Dynamic Transcriptome Changes Related to Oil Accumulation in Developing Soybean Seeds




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Dynamic Changes of Oil Content in Developing Soybean Seeds
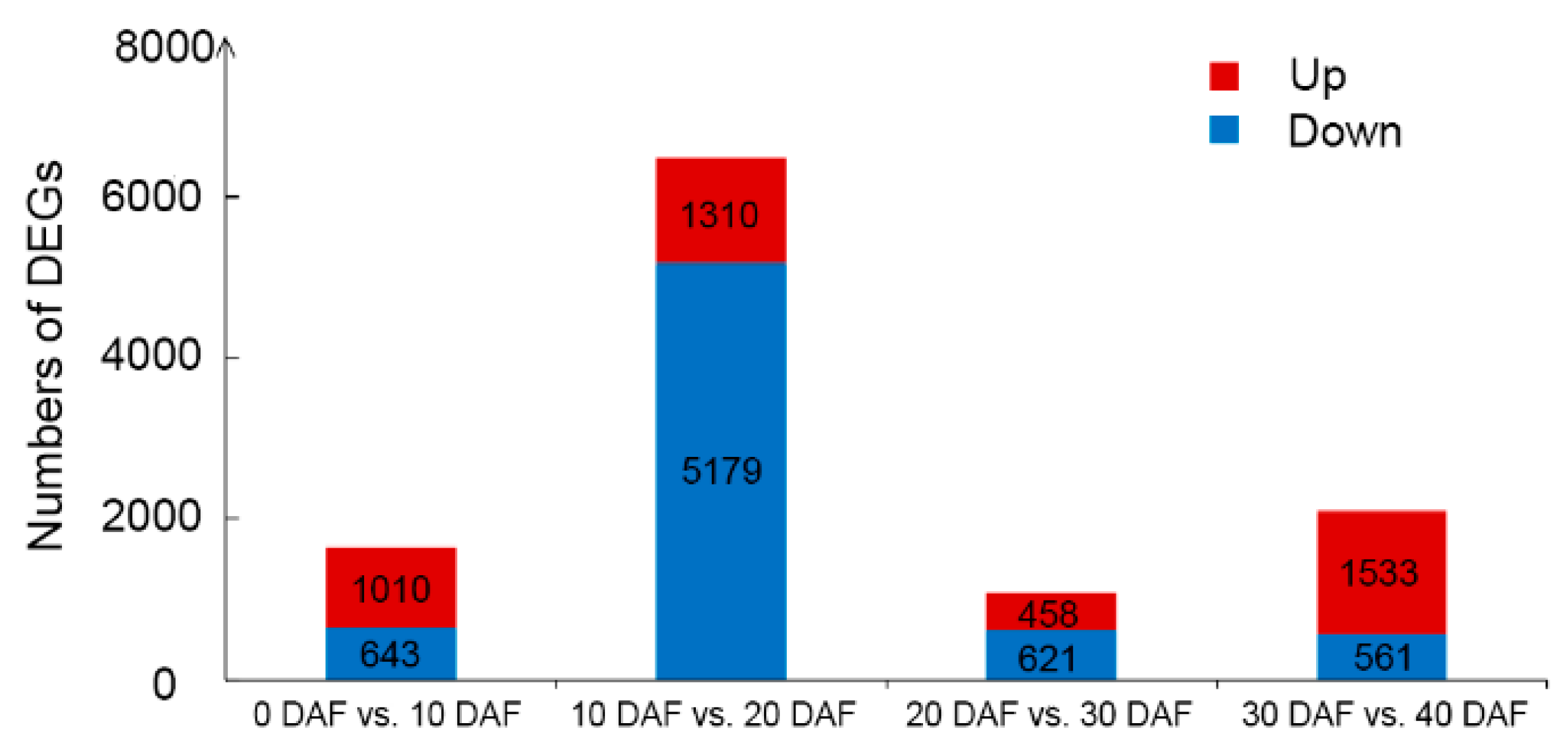
2.2. DEGs Between Adjacent Developmental Stages and Validation of RNA-Seq by Quantitative RT-PCR
2.3. Dynamic Changes in Expression Levels of Genes in the Fatty Acid Metabolic Pathways in Developing Soybean Seeds
2.4. Functional Analysis of DEGs Comparing 20 DAF with 10 DAF
2.5. Co-Expression Gene Networks and Their Correlations with Soybean Seed Oil Content
2.6. Identification of Hub Genes in the Co-Expression Modules Related to Soybean Seed Oil Content
2.7. Identification of Candidate Genes Related to Soybean Seed Oil Synthesis
3. Discussion
3.1. Pathways Associated with Soybean Seed Oil Synthesis
3.2. Screening Soybean Seed Oil Related Candidate Genes Based on WGCNA Analysis
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. Quantitation of Seed Oil Content
4.3. Library Construction and RNA Sequence
4.4. Raw Data Filtering
4.5. Transcriptome Analysis and Data Normalization
4.6. Quantitative RT-PCR Analysis (qRT-PCR)
4.7. Pathway and GO Enrichment Analysis and MapMan Metablic Map
4.8. Co-Expression Network Analysis
4.9. Heatmap Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABI3 | Abscisic acid-insensitive 3 |
| ACCase | Acetyl-CoA carboxylase |
| BP | Biology process |
| BC | Biotin carboxylase subunit |
| BCCP | Biotin carboxyl carrier protein |
| DAF | Days after flowering |
| DAG | Diacylglycerol |
| DGK | Diacylglycerol kinase |
| DEG | Differently expressed gene |
| DW | Dry weight |
| EAR | Enoyl-ACP reductase |
| FDR | False discovery rate |
| FAD2 | Fatty acid desaturase 2 |
| FAD3 | Fatty acid desaturase 3 |
| FPKM | Fragments per kilobase per million |
| GS | Gene significance |
| HAD | Hydroxyacyl-ACP dehydrase |
| HSD | 11-β-Hydroxysteroid dehydrogenase-like |
| KAR | Ketoacyl-ACP reductase |
| KAS I | Ketoacyl-ACP synthase I |
| KAS II | Ketoacyl-ACP synthase II |
| KAS III | Ketoacyl-ACP synthase III |
| MCMT | Malonyl-CoA: ACP malonyltransferase |
| ME | Module eigengene |
| MM | Module membership |
| PDAT | Phospholipid: diacylglycerol acyltransferase |
| RNA-Seq | RNA sequencing |
| TAGs | Triacylglycerols |
| WGCNA | Weighted correlation network analysis |
References
- Bates, P.D.; Stymne, S.; Ohlrogge, J. Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 2013, 16, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Baud, S.; Lepiniec, L. Regulation of de novo fatty acid synthesis in maturing oilseeds of Arabidopsis. Plant Physiol. Biochem. 2009, 47, 448–455. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.M.; Kong, X.D.; Chen, F.; Huang, J.X.; Lou, X.Y.; Zhao, J.Y. Transcriptome analysis of Brassica napus pod using RNA-Seq and identification of lipid-related candidate genes. BMC Genom. 2015, 16, 858. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Horvath, S.; Dong, J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar] [CrossRef]
