Molecular Modeling-Guided Design of Phospholipid-Based Prodrugs

 ,
,  , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
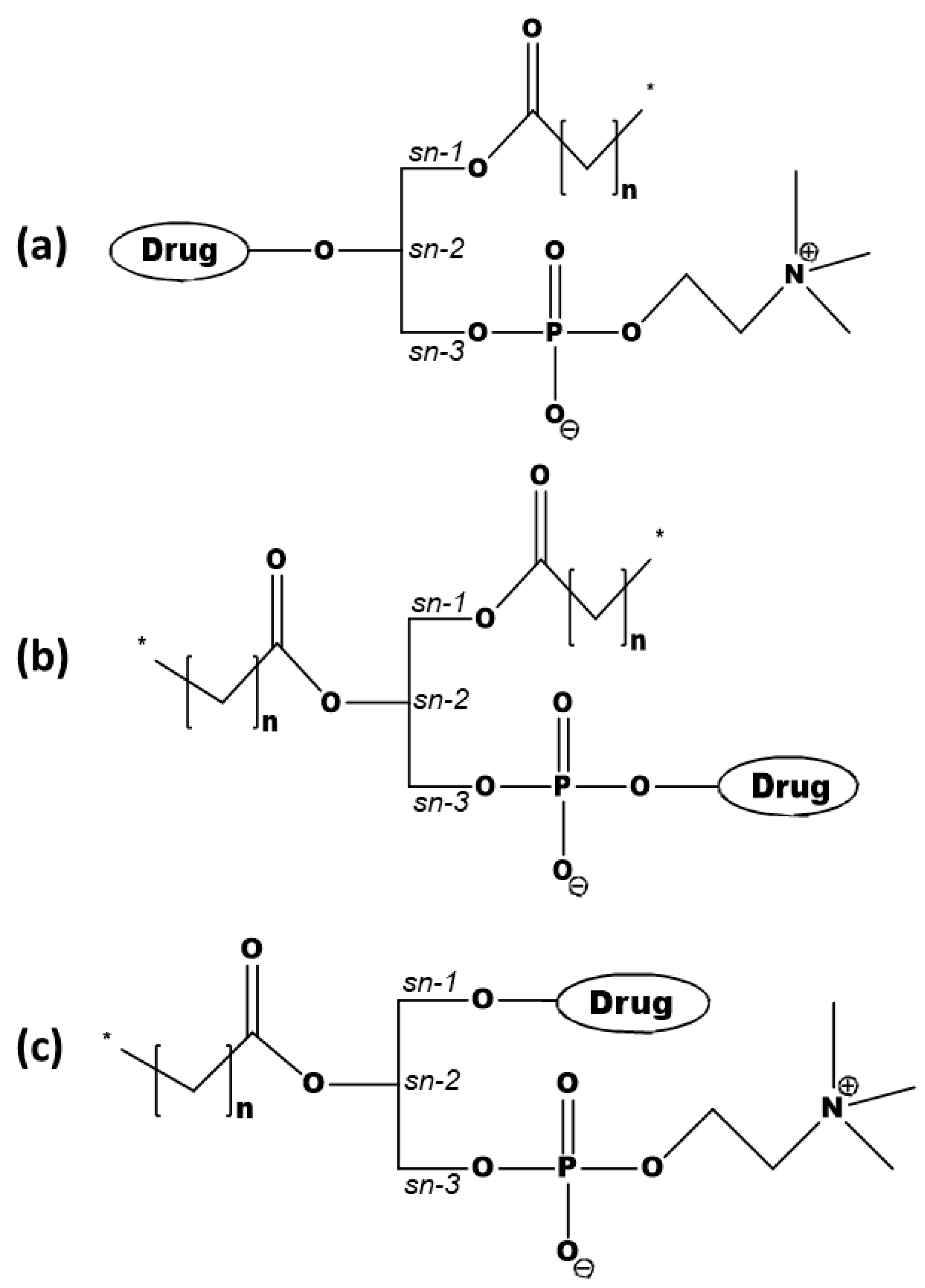
2. Overview of Phospholipid-Based Prodrugs
3. Computational Optimization of PL-Prodrug Design
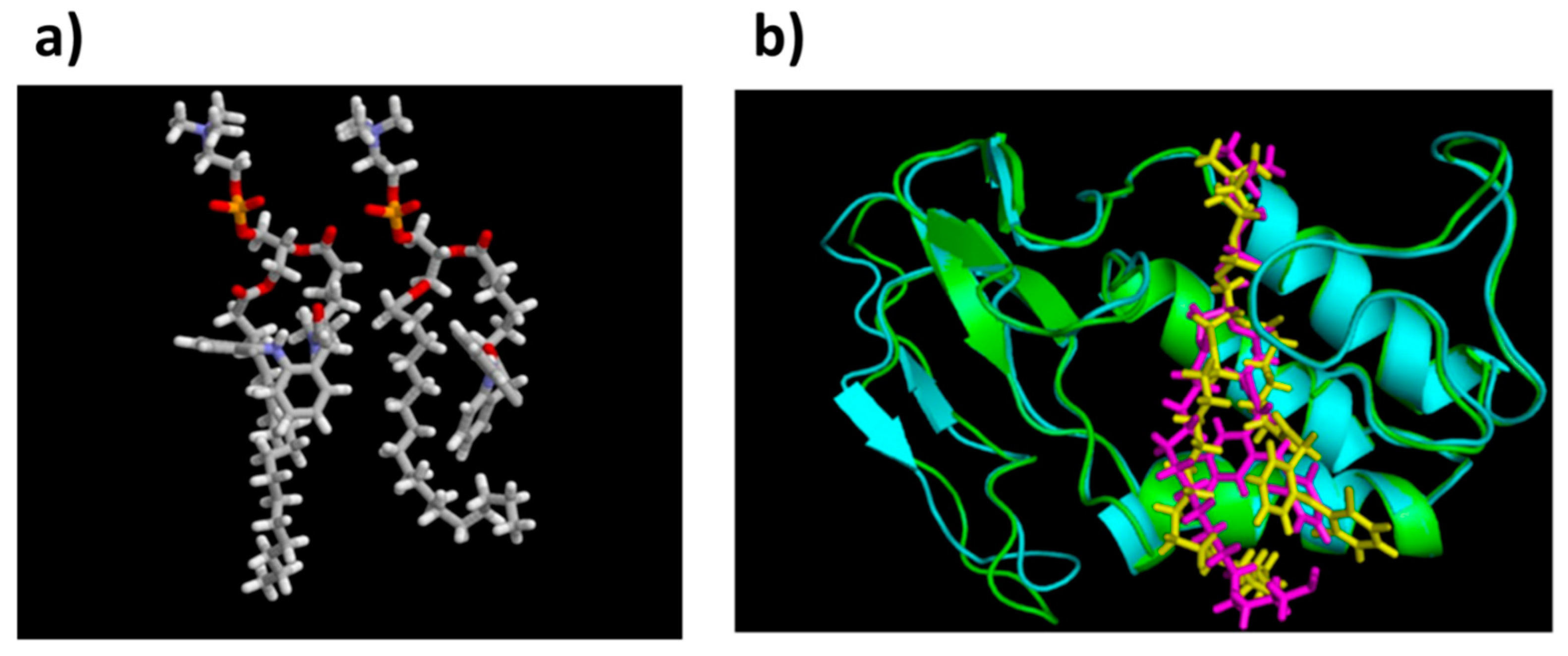
3.1. Molecular Docking
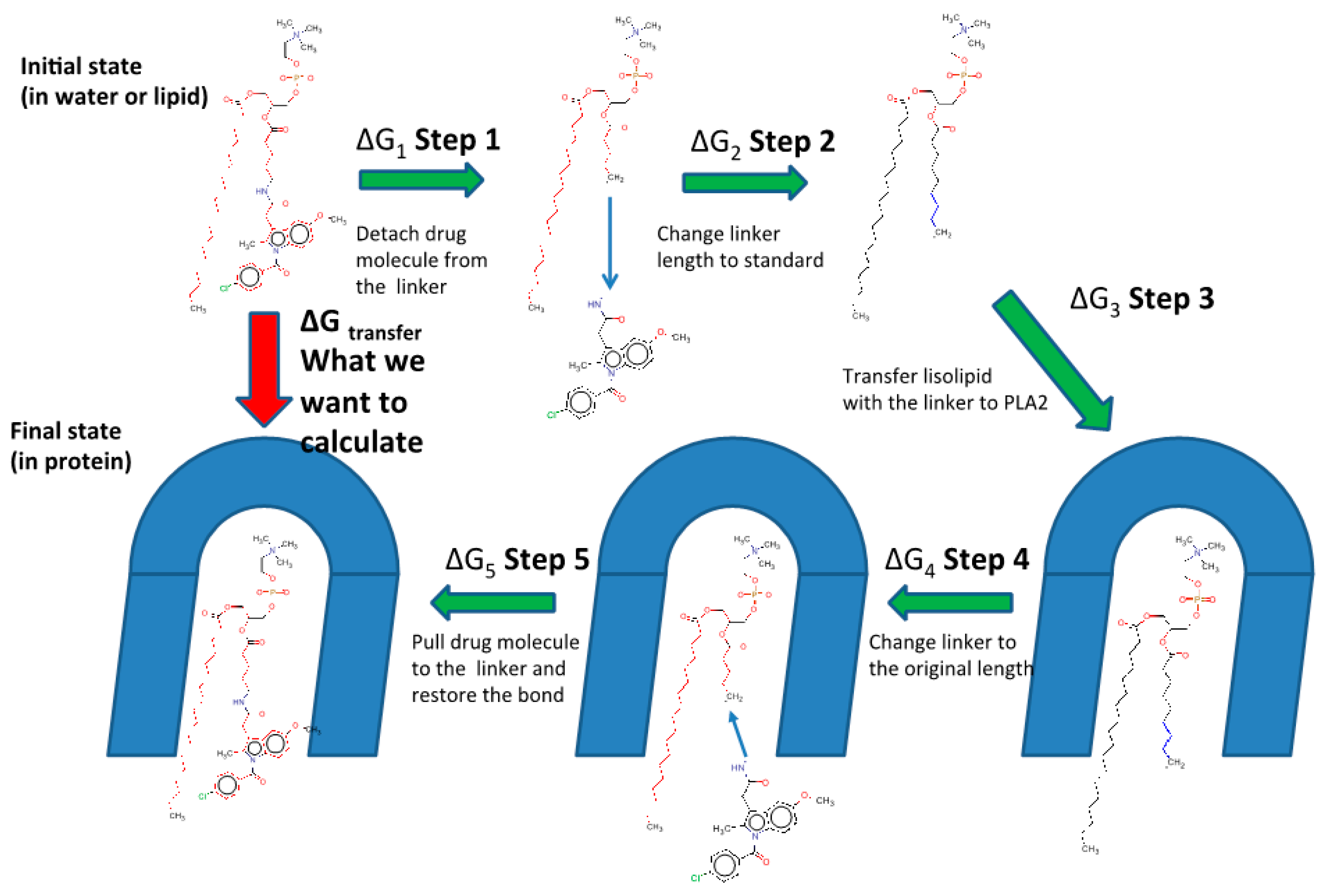
3.2. Free Energy Perturbation (FEP) and Molecular Dynamics (MD) Simulation: Linker Length Dependent PLA2-Mediated Activation
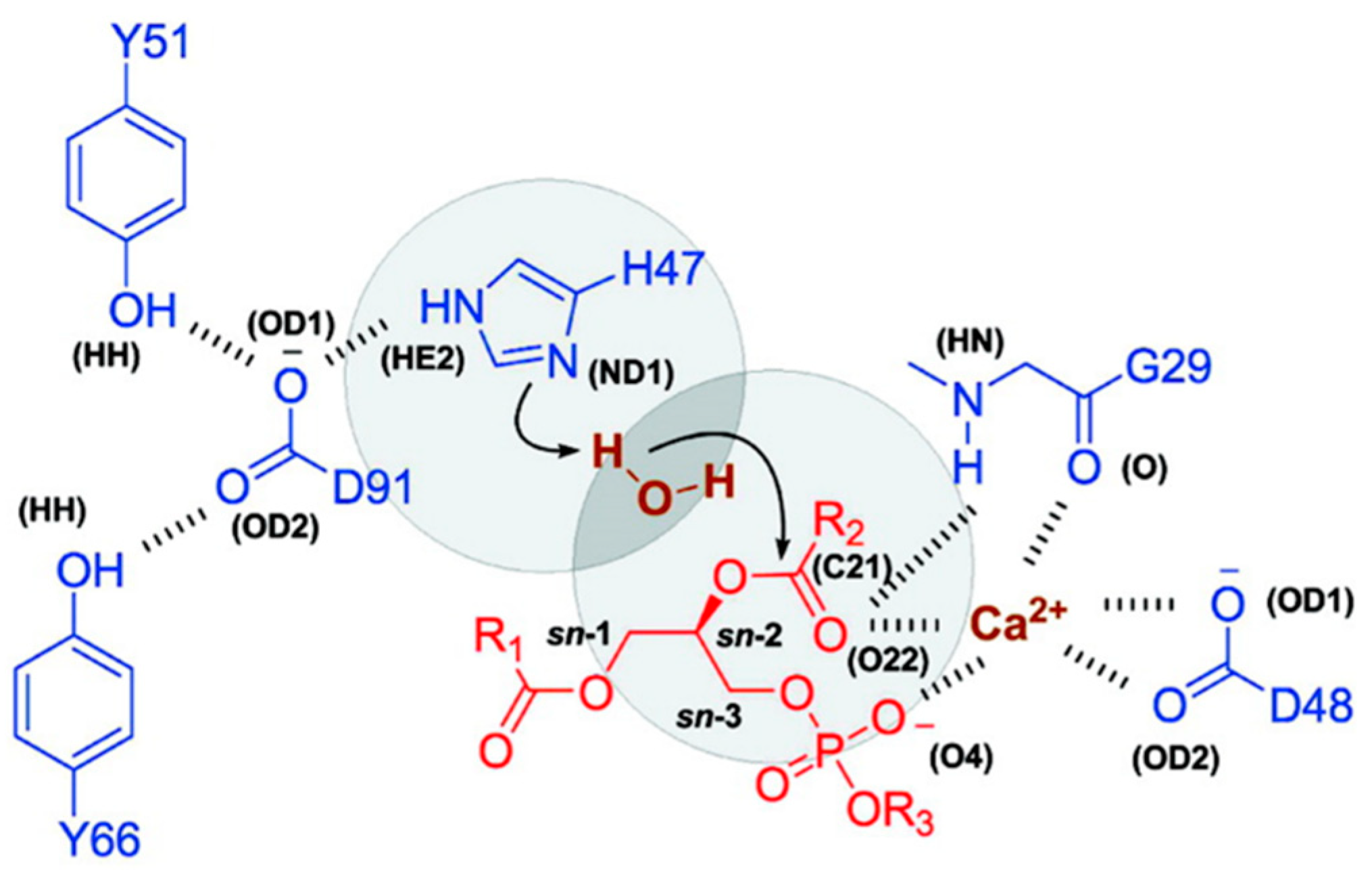
3.3. MD Simulation: Michaelis–Menten Complex and Water Availability in PLA2-Mediated Activation
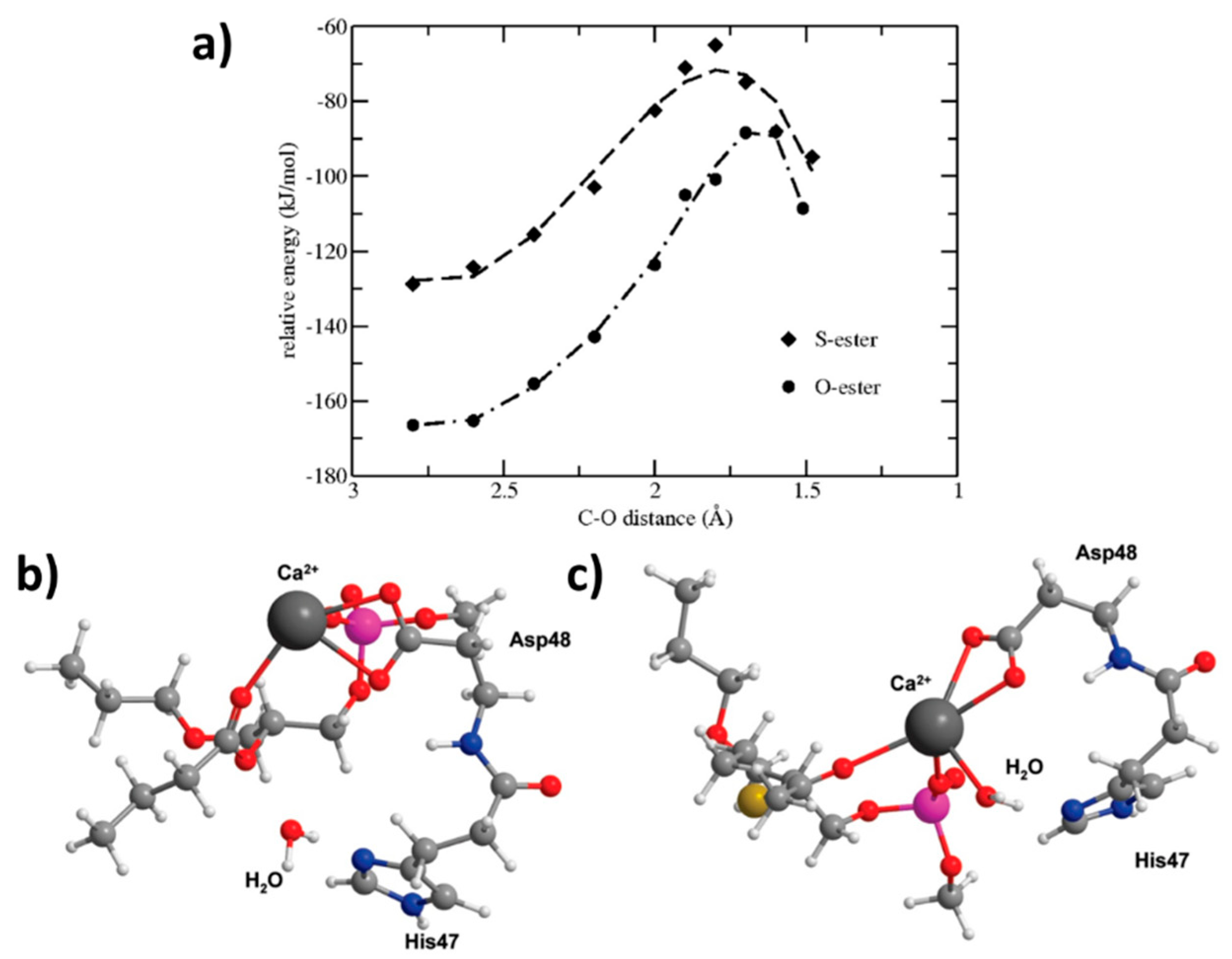
3.4. MD Simulation and Density Functional Theory: Side Group Influence, S-Ester vs. O-Ester
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559. [Google Scholar] [CrossRef]
- Stella, V.J. Prodrugs as therapeutics. Expert Opin. Ther. Pat. 2004, 14, 277–280. [Google Scholar] [CrossRef]
- Charman, W.N.; Porter, C.J. Lipophilic prodrugs designed for intestinal lymphatic transport. Adv. Drug Deliv. Rev. 1996, 19, 149–169. [Google Scholar] [CrossRef]
- Dahan, A.; Khamis, M.; Agbaria, R.; Karaman, R. Targeted prodrugs in oral drug delivery: The modern molecular biopharmaceutical approach. Expert Opin. Drug Deliv. 2012, 9, 1001–1013. [Google Scholar] [CrossRef]
- Dahan, A.; Zimmermann, E.M.; Ben-Shabat, S.; Eynde, J.J.V. Modern Prodrug Design for Targeted Oral Drug Delivery. Molecules 2014, 19, 16489–16505. [Google Scholar] [CrossRef]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef]
