Targeted Therapy Against the Cell of Origin in Cutaneous Squamous Cell Carcinoma



Abstract
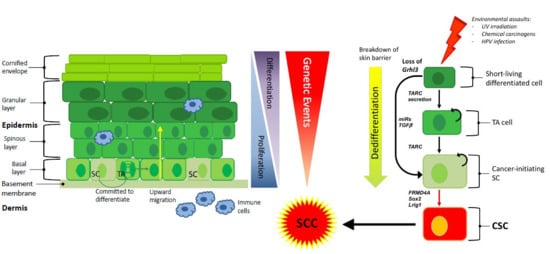
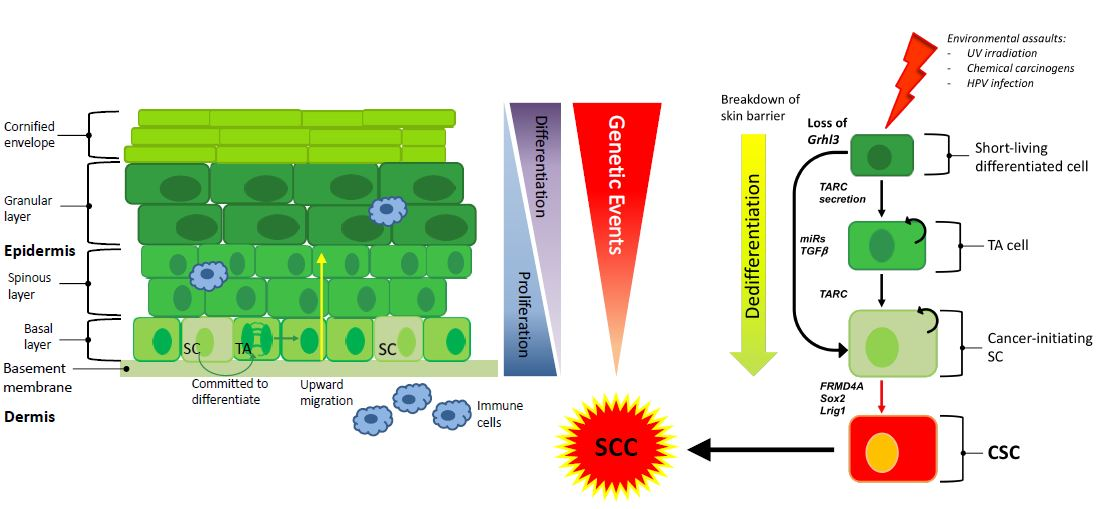
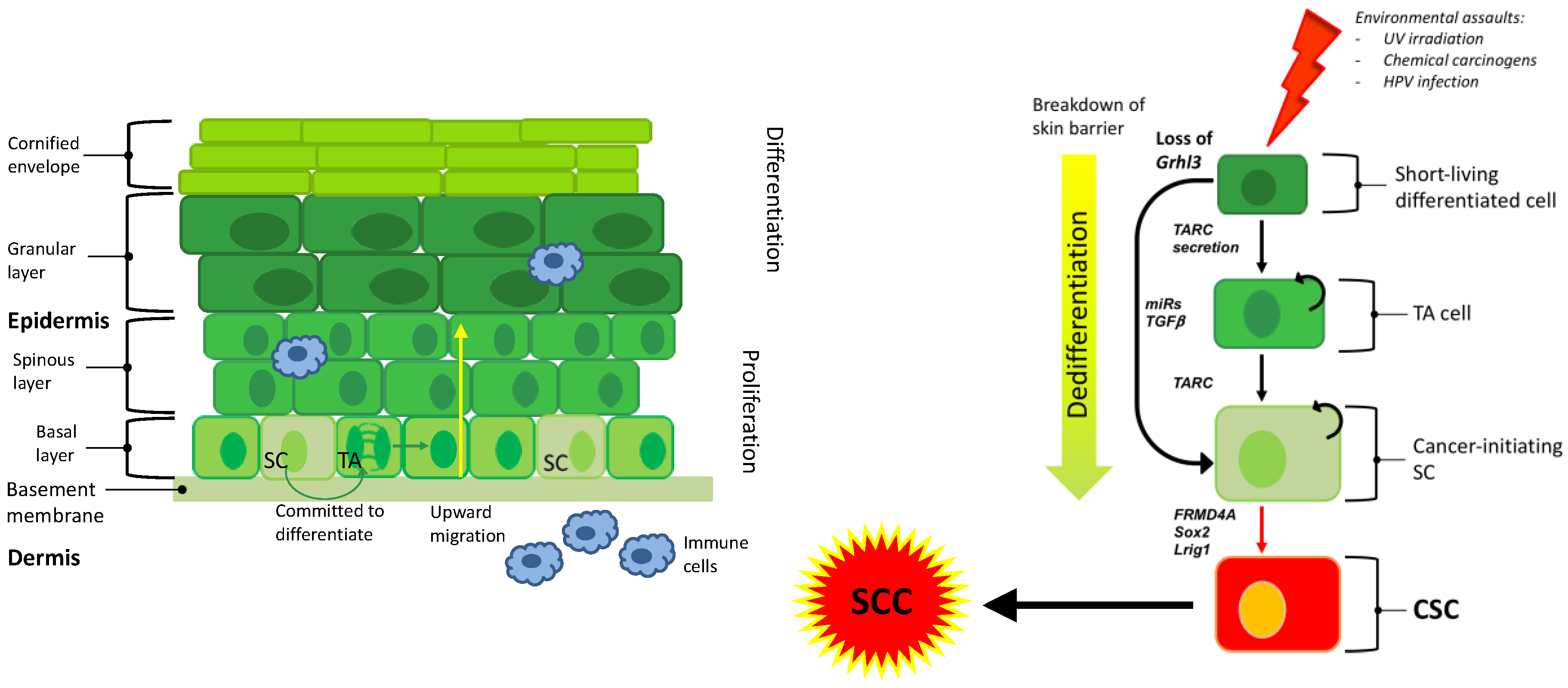
1. Introduction
2. Stem Cells in Cutaneous SCC
3. Heterogeneity of Cutaneous SCC
4. The Cell of Origin in Cutaneous SCC
5. Evaluation of Normal Stem Cell Markers in SCC
6. Pro-inflammatory Secreted Factors in SCC Development
7. Genome Wide Association Studies and Cutaneous SCC
8. Targeted Therapy and the Cell of Origin in Cutaneous SCC
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Alam, M.; Ratner, D. Cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2001, 344, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Staples, M.P.; Elwood, M.; Burton, R.C.; Williams, J.L.; Marks, R.; Giles, G.G. Non-melanoma skin cancer in Australia: The 2002 national survey and trends since 1985. Med. J. Aust. 2006, 184, 6–10. [Google Scholar] [PubMed]
- Pondicherry, A.; Martin, R.; Meredith, I.; Rolfe, J.; Emanuel, P.; Elwood, M. The burden of non-melanoma skin cancers in Auckland, New Zealand. Australas J. Dermatol. 2018, 59, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.A.; Miller, S.J. Nonmelanoma skin cancer. Facial Plast. Surg. Clin. N. Am. 2009, 17, 309–324. [Google Scholar] [CrossRef]
- Patel, R.V.; Frankel, A.; Goldenberg, G. An update on nonmelanoma skin cancer. J. Clin Aesthet. Dermatol. 2011, 4, 20–27. [Google Scholar]
- Cangkrama, M.; Ting, S.B.; Darido, C. Stem cells behind the barrier. Int. J. Mol. Sci. 2013, 14, 13670–13686. [Google Scholar] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Hahn, W.C.; Weinberg, R.A. Rules for making human tumor cells. N. Engl. J. Med. 2002, 347, 1593–1603. [Google Scholar] [CrossRef]
- Mascre, G.; Dekoninck, S.; Drogat, B.; Youssef, K.K.; Brohee, S.; Sotiropoulou, P.A.; Simons, B.D.; Blanpain, C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 2012, 489, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Sada, A.; Jacob, F.; Leung, E.; Wang, S.; White, B.S.; Shalloway, D.; Tumbar, T. Defining the cellular lineage hierarchy in the interfollicular epidermis of adult skin. Nat. Cell Biol. 2016, 18, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Danes, A.; Blanpain, C. Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer 2018, 18, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.B.; Jones, J.; Watt, F.M. A stem cell gene expression profile of human squamous cell carcinomas. Cancer Lett. 2008, 272, 23–31. [Google Scholar] [CrossRef]
- Locke, M.; Heywood, M.; Fawell, S.; Mackenzie, I.C. Retention of intrinsic stem cell hierarchies in carcinoma-derived cell lines. Cancer Res. 2005, 65, 8944–8950. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef]
