevated in the hypothalamus in a serotonin syndrome animal model induced by tranylcypromine and fluoxetine. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 633–640. [Google Scholar] [CrossRef]
- Gobert, A.; Millan, M.J. Serotonin (5-ht)2a receptor activation enhances dialysate levels of dopamine and noradrenaline, but not 5-ht, in the frontal cortex of freely-moving rats. Neuropharmacology 1999, 38, 315–317. [Google Scholar] [CrossRef]
- Nisijima, K.; Shioda, K.; Yoshino, T.; Takano, K.; Kato, S. Memantine, an nmda antagonist, prevents the development of hyperthermia in an animal model for serotonin syndrome. Pharmacopsychiatry 2004, 37, 57–62. [Google Scholar]
- Dvir, Y.; Smallwood, P. Serotonin syndrome: A complex but easily avoidable condition. Gen. Hosp. Psychiatry 2008, 30, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Frazer, A.; Hensler, J. Basic neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Lippincott-Raven: Philadelphia, PA, USA, 1999. [Google Scholar]
- Fernstrom, J.D. Effects and side effects associated with the non-nutritional use of tryptophan by humans. J. Nutr. 2012, 142, 2236S–2244S. [Google Scholar] [CrossRef]
- Fidalgo, S.; Ivanov, D.K.; Wood, S.H. Serotonin: From top to bottom. Biogerontology 2013, 14, 21–45. [Google Scholar] [CrossRef]
- Pithadia, A.B.; Jain, S.M. 5-hydroxytryptamine receptor subtypes and their modulators with therapeutic potentials. J. Clin. Med. Res. 2009, 1, 72–80. [Google Scholar] [CrossRef]
- Hensler, J.G. Basic Neurochemistry: Principles of Molecular, Cellular, and Medical Neurobiology, 8th ed.; Academic Press (Elsevier): Waltham, MA, USA, 2012; Volume 15. [Google Scholar]
- Remick, R.A.; Froese, C. Monoamine oxidase inhibitors: Clinical review. Can. Fam. Physician. 1990, 36, 1151–1155. [Google Scholar]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295. [Google Scholar] [CrossRef] [PubMed]
- Asch, D.A.; Parker, R.M. The libby zion case. N. Engl. J. Med. 1988, 318, 771–775. [Google Scholar] [CrossRef]
- Isbister, G.K.; Bowe, S.J.; Dawson, A.; Whyte, I.M. Relative toxicity of selective serotonin reuptake inhibitors (ssris) in overdose. J. Toxicol. Clin. Toxicol. 2004, 42, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Parrott, A.C. Recreational ecstasy/mdma, the serotonin syndrome, and serotonergic neurotoxicity. Pharm. Biochem. Behav. 2002, 71, 837–844. [Google Scholar] [CrossRef]
- Demirkiran, M.; Jankovic, J.; Dean, J.M. Ecstasy intoxication: An overlap between serotonin syndrome and neuroleptic malignant syndrome. Clin. Neuropharmacol. 1996, 19, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Gillman, P.K. Ecstasy, serotonin syndrome and the treatment of hyperpyrexia. Med. J. Aust. 1997, 167, 109–111. [Google Scholar]
- Sun-Edelstein, C.; Tepper, S.J.; Shapiro, R.E. Drug-induced serotonin syndrome: A review. Expert Opin. Drug Saf. 2008, 7, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Newby, D.; Whyte, I.; Carter, G.; Jones, A.; Isbister, G. Serotonin toxicity from antidepressant overdose and its association with the t102c polymorphism of the 5-ht2a receptor. Pharm. J. 2014, 14, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Foong, A.L.; Grindrod, K.A.; Patel, T.; Kellar, J. Demystifying serotonin syndrome (or serotonin toxicity). Can. Fam. Physician. 2018, 64, 720–727. [Google Scholar]
- Report of the Psychiatric Drug Safety Expert Advisory Panel; TG Administration, Ed.; Department of Health Australian Government: Woden, ACT, Australia, 2009.