- Wolk, O.; Epstein, S.; Ioffe-Dahan, V.; Ben-Shabat, S.; Dahan, A. New targeting strategies in drug therapy of inflammatory bowel disease: Mechanistic approaches and opportunities. Expert Opin. Drug Deliv. 2013, 10, 1275–1286. [Google Scholar]
- Hoffman, A.; Dahan, A. Enhanced Gastrointestinal Absorption of Lipophilic Drugs. In Enhancement in Drug Delivery; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar] [CrossRef]
- Irby, D.; Du, C.; Li, F. Lipid–Drug Conjugate for Enhancing Drug Delivery. Mol. Pharm. 2017, 14, 1325–1338. [Google Scholar] [CrossRef]
- Lambert, D.M. Rationale and applications of lipids as prodrug carriers. Eur. J. Pharm. Sci. 2000, 11, S15–S27. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Lipidic prodrug approach for improved oral drug delivery and therapy. Med. Res. Rev. 2018, 39, 579–607. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Prospects and Challenges of Phospholipid-Based Prodrugs. Pharmaceutics 2018, 10, 210. [Google Scholar] [CrossRef]
- Arouri, A.; Hansen, A.H.; Rasmussen, T.E.; Mouritsen, O.G. Lipases, liposomes and lipid-prodrugs. Curr. Opin. Coll. Interface Sci. 2013, 18, 419–431. [Google Scholar] [CrossRef]
- Zaro, J.L. Lipid-based drug carriers for prodrugs to enhance drug delivery. AAPS J. 2015, 17, 83–92. [Google Scholar] [CrossRef]