- El-Sharkawy, I.; Liang, D.; Xu, K. Transcriptome analysis of an apple (Malus × domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 2015, 66, 7359–7376. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Rajkumar, M.S.; Garg, R.; Jain, M. Genome-wide identification and co-expression network analysis provide insights into the roles of auxin response factor gene family in chickpea. Sci. Rep. 2017, 7, 10895. [Google Scholar] [CrossRef]
- Hollender, C.A.; Kang, C.; Darwish, O.; Geretz, A.; Matthews, B.F.; Slovin, J.; Alkharouf, N.; Liu, Z. Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks. Plant Physiol. 2014, 165, 1062–1075. [Google Scholar] [CrossRef]
- Hopper, D.W.; Ghan, R.; Schlauch, K.A.; Cramer, G.R. Transcriptomic network analyses of leaf dehydration responses identify highly connected ABA and ethylene signaling hubs in three grapevine species differing in drought tolerance. BMC Plant Biol. 2016, 16, 118. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Bankhead, A., 3rd; Eisfeld, A.J.; Hatta, Y.; Jeng, S.; Chang, J.H.; Aicher, L.D.; Proll, S.; Ellis, A.L.; Law, G.L.; et al. Host regulatory network response to infection with highly pathogenic H5N1 avian influenza virus. J. Virol. 2011, 85, 10955–10967. [Google Scholar] [CrossRef]
- Van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Butte, A.J.; Kohane, I.S. Mutual information relevance networks: Functional genomic clustering using pairwise entropy measurements. Biocomputing 2000, 1999, 418–429. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, D.O.; Yang, H.; Chen, M.; Zhang, W.; Zhang, L. Metabolome and transcriptome sequencing analysis reveals anthocyanin metabolism in pink flowers of anthocyanin-rich tea (Camellia sinensis). Molecules 2019, 24, 1064. [Google Scholar] [CrossRef]
- Chen, H.; Wang, F.W.; Dong, Y.Y.; Wang, N.; Sun, Y.P.; Li, X.Y.; Liu, L.; Fan, X.D.; Yin, H.L.; Jing, Y.Y.; et al. Sequence mining and transcript profiling to explore differentially expressed genes associated with lipid biosynthesis during soybean seed development. BMC Plant Biol. 2012, 12, 122. [Google Scholar] [CrossRef]
- Jones, S.I.; Vodkin, L.O. Using RNA-Seq to profile soybean seed development from fertilization to maturity. PLoS ONE 2013, 8, e59270. [Google Scholar] [CrossRef]
- Goettel, W.; Xia, E.; Upchurch, R.; Wang, M.L.; Chen, P.Y.; An, Y.Q.C. Identification and characterization of transcript polymorphisms in soybean lines varying in oil composition and content. BMC Genom. 2014, 15, 299. [Google Scholar] [CrossRef] [PubMed]
- Goettel, W.; Ramirez, M.; Upchurch, R.G.; An, Y.Q. Identification and characterization of large DNA deletions affecting oil quality traits in soybean seeds through transcriptome sequencing analysis. Appl. Genet. 2016, 129, 1577–1593. [Google Scholar] [CrossRef]
- Liu, Y.F.; Li, Q.T.; Lu, X.; Song, Q.X.; Lam, S.M.; Zhang, W.K.; Ma, B.; Lin, Q.; Man, W.Q.; Du, W.G.; et al. Soybean GmMYB73 promotes lipid accumulation in transgenic plants. BMC Plant Biol. 2014, 14, 73. [Google Scholar] [CrossRef]