- Baldo, B.A. Opioid analgesic drugs and serotonin toxicity (syndrome): Mechanisms, animal models, and links to clinical effects. Arch. Toxicol. 2018, 92, 2457–2473. [Google Scholar] [CrossRef]
- Volpi-Abadie, J.; Kaye, A.M.; Kaye, A.D. Serotonin syndrome. Ochsner. J. 2013, 13, 533–540. [Google Scholar]
- Bijl, D. The serotonin syndrome. Neth. J. Med. 2004, 62, 309–313. [Google Scholar]
- Rastogi, R.; Swarm, R.A.; Patel, T.A. Case scenario: Opioid association with serotonin syndrome: Implications to the practitioners. Anesthesiology 2011, 115, 1291–1298. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Lee, J.; Franz, L.; Goforth, H.W. Serotonin syndrome in a chronic-pain patient receiving concurrent methadone, ciprofloxacin, and venlafaxine. Psychosomatics 2009, 50, 638–639. [Google Scholar] [CrossRef]
- Levin, T.T.; Cortes-Ladino, A.; Weiss, M.; Palomba, M.L. Life-threatening serotonin toxicity due to a citalopram-fluconazole drug interaction: Case reports and discussion. Gen. Hosp. Psychiatry 2008, 30, 372–377. [Google Scholar] [CrossRef]
- Mitchell, P.B. Drug interactions of clinical significance with selective serotonin reuptake inhibitors. Drug Saf. 1997, 17, 390–406. [Google Scholar] [CrossRef]
- Steinkellner, T.; Montgomery, T.R.; Hofmaier, T.; Kudlacek, O.; Yang, J.W.; Rickhag, M.; Jung, G.; Lubec, G.; Gether, U.; Freissmuth, M.; et al. Amphetamine action at the cocaine- and antidepressant-sensitive serotonin transporter is modulated by alphacamkii. J. Neurosci. 2015, 35, 8258–8271. [Google Scholar] [CrossRef] [PubMed]
- Zolkowska, D.; Rothman, R.B.; Baumann, M.H. Amphetamine analogs increase plasma serotonin: Implications for cardiac and pulmonary disease. J. Pharm. Exp. 2006, 318, 604–610. [Google Scholar] [CrossRef]
- Sandtner, W.; Schmid, D.; Schicker, K.; Gerstbrein, K.; Koenig, X.; Mayer, F.P.; Boehm, S.; Freissmuth, M.; Sitte, H.H. A quantitative model of amphetamine action on the 5-ht transporter. Br. J. Pharm. 2014, 171, 1007–1018. [Google Scholar] [CrossRef]
- Duthie, D.J.; Nimmo, W.S. Adverse effects of opioid analgesic drugs. Br. J. Anaesth. 1987, 59, 61–77. [Google Scholar] [CrossRef]
- Tao, R.; Auerbach, S.B. Opioid receptor subtypes differentially modulate serotonin efflux in the rat central nervous system. J. Pharm. Exp. 2002, 303, 549–556. [Google Scholar] [CrossRef]
- Haigler, H.J. Morphine: Effects on serotonergic neurons and neurons in areas with a serotonergic input. Eur. J. Pharm. 1978, 51, 361–376. [Google Scholar] [CrossRef]
- Tao, R.; Auerbach, S.B. Gabaergic and glutamatergic afferents in the dorsal raphe nucleus mediate morphine-induced increases in serotonin efflux in the rat central nervous system. J. Pharm. Exp. 2002, 303, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, G. Nhs prescribed record number of antidepressants last year. BMJ 2019, 364, l1508. [Google Scholar] [CrossRef] [PubMed]
- Fda Warns about Several Safety Issues with Opioid Pain Medicines; Requires Label Changes. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-about-several-safety-issues-opioid-pain-medicines-requires website (accessed on 27 April 2019).