- Arouri, A.; Mouritsen, O.G. Anticancer double lipid prodrugs: Liposomal preparation and characterization. J. Liposome Res. 2011, 21, 296–305. [Google Scholar] [CrossRef]
- Dahan, A.; Markovic, M.; Epstein, S.; Cohen, N.; Zimmermann, E.M.; Aponick, A.; Ben-Shabat, S. Phospholipid-drug conjugates as a novel oral drug targeting approach for the treatment of inflammatory bowel disease. Eur. J. Pharm. Sci. 2017, 108, 78–85. [Google Scholar] [CrossRef]
- Jensen, S.S.; Andresen, T.L.; Davidsen, J.; Høyrup, P.; Shnyder, S.D.; Bibby, M.C.; Gill, J.H.; Jørgensen, K. Secretory phospholipase A2 as a tumor-specific trigger for targeted delivery of a novel class of liposomal prodrug anticancer etherlipids. Mol. Cancer Ther. 2004, 3, 1451–1458. [Google Scholar]
- Dahan, A.; Duvdevani, R.; Shapiro, I.; Elmann, A.; Finkelstein, E.; Hoffman, A. The oral absorption of phospholipid prodrugs: In vivo and in vitro mechanistic investigation of trafficking of a lecithin-valproic acid conjugate following oral administration. J. Control Release 2008, 126, 1–9. [Google Scholar] [CrossRef]
- Dahan, A.; Duvdevani, R.; Dvir, E.; Elmann, A.; Hoffman, A. A novel mechanism for oral controlled release of drugs by continuous degradation of a phospholipid prodrug along the intestine: In-vivo and in-vitro evaluation of an indomethacin–lecithin conjugate. J. Control Release 2007, 119, 86–93. [Google Scholar] [CrossRef]
- Keinan, S.; Frush, E.H.; Shipman, W.J. Leveraging Cloud Computing for In-Silico Drug Design Using the Quantum Molecular Design (QMD) Framework. Comput. Sci. Eng. 2018, 20, 66–73. [Google Scholar] [CrossRef]