- Van Duuren, B.L.; Sivak, A.; Katz, C.; Seidman, I.; Melchionne, S. The effect of aging and interval between primary and secondary treatment in two-stage carcinogenesis on mouse skin. Cancer Res. 1975, 35, 502–505. [Google Scholar]
- Morris, R.J.; Fischer, S.M.; Slaga, T.J. Evidence that a slowly cycling subpopulation of adult murine epidermal cells retains carcinogen. Cancer Res. 1986, 46, 3061–3066. [Google Scholar] [PubMed]
- Brown, K.; Strathdee, D.; Bryson, S.; Lambie, W.; Balmain, A. The malignant capacity of skin tumours induced by expression of a mutant H-ras transgene depends on the cell type targeted. Curr. Biol. 1998, 8, 516–524. [Google Scholar] [CrossRef]
- White, A.C.; Tran, K.; Khuu, J.; Dang, C.; Cui, Y.; Binder, S.W.; Lowry, W.E. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 7425–7430. [Google Scholar] [CrossRef] [PubMed]
- Lapouge, G.; Youssef, K.K.; Vokaer, B.; Achouri, Y.; Michaux, C.; Sotiropoulou, P.A.; Blanpain, C. Identifying the cellular origin of squamous skin tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 7431–7436. [Google Scholar] [CrossRef]
- Argyris, T.S.; Slaga, T.J. Promotion of carcinomas by repeated abrasion in initiated skin of mice. Cancer Res. 1981, 41, 5193–5195. [Google Scholar] [PubMed]
- Caulin, C.; Nguyen, T.; Lang, G.A.; Goepfert, T.M.; Brinkley, B.R.; Cai, W.W.; Lozano, G.; Roop, D.R. An inducible mouse model for skin cancer reveals distinct roles for gain- and loss-of-function p53 mutations. J. Clin. Invest. 2007, 117, 1893–1901. [Google Scholar] [CrossRef]
- Nassar, D.; Latil, M.; Boeckx, B.; Lambrechts, D.; Blanpain, C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat. Med. 2015, 21, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Danes, A.; Hannezo, E.; Larsimont, J.C.; Liagre, M.; Youssef, K.K.; Simons, B.D.; Blanpain, C. Defining the clonal dynamics leading to mouse skin tumour initiation. Nature 2016, 536, 298–303. [Google Scholar] [CrossRef]
- Patel, G.K.; Yee, C.L.; Terunuma, A.; Telford, W.G.; Voong, N.; Yuspa, S.H.; Vogel, J.C. Identification and characterization of tumor-initiating cells in human primary cutaneous squamous cell carcinoma. J. Invest. Dermatol. 2012, 132, 401–409. [Google Scholar] [CrossRef]
- Siegle, J.M.; Basin, A.; Sastre-Perona, A.; Yonekubo, Y.; Brown, J.; Sennett, R.; Rendl, M.; Tsirigos, A.; Carucci, J.A.; Schober, M. SOX2 is a cancer-specific regulator of tumour initiating potential in cutaneous squamous cell carcinoma. Nat. Commun. 2014, 5, 4511. [Google Scholar] [CrossRef]