- Gillman, P.K. Monoamine oxidase inhibitors, opioid analgesics and serotonin toxicity. Br. J. Anaesth. 2005, 95, 434–441. [Google Scholar] [CrossRef]
- Abadie, D.; Rousseau, V.; Logerot, S.; Cottin, J.; Montastruc, J.L.; Montastruc, F. Serotonin syndrome: Analysis of cases registered in the french pharmacovigilance database. J. Clin. Psychopharmacol. 2015, 35, 382–388. [Google Scholar] [CrossRef]
- Gillman, P.K. Extracting value from case reports: Lessons from serotonin toxicity. Anaesthesia 2006, 61, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Riblet, L.A.; Eison, A.S.; Eison, M.S.; Taylor, D.P.; Temple, D.L.; van der Maelen, C.P. Neuropharmacology of buspirone. Psychopathology 1984, 17, 69–78. [Google Scholar] [CrossRef]
- Peroutka, S.J.; Snyder, S.H. Multiple serotonin receptors: Differential binding of [3h]5-hydroxytryptamine, [3h]lysergic acid diethylamide and [3h]spiroperidol. Mol. Pharm. 1979, 16, 687–699. [Google Scholar]
- Goodwin, G.M.; De Souza, R.J.; Wood, A.J.; Green, A.R. The enhancement by lithium of the 5-ht1a mediated serotonin syndrome produced by 8-oh-dpat in the rat: Evidence for a post-synaptic mechanism. Psychopharmacology 1986, 90, 488–493. [Google Scholar] [CrossRef]
- Ohman, R.; Spigset, O. Serotonin syndrome induced by fluvoxamine-lithium interaction. Pharmacopsychiatry 1993, 26, 263–264. [Google Scholar] [CrossRef]
- Muly, E.C.; McDonald, W.; Steffens, D.; Book, S. Serotonin syndrome produced by a combination of fluoxetine and lithium. Am. J. Psychiatry 1993, 150, 1565. [Google Scholar]
- Norton, N.; Owen, M.J. Htr2a: Association and expression studies in neuropsychiatric genetics. Ann. Med. 2005, 37, 121–129. [Google Scholar] [CrossRef]
- Murphy, G.M., Jr.; Kremer, C.; Rodrigues, H.E.; Schatzberg, A.F. Pharmacogenetics of antidepressant medication intolerance. Am. J. Psychiatry 2003, 160, 1830–1835. [Google Scholar] [CrossRef] [PubMed]
- Kasper, S.; Praschak-Rieder, N.; Tauscher, J.; Wolf, R. A risk-benefit assessment of mirtazapine in the treatment of depression. Drug Saf. 1997, 17, 251–264. [Google Scholar] [CrossRef]
- Anttila, S.A.; Leinonen, E.V. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001, 7, 249–264. [Google Scholar] [CrossRef]
- Lattanzi, L.; Danesi, R.; Lastella, M.; Mungai, F.; Di Paolo, A.; Tuccori, M.; Cassano, G.; Del Tacca, M. Serotonin syndrome and the t102 to c polymorphism of the 5-ht2a receptor: A case report. Bipolar. Disord. 2008, 10, 655–656. [Google Scholar] [CrossRef] [PubMed]
- Hegazi, A.; Mc Keown, D.; Doerholt, K.; Donaghy, S.; Sadiq, S.T.; Hay, P. Serotonin syndrome following drug–drug interactions and cyp2d6 and cyp2c19 genetic polymorphisms inan hiv-infected patient. AIDS 2012, 26, 2417–2423. [Google Scholar]
- Kaneda, Y.; Kawamura, I.; Fujii, A.; Ohmori, T. Serotonin syndrome—‘potential’ role of the cyp2d6 genetic polymorphism in asians. Int. J. Neuropsychopharmacol. 2002, 5, 105–106. [Google Scholar]
- Piatkov, I.; Mann, G.; Jones, T.; McLean, M.; Gunja, N. Serotonin toxicity and cytochrome p450 poor metaboliser genotype patient case. J. Investig. Genom. 2017, 4, 1–5. [Google Scholar] [CrossRef]
- Fox, M.A.; Jensen, C.L.; Gallagher, P.S.; Murphy, D.L. Receptor mediation of exaggerated responses to serotonin-enhancing drugs in serotonin transporter (sert)-deficient mice. Neuropharmacology 2007, 53, 643–656. [Google Scholar] [CrossRef]