- Lee, A.; Lee, K.; Kim, D. Using reverse docking for target identification and its applications for drug discovery. Expert Opin. Drug Discov. 2016, 11, 707–715. [Google Scholar] [CrossRef]
- Atkovska, K.; Samsonov, S.A.; Paszkowski-Rogacz, M.; Pisabarro, M.T. Multipose binding in molecular docking. Int. J. Mol. Sci. 2014, 15, 2622–2645. [Google Scholar] [CrossRef]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Dahan, A.; Ben-Shabat, S.; Cohen, N.; Keinan, S.; Kurnikov, I.; Aponick, A.; Zimmermann, E.M. Phospholipid-Based Prodrugs for Drug Targeting in Inflammatory Bowel Disease: Computational Optimization and in-Vitro Correlation. Curr. Top. Med. Chem. 2016, 16, 2543–2548. [Google Scholar] [CrossRef]
- Dahan, A.; Markovic, M.; Keinan, S.; Kurnikov, I.; Aponick, A.; Zimmermann, E.M.; Ben-Shabat, S. Computational modeling and in-vitro/in-silico correlation of phospholipid-based prodrugs for targeted drug delivery in inflammatory bowel disease. J. Comput. Aided Mol. Des. 2017, 31, 1021–1028. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Tso, P. Gastrointestinal Digestion and Absorption of Lipid. Adv. Lipid Res. 1985, 21, 143–186. [Google Scholar]
- Alexander, P.; Kucera, G.; Pardee, T.S. Improving nucleoside analogs via lipid conjugation: Is fatter any better? Crit. Rev. Oncol. Hematol. 2016, 100, 46–56. [Google Scholar] [CrossRef]