- Boumahdi, S.; Driessens, G.; Lapouge, G.; Rorive, S.; Nassar, D.; Le Mercier, M.; Delatte, B.; Caauwe, A.; Lenglez, S.; Nkusi, E.; et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 2014, 511, 246–250. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science 2014, 344, 1242281. [Google Scholar] [CrossRef]
- Lotti, R.; Palazzo, E.; Petrachi, T.; Dallaglio, K.; Saltari, A.; Truzzi, F.; Quadri, M.; Puviani, M.; Maiorana, A.; Marconi, A.; et al. Survivin Modulates Squamous Cell Carcinoma-Derived Stem-Like Cell Proliferation, Viability and Tumor Formation in Vivo. Int. J. Mol. Sci. 2016, 17, 89. [Google Scholar] [CrossRef]
- Santarelli, A.; Mascitti, M.; Rubini, C.; Bambini, F.; Giannatempo, G.; Lo Russo, L.; Sartini, D.; Emanuelli, M.; Procaccini, M.; Lo Muzio, L. Nuclear Survivin as a Prognostic Factor in Squamous-Cell Carcinoma of the Oral Cavity. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, A.; Mascitti, M.; Lo Russo, L.; Sartini, D.; Troiano, G.; Emanuelli, M.; Lo Muzio, L. Survivin-Based Treatment Strategies for Squamous Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 971. [Google Scholar] [CrossRef]
- Jensen, K.B.; Watt, F.M. Single-cell expression profiling of human epidermal stem and transit-amplifying cells: Lrig1 is a regulator of stem cell quiescence. Proc. Natl. Acad. Sci. USA 2006, 103, 11958–11963. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Umeda, M. FRMD4A regulates epithelial polarity by connecting Arf6 activation with the PAR complex. Proc. Natl. Acad. Sci. USA 2010, 107, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Christofori, G. New signals from the invasive front. Nature 2006, 441, 444–450. [Google Scholar] [CrossRef]
- Goldie, S.J.; Mulder, K.W.; Tan, D.W.; Lyons, S.K.; Sims, A.H.; Watt, F.M. FRMD4A upregulation in human squamous cell carcinoma promotes tumor growth and metastasis and is associated with poor prognosis. Cancer Res. 2012, 72, 3424–3436. [Google Scholar] [CrossRef] [PubMed]
- Arwert, E.N.; Mentink, R.A.; Driskell, R.R.; Hoste, E.; Goldie, S.J.; Quist, S.; Watt, F.M. Upregulation of CD26 expression in epithelial cells and stromal cells during wound-induced skin tumour formation. Oncogene 2012, 31, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cai, G.; Liu, C.; Zhao, J.; Gu, C.; Wu, L.; Hamilton, T.A.; Zhang, C.J.; Ko, J.; Zhu, L.; et al. IL-17R-EGFR axis links wound healing to tumorigenesis in Lrig1(+) stem cells. J. Exp. Med. 2019, 216, 195–214. [Google Scholar] [CrossRef]
- Darido, C.; Jane, S.M. Grhl3 and GEF19 in the front rho. Small GTPases 2010, 1, 104–107. [Google Scholar] [CrossRef]
- Dworkin, S.; Jane, S.M.; Darido, C. The planar cell polarity pathway in vertebrate epidermal development, homeostasis and repair. Organogenesis 2011, 7, 202–208. [Google Scholar] [CrossRef]