- Lesch, K.P.; Bengel, D.; Heils, A.; Sabol, S.Z.; Greenberg, B.D.; Petri, S.; Benjamin, J.; Muller, C.R.; Hamer, D.H.; Murphy, D.L. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 1996, 274, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.C. Serotonin syndrome. Am. Fam. Physician. 1995, 52, 1475–1482. [Google Scholar] [CrossRef]
- Francescangeli, J.; Vaida, S.; Bonavia, A.S. Perioperative diagnosis and treatment of serotonin syndrome following administration of methylene blue. Am. J. Case Rep. 2016, 17, 347–351. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shakoor, M.; Ayub, S.; Ahad, A.; Ayub, Z. Transient serotonin syndrome caused by concurrent use of tramadol and selective serotonin reuptake inhibitor. Am. J. Case Rep. 2014, 15, 562–564. [Google Scholar]
- Ng, B.K.; Cameron, A.J.; Liang, R.; Rahman, H. serotonin syndrome following methylene blue infusion during parathyroidectomy: A case report and literature review. Can. J. Anaesth 2008, 55, 36–41. [Google Scholar] [CrossRef]
- Graudins, A.; Stearman, A.; Chan, B. Treatment of the serotonin syndrome with cyproheptadine. J. Emerg. Med. 1998, 16, 615–619. [Google Scholar] [CrossRef]
- McDaniel, W.W. Serotonin syndrome: Early management with cyproheptadine. Ann. Pharm. 2001, 35, 870–873. [Google Scholar] [CrossRef]
- Maryland Poison Center: Antidote List. Available online: https://mdpoison.com/media/SOP/mdpoisoncom/healthcareprofessionals/antidote-facts/MPC%20Antidote%20List%202016.pdf (accessed on 17 March 2019).
- Gillman, P. The serotonin syndrome and its treatment. J. Psychopharmacol. 1999, 13, 100–109. [Google Scholar] [CrossRef]
- Rosebush, P.I. Serotonin Syndrome-Mhaus. Available online: https://www.mhaus.org/nmsis/medical-education-programs/serotonin-syndrome/ (accessed on 17 March 2019).
- Ramos, M.; Berrogain, C.; Concha, J.; Lomba, L.; Garcia, C.B.; Ribate, M.P. Pharmacogenetic studies: A tool to improve antidepressant therapy. Drug Metab. Pers. 2016, 31, 197–204. [Google Scholar] [CrossRef]
- Bousman, C.A.; Forbes, M.; Jayaram, M.; Eyre, H.; Reynolds, C.F.; Berk, M.; Hopwood, M.; Ng, C. Antidepressant prescribing in the precision medicine era: A prescriber’s primer on pharmacogenetic tools. BMC Psychiatry 2017, 17, 60. [Google Scholar] [CrossRef]
- Lett, T.A.; Walter, H.; Brandl, E.J. Pharmacogenetics and imaging-pharmacogenetics of antidepressant response: Towards translational strategies. CNS Drugs 2016, 30, 1169–1189. [Google Scholar] [CrossRef] [PubMed]
- Stern, S.; Linker, S.; Vadodaria, K.C.; Marchetto, M.C.; Gage, F.H. Prediction of response to drug therapy in psychiatric disorders. Open Biol. 2018, 8. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Sternbach Criteria | Radomski Criteria | Hunter Criteria | |
|---|---|---|---|
| Inclusion Criteria | Presence of serotonergic medication | Presence of serotonergic medication | Presence of serotonergic medication |
| Exclusion Criteria | Presence of other possible disease etiologies (e.g., infection, substance abuse, and withdrawal) and/or recent addition (or increase in dose) of neuroleptic medication. | None | None |
| Signs and Symptoms | At least three of the following signs/symptoms: | Either four major, or three major plus two minor signs/symptoms: | Any of the following combinations of primary (1°) ± secondary (2°) signs/symptoms: |
| Major: | |||
| Mental status changes (confusion, hypomania) | Impaired consciousness | ||
| Elevated mood | |||
| Agitation | Semicoma/coma | 1°: Spontaneous clonus alone | |
| Myoclonus | |||
| Myoclonus | Tremor | 1°: Inducible clonus AND | |
| Shivering | |||
| Hyperreflexia | Rigidity | 2°: Agitation or diaphoresis | |
| Hyperreflexia | |||