- Alexander, P.M.; Caudell, D.L.; Kucera, G.L.; Pladna, K.M.; Pardee, T.S. The novel phospholipid mimetic KPC34 is highly active against preclinical models of Philadelphia chromosome positive acute lymphoblastic leukemia. PLoS ONE 2017, 12, e0179798. [Google Scholar] [CrossRef]
- Alexander, R.L.; Greene, B.T.; Torti, S.V.; Kucera, G.L. A novel phospholipid gemcitabine conjugate is able to bypass three drug-resistance mechanisms. Cancer Chemother. Pharmacol. 2005, 56, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kucera, G.L.; Alexander, P.; Pladna, K.; Pardee, T.S. Abstract 4059: KPC34: A co-drug that combines a DNA damaging agent with a targeted therapy for the treatment of AML. Cancer Res. 2017, 77, 4059. [Google Scholar] [CrossRef]
- Arouri, A.; Mouritsen, O.G. Phospholipase A2-susceptible liposomes of anticancer double lipid-prodrugs. Eur. J. Pharm. Sci. 2012, 45, 408–420. [Google Scholar] [CrossRef]
- Dvir, E.; Elman, A.; Simmons, D.; Shapiro, I.; Duvdevani, R.; Dahan, A.; Hoffman, A.; Friedman, J.E. DP-155, a Lecithin Derivative of Indomethacin, is a Novel Nonsteroidal Antiinflammatory Drug for Analgesia and Alzheimer’s Disease Therapy. CNS Drug Rev. 2007, 13, 260–277. [Google Scholar] [CrossRef]
- Pan, D.; Sanyal, N.; Schmieder, A.H.; Senpan, A.; Kim, B.; Yang, X.; Hu, G.; Allen, J.S.; Gross, R.W.; Wickline, S.A.; et al. Antiangiogenic nanotherapy with lipase-labile Sn-2 fumagillin prodrug. Nanomedicine 2012, 7, 1507–1519. [Google Scholar] [CrossRef]