- Song, Q.X.; Li, Q.T.; Liu, Y.F.; Zhang, F.X.; Ma, B.; Zhang, W.K.; Man, W.Q.; Du, W.G.; Wang, G.D.; Chen, S.Y.; et al. Soybean GmbZIP123 gene enhances lipid content in the seeds of transgenic Arabidopsis plants. J. Exp. Bot. 2013, 64, 4329–4341. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, Q.T.; Xiong, Q.; Li, W.; Bi, Y.D.; Lai, Y.C.; Liu, X.L.; Man, W.Q.; Zhang, W.K.; Ma, B.; et al. The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 2016, 86, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wang, Y.; Li, W.; Gu, Y.; Lai, Y.; Bi, Y.; He, C. Transcriptomic comparison reveals genetic variation potentially underlying seed developmental evolution of soybeans. J. Exp. Bot. 2018, 69, 5089–5104. [Google Scholar] [CrossRef]
- Sarmiento, C.; Ross, J.H.; Herman, E.; Murphy, D.J. Expression and subcellular targeting of a soybean oleosin in transgenic rapeseed. Implications for the mechanism of oil-body formation in seeds. Plant J. 1997, 11, 783–796. [Google Scholar] [CrossRef]
- Sandhu, D.; Alt, J.L.; Scherder, C.W.; Fehr, W.R.; Bhattacharyya, M.K. Enhanced oleic acid content in the soybean mutant M23 is associated with the deletion in the Fad2-1a gene encoding a fatty acid desaturase. J. Am. Oil Chem. Soc. 2007, 84, 229–235. [Google Scholar] [CrossRef]
- Porta, H.; Rocha-Sosa, M. Plant lipoxygenases. Physiological and molecular features. Plant Physiol. 2002, 130, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, D.; Qiu, W.; Shi, Y.; Yang, J.J.; Chen, S.; Wang, Q.; Pan, H. Application of weighted gene co-expression network analysis for data from paired design. Sci. Rep. 2018, 8, 622. [Google Scholar] [CrossRef] [PubMed]
- Galili, G.; Avin-Wittenberg, T.; Angelovici, R.; Fernie, A.R. The role of photosynthesis and amino acid metabolism in the energy status during seed development. Front. Plant Sci. 2014, 5, 447. [Google Scholar] [CrossRef]
- Marmon, S.; Sturtevant, D.; Herrfurth, C.; Chapman, K.; Stymne, S.; Feussner, I. Two acyltransferases contribute differently to linolenic acid levels in seed oil. Plant Physiol. 2017, 173, 2081–2095. [Google Scholar] [CrossRef]
- Tan, W.J.; Yang, Y.C.; Zhou, Y.; Huang, L.P.; Xu, L.; Chen, Q.F.; Yu, L.J.; Xiao, S. Diacylglycerol acyltransferase and diacylglycerol kinase modulate triacylglycerol and phosphatidic acid production in the plant response to freezing stress. Plant Physiol. 2018, 177, 1303–1318. [Google Scholar] [CrossRef]
- Pham, A.T.; Lee, J.D.; Shannon, J.G.; Bilyeu, K.D. Mutant alleles of FAD2-1A and FAD2-1B combine to produce soybeans with the high oleic acid seed oil trait. BMC Plant Biol. 2010, 10, 195. [Google Scholar] [CrossRef]
- Jolivet, P.; Roux, E.; D’Andrea, S.; Davanture, M.; Negroni, L.; Zivy, M.; Chardot, T. Protein composition of oil bodies in Arabidopsis thaliana ecotype WS. Plant Physiol. Biochem. 2004, 42, 501–509. [Google Scholar] [CrossRef]
- Li, F.; Asami, T.; Wu, X.; Tsang, E.W.; Cutler, A.J. A putative hydroxysteroid dehydrogenase involved in regulating plant growth and development. Plant Physiol. 2007, 145, 87–97. [Google Scholar] [CrossRef]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef]