| Diaphoresis | Fever | 1°: Ocular clonus AND | |
| Sweating | |||
| Shivering | Minor: | 2°: Agitation or diaphoresis | |
| Restlessness | |||
| Tremor | Insomnia | 1°: Tremor AND | |
| Incoordination | |||
| Diarrhea | Dilated pupils | 2°: Hyperreflexia | |
| Akathisia | |||
| Incoordination | Tachycardia | 1°: Hypertonicity AND fever (temperature >38 °C) AND | |
| Tachypnea/Dyspnea | |||
| Fever | Diarrhea | 2°: Ocular clonus or inducible clonus | |
| Hypertension/hypotension |
| Disease | Medication Exposure | Shared Clinical Features | Distinguishing Clinical Features |
|---|---|---|---|
| Serotonin Syndrome | Serotonergic medications | Hypertension | Clonus, hyperreflexia Hyperactive bowel sounds |
| Neuroleptic Malignant Syndrome | Dopamine antagonists | Tachycardia | No clonus or hyperreflexia Bradykinesia |
| Anticholinergic Toxicity | Acetylcholine antagonist | Hyperthermia | No clonus or hyperreflexia Dry skin Absent bowel sounds |
| Malignant Hyperthermia | Halogenated anesthetics Succinylcholine | Altered mental Status | No clonus or hyperreflexia Extreme muscular rigidity |
| Synthesis and Release | |||
| Increase Serotonin Synthesis | Dietary supplements: L-tryptophan | ||
| Increase Serotonin Release | Psychostimulants: Amphetamines, phentermine, MDMA Antidepressants: mirtazapine Opioids: meperidine, oxycodone, tramadol Cough suppressants: dextromethorphan | ||
| Metabolism | |||
| Inhibit Serotonin Uptake | Psychostimulants: Amphetamines, MDMA, cocaine Antidepressants: trazodone SNRI: desvenlafaxine, duloxetine, venlafaxine SSRI: citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline TCA: amitriptyline, amoxapine, clomipramine, desipramine, doxepin, imipramine, maprotiline, nortriptyline, protriptyline, trimipramine Opioids: meperidine, methadone, tramadol Cough suppressants: dextromethorphan | ||
| Inhibit Serotonin Metabolism | Anxiolytics: buspirone MAOI: furazolidone, isocarboxazid, linezolid, methylene blue, phenelzine, selegiline, tranylcypromine | ||
| Inhibit Cytochrome P450 Microsomal Oxidases | CYP2D6 | CYP3A4 | CYP2C19 |
| Inhibitors: fluoxetine, sertraline Substrates: dextromethorphan, oxycodone, risperidone, tramadol | Inhibitors: ciprofloxacin, ritonavir Substrates: methadone, oxycodone, venlafaxine | Inhibitors: fluconazole Substrates: citalopram | |
| Receptor Activation | |||
| Activate Serotonin Receptors | Hallucinogen: LSD Anxiolytics: buspirone Antidepressants: trazodone Opioids*: fentanyl, meperidine Mood stabilizers: lithium | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francescangeli, J.; Karamchandani, K.; Powell, M.; Bonavia, A. The Serotonin Syndrome: From Molecular Mechanisms to Clinical Practice. Int. J. Mol. Sci. 2019, 20, 2288. https://doi.org/10.3390/ijms20092288
Francescangeli J, Karamchandani K, Powell M, Bonavia A. The Serotonin Syndrome: From Molecular Mechanisms to Clinical Practice. International Journal of Molecular Sciences. 2019; 20(9):2288. https://doi.org/10.3390/ijms20092288
Chicago/Turabian StyleFrancescangeli, James, Kunal Karamchandani, Meghan Powell, and Anthony Bonavia. 2019. "The Serotonin Syndrome: From Molecular Mechanisms to Clinical Practice" International Journal of Molecular Sciences 20, no. 9: 2288. https://doi.org/10.3390/ijms20092288
APA StyleFrancescangeli, J., Karamchandani, K., Powell, M., & Bonavia, A. (2019). The Serotonin Syndrome: From Molecular Mechanisms to Clinical Practice. International Journal of Molecular Sciences, 20(9), 2288. https://doi.org/10.3390/ijms20092288