- Minami, T.; Tojo, H.; Shinomura, Y.; Komatsubara, T.; Matsuzawa, Y.; Okamoto, M. Elevation of phospholipase A2 protein in sera of patients with Crohn’s disease and ulcerative colitis. Am. J. Gastroenterol. 1993, 88, 1076–1080. [Google Scholar]
- Peterson, J.W.; Dickey, W.D.; Saini, S.S.; Gourley, W.; Klimpel, G.R.; Chopra, A.K. Phospholipase A2 activating protein and idiopathic inflammatory bowel disease. Gut 1996, 39, 698–704. [Google Scholar] [CrossRef]
- Pruzanski, W.; Vadas, P.; Stefanski, E.; Urowitz, M.B. Phospholipase A2 activity in sera and synovial fluids in rheumatoid arthritis and osteoarthritis. Its possible role as a proinflammatory enzyme. J. Rheumatol. 1985, 12, 211–216. [Google Scholar]
- Laye, J.P.; Gill, J.H. Phospholipase A2 expression in tumours: A target for therapeutic intervention? Drug Discov. Today 2003, 8, 710–716. [Google Scholar] [CrossRef]
- Thunnissen, M.M.G.M.; Ab, E.; Kalk, K.H.; Drenth, J.; Dijkstra, B.W.; Kuipers, O.P.; Dijkman, R.; De Haas, G.H.; Verheij, H.M. X-ray structure of phospholipase A2 complexed with a substrate-derived inhibitor. Nature 1990, 347, 689–691. [Google Scholar] [CrossRef]
- Sakai, A.; Mori, N.; Shuto, S.; Suzuki, T.; Suzuki, T. Deacylation-Reacylation Cycle: A Possible Absorption Mechanism for the Novel Lymphotropic Antitumor Agent Dipalmitoylphosphatidylfluorouridine in Rats. J. Pharm. Sci. 1993, 82, 575–578. [Google Scholar] [CrossRef]
- Porter, C. Intestinal lymphatic drug transport: An update. Adv. Drug Deliv. Rev. 2001, 50, 61–80. [Google Scholar] [CrossRef]