- Darido, C.; Jane, S.M. Golgi Feels Its Own Wound. Adv. Wound Care 2013, 2, 87–92. [Google Scholar] [CrossRef]
- Cangkrama, M.; Darido, C.; Georgy, S.R.; Partridge, D.; Auden, A.; Srivastava, S.; Wilanowski, T.; Jane, S.M. Two Ancient Gene Families Are Critical for Maintenance of the Mammalian Skin Barrier in Postnatal Life. J. Invest. Dermatol. 2016, 136, 1438–1448. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Jane, S.M. The role of barrier genes in epidermal malignancy. Oncogene 2016, 35, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Youssef, M.; Cuddihy, A.; Darido, C. Long-Lived Epidermal Cancer-Initiating Cells. Int. J. Mol. Sci. 2017, 18, 1369. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Wilanowski, T.; Dworkin, S.; Auden, A.; Zhao, Q.; Rank, G.; Srivastava, S.; Finlay, M.J.; Papenfuss, A.T.; et al. Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto-oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell 2011, 20, 635–648. [Google Scholar] [CrossRef]
- Georgy, S.R.; Cangkrama, M.; Srivastava, S.; Partridge, D.; Auden, A.; Dworkin, S.; McLean, C.A.; Jane, S.M.; Darido, C. Identification of a Novel Proto-oncogenic Network in Head and Neck Squamous Cell Carcinoma. J. Natl. Cancer Inst. 2015, 107, djv152. [Google Scholar] [CrossRef] [PubMed]
- Darido, C.; Georgy, S.R.; Cullinane, C.; Partridge, D.D.; Walker, R.; Srivastava, S.; Roslan, S.; Carpinelli, M.R.; Dworkin, S.; Pearson, R.B. Stage-dependent therapeutic efficacy in PI3K/mTOR-driven squamous cell carcinoma of the skin. Cell Death Differ. 2018, 25, 1146–1159. [Google Scholar] [CrossRef]
- Goldie, S.J.; Cottle, D.L.; Tan, F.H.; Roslan, S.; Srivastava, S.; Brady, R.; Partridge, D.D.; Auden, A.; Smyth, I.M.; Jane, S.M.; et al. Loss of GRHL3 leads to TARC/CCL17-mediated keratinocyte proliferation in the epidermis. Cell Death Dis. 2018, 9, 1072. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, Y.; Saeki, H.; Nakamura, K.; Nagakubo, D.; Nakayama, T.; Yoshie, O.; Kagami, S.; Shimazu, K.; Kadono, T.; Sugaya, M.; et al. CCL17 transgenic mice show an enhanced Th2-type response to both allergic and non-allergic stimuli. Eur. J. Immunol. 2006, 36, 2116–2127. [Google Scholar] [CrossRef]
- Yoshie, O.; Matsushima, K. CCR4 and its ligands: From bench to bedside. Int. Immunol. 2015, 27, 11–20. [Google Scholar] [CrossRef]
- Zhang, L.; Ge, Y.; Fuchs, E. miR-125b can enhance skin tumor initiation and promote malignant progression by repressing differentiation and prolonging cell survival. Genes Dev. 2014, 28, 2532–2546. [Google Scholar] [CrossRef]