- Zhang, D.; Zhao, M.; Li, S.; Sun, L.; Wang, W.; Cai, C.; Dierking, E.C.; Ma, J. Plasticity and innovation of regulatory mechanisms underlying seed oil content mediated by duplicated genes in the palaeopolyploid soybean. Plant J. 2017, 90, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, J.; Lambert, K.N.; Lin, Y. Developmental control of Arabidopsis seed oil biosynthesis. Planta 2007, 226, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Luthra, R.; Munshi, S.K.; Sukhija, P.S. Relationship of carbohydrate metabolism with lipid biosynthesis in developing sunflower (Helianthus annuus L.) seeds. J. Plant Physiol. 1991, 137, 312–318. [Google Scholar] [CrossRef]
- Tschiersch, H.; Borisjuk, L.; Rutten, T.; Rolletschek, H. Gradients of seed photosynthesis and its role for oxygen balancing. Biosystems 2011, 103, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Rolletschek, H.; Weber, H.; Borisjuk, L. Energy status and its control on embryogenesis of legumes. Embryo photosynthesis contributes to oxygen supply and is coupled to biosynthetic fluxes. Plant Physiol. 2003, 132, 1196–1206. [Google Scholar] [CrossRef]
- Dong, Z.; Zhao, H.; He, J.; Huai, J.; Lin, H.; Zheng, J.; Liu, Y.; Wang, G. Overexpression of a foxtail millet Acetyl-CoA carboxylase gene in maize increases sethoxydim resistance and oil content. Afr. J. Biotechnol. 2011, 10, 3986–3995. [Google Scholar]
- Klaus, D.; Ohlrogge, J.B.; Neuhaus, H.E.; Dormann, P. Increased fatty acid production in potato by engineering of Acetyl-CoA carboxylase. Planta 2004, 219, 389–396. [Google Scholar] [CrossRef]
- Pidkowich, M.S.; Nguyen, H.T.; Heilmann, I.; Ischebeck, T.; Shanklin, J. Modulating seed β-ketoacyl-acyl carrier protein synthase II level converts the composition of a temperate seed oil to that of a palm-like tropical oil. Proc. Natl. Acad. Sci. Biol. 2007, 104, 4742–4747. [Google Scholar] [CrossRef]
- Chen, B.; Wang, J.; Zhang, G.; Liu, J.; Manan, S.; Hu, H.; Zhao, J. Two types of soybean diacylglycerol acyltransferases are differentially involved in triacylglycerol biosynthesis and response to environmental stresses and hormones. Sci. Rep. 2016, 6, 28541. [Google Scholar] [CrossRef]
- Greenham, K.; Guadagno, C.R.; Gehan, M.A.; Mockler, T.C.; Weinig, C.; Ewers, B.E.; McClung, C.R. Temporal network analysis identifies early physiological and transcriptomic indicators of mild drought in Brassica rapa. Elife 2017, 6, e29655. [Google Scholar] [CrossRef]
- Zhou, Z.; Cheng, Y.; Jiang, Y.; Liu, S.; Zhang, M.; Liu, J.; Zhao, Q. Ten hub genes associated with progression and prognosis of pancreatic carcinoma identified by co-expression analysis. Int. J. Biol. Sci. 2018, 14, 124–136. [Google Scholar] [CrossRef]
- Lee, S.; Van, K.; Sung, M.; Nelson, R.; LaMantia, J.; McHale, L.K.; Mian, M.A.R. Genome-wide association study of seed protein, oil and amino acid contents in soybean from maturity groups I to IV. Appl. Genet. 2019. [Google Scholar] [CrossRef]
- Zhang, J.; Ge, Y.; Han, F.; Li, B.; Yan, S.; Sun, J.; Wang, L. Isoflavone content of soybean cultivars from maturity group 0 to VI grown in northern and southern China. J. Am. Oil Chem. Soc. 2014, 91, 1019–1028. [Google Scholar] [CrossRef]