- Andresen, T.L.; Davidsen, J.; Begtrup, M.; Mouritsen, O.G.; Jørgensen, K. Enzymatic Release of Antitumor Ether Lipids by Specific Phospholipase A2Activation of Liposome-Forming Prodrugs. J. Med. Chem. 2004, 47, 1694–1703. [Google Scholar] [CrossRef]
- Andresen, T.L.; Jensen, S.S.; Madsen, R.; Jørgensen, K. Synthesis and Biological Activity of Anticancer Ether Lipids That Are Specifically Released by Phospholipase A2in Tumor Tissue. J. Med. Chem. 2005, 48, 7305–7314. [Google Scholar] [CrossRef]
- Linderoth, L.; Fristrup, P.; Hansen, M.; Melander, F.; Madsen, R.; Andresen, T.L.; Peters, G.H. Mechanistic Study of the sPLA2-Mediated Hydrolysis of a Thio-ester Pro Anticancer Ether Lipid. J. Am. Chem. Soc. 2009, 131, 12193–12200. [Google Scholar] [CrossRef] [PubMed]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L.; Goni, R. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar]
- Beg, S.; Raza, K.; Kumar, R.; Chadha, R.; Katare, O.P.; Singh, B. Improved Intestinal Lymphatic Drug Targeting via Phospholipid Complex-loaded Nanolipospheres of Rosuvastatin Calcium. RSC Adv. 2016, 6, 8173–8187. [Google Scholar] [CrossRef]
- Linderoth, L.; Andresen, T.L.; Jørgensen, K.; Madsen, R.; Peters, G.H. Molecular Basis of Phospholipase A(2) Activity toward Phospholipids with sn-1 Substitutions. Biophys. J. 2008, 94, 14–26. [Google Scholar] [CrossRef]
- Straatsma, T.P.; Berendsen, H.J.C. Free energy of ionic hydration: Analysis of a thermodynamic integration technique to evaluate free energy differences by molecular dynamics simulations. J. Chem. Phys. 1988, 89, 5876–5886. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Pedersen, P.J.; Adolph, S.K.; Subramanian, A.K.; Arouri, A.; Andresen, T.L.; Mouritsen, O.G.; Madsen, R.; Madsen, M.W.; Peters, G.H.; Clausen, M.H. Liposomal Formulation of Retinoids Designed for Enzyme Triggered Release. J. Med. Chem. 2010, 53, 3782–3792. [Google Scholar] [CrossRef]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chemico-Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Molecular Modeling-Guided Design of Phospholipid-Based Prodrugs. Int. J. Mol. Sci. 2019, 20, 2210. https://doi.org/10.3390/ijms20092210
Markovic M, Ben-Shabat S, Keinan S, Aponick A, Zimmermann EM, Dahan A. Molecular Modeling-Guided Design of Phospholipid-Based Prodrugs. International Journal of Molecular Sciences. 2019; 20(9):2210. https://doi.org/10.3390/ijms20092210
Chicago/Turabian StyleMarkovic, Milica, Shimon Ben-Shabat, Shahar Keinan, Aaron Aponick, Ellen M. Zimmermann, and Arik Dahan. 2019. "Molecular Modeling-Guided Design of Phospholipid-Based Prodrugs" International Journal of Molecular Sciences 20, no. 9: 2210. https://doi.org/10.3390/ijms20092210
APA StyleMarkovic, M., Ben-Shabat, S., Keinan, S., Aponick, A., Zimmermann, E. M., & Dahan, A. (2019). Molecular Modeling-Guided Design of Phospholipid-Based Prodrugs. International Journal of Molecular Sciences, 20(9), 2210. https://doi.org/10.3390/ijms20092210