- Dhungana, S.K.; Kulkarni, K.P.; Kim, M.; Ha, B.-K.; Kang, S.; Song, J.T.; Shin, D.-H.; Lee, J.-D. Environmental stability and correlation of soybean seed starch with protein and oil contents. Plant Breed. Biotech. 2017, 5, 293–303. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Park, H.M.; Hwang, T.Y.; Kim, S.L.; Kim, M.J.; Lee, S.K.; Seo, M.J.; Kim, K.J.; Kwon, Y.U.; Lee, S.C.; et al. A correlation between tocopherol content and antioxidant activity in seeds and germinating seeds of soybean cultivars. J. Sci. Food Agric. 2015, 95, 819–827. [Google Scholar] [CrossRef]
- Zhang, D.J.; Sun, L.J.; Li, S.; Wang, W.D.; Ding, Y.H.; Swarm, S.A.; Li, L.H.; Wang, X.T.; Tang, X.M.; Zhang, Z.F.; et al. Elevation of soybean seed oil content through selection for seed coat shininess. Nat. Plants 2018, 4, 30–35. [Google Scholar] [CrossRef]
- Maughan, P.J.; Maroof, M.A.S.; Buss, G.R. Identification of quantitative trait loci controlling sucrose content in soybean (Glycine max). Mol. Breed. 2000, 6, 105–111. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Macrander, J.; Brugler, M.R.; Daly, M. A RNA-Seq approach to identify putative toxins from acrorhagi in aggressive and non-aggressive anthopleura elegantissima polyps. BMC Genom. 2015, 16, 221. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.Q.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, 1178–1186. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 25. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with Tophat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Hu, R.; Fan, C.; Li, H.; Zhang, Q.; Fu, Y.F. Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Mol. Biol. 2009, 10, 93. [Google Scholar] [CrossRef]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. Clusterprofiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. Agrigo: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, 64–70. [Google Scholar] [CrossRef]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Muller, L.A.; Rhee, S.Y.; Stitt, M. Mapman: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Howe, E.; Holton, K.; Nair, S.; Schlauch, D.; Sinha, R.; Quackenbush, J. Mev: Multiexperiment Viewer; Springer: Boston, MA, USA, 2010; pp. 267–277. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Miao, L.; He, J.; Zhang, K.; Li, Y.; Gai, J. Dynamic Transcriptome Changes Related to Oil Accumulation in Developing Soybean Seeds. Int. J. Mol. Sci. 2019, 20, 2202. https://doi.org/10.3390/ijms20092202
Yang S, Miao L, He J, Zhang K, Li Y, Gai J. Dynamic Transcriptome Changes Related to Oil Accumulation in Developing Soybean Seeds. International Journal of Molecular Sciences. 2019; 20(9):2202. https://doi.org/10.3390/ijms20092202
Chicago/Turabian StyleYang, Songnan, Long Miao, Jianbo He, Kai Zhang, Yan Li, and Junyi Gai. 2019. "Dynamic Transcriptome Changes Related to Oil Accumulation in Developing Soybean Seeds" International Journal of Molecular Sciences 20, no. 9: 2202. https://doi.org/10.3390/ijms20092202
APA StyleYang, S., Miao, L., He, J., Zhang, K., Li, Y., & Gai, J. (2019). Dynamic Transcriptome Changes Related to Oil Accumulation in Developing Soybean Seeds. International Journal of Molecular Sciences, 20(9), 2202. https://doi.org/10.3390/ijms20092202