- Toll, A.; Salgado, R.; Espinet, B.; Diaz-Lagares, A.; Hernandez-Ruiz, E.; Andrades, E.; Sandoval, J.; Esteller, M.; Pujol, R.M.; Hernandez-Munoz, I. MiR-204 silencing in intraepithelial to invasive cutaneous squamous cell carcinoma progression. Mol. Cancer 2016, 15, 53. [Google Scholar] [CrossRef]
- Macias, E.; Rao, D.; Digiovanni, J. Role of stat3 in skin carcinogenesis: Insights gained from relevant mouse models. J. Skin Cancer 2013, 2013, 684050. [Google Scholar] [CrossRef]
- Chahal, H.S.; Lin, Y.; Ransohoff, K.J.; Hinds, D.A.; Wu, W.; Dai, H.J.; Qureshi, A.A.; Li, W.Q.; Kraft, P.; Tang, J.Y.; et al. Genome-wide association study identifies novel susceptibility loci for cutaneous squamous cell carcinoma. Nat. Commun. 2016, 7, 12048. [Google Scholar] [CrossRef]
- Walko, G.; Woodhouse, S.; Pisco, A.O.; Rognoni, E.; Liakath-Ali, K.; Lichtenberger, B.M.; Mishra, A.; Telerman, S.B.; Viswanathan, P.; Logtenberg, M.; et al. A genome-wide screen identifies YAP/WBP2 interplay conferring growth advantage on human epidermal stem cells. Nat. Commun. 2017, 8, 14744. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J. Clin. Oncol. 2015, 33, 3817–3825. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Schwaederle, M.; Wei, C.; Lee, J.J.; Hong, D.S.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of a Biomarker-Based Strategy on Oncology Drug Development: A Meta-analysis of Clinical Trials Leading to FDA Approval. J. Natl. Cancer Inst. 2015, 107, djv253. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.C.; Heiser, L.M.; Georgii, E.; Gonen, M.; Menden, M.P.; Wang, N.J.; Bansal, M.; Ammad-ud-din, M.; Hintsanen, P.; Khan, S.A.; et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014, 32, 1202–1212. [Google Scholar] [CrossRef]
- Way, G.P.; Sanchez-Vega, F.; La, K.; Armenia, J.; Chatila, W.K.; Luna, A.; Sander, C.; Cherniack, A.D.; Mina, M.; Ciriello, G.; et al. Machine Learning Detects Pan-cancer Ras Pathway Activation in The Cancer Genome Atlas. Cell Rep. 2018, 23, 172–180. [Google Scholar] [CrossRef]
- Sengupta, S.; Sun, S.Q.; Huang, K.L.; Oh, C.; Bailey, M.H.; Varghese, R.; Wyczalkowski, M.A.; Ning, J.; Tripathi, P.; McMichael, J.F.; et al. Integrative omics analyses broaden treatment targets in human cancer. Genome Med 2018, 10, 60. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Factor | Potential Mechanism | References |
|---|---|---|
| CD44 | Cell surface marker elevated in subpopulations of SCC, i.e., putative tumour stem cell marker | [15] |
| CD133 | Putative tumour stem cell marker in SCC | [28] |
| TGF-β | Promotes chemotherapy resistance in SCC | [18] |
| BMI1 | Tumour self-renewal and tumourigenesis | [15] |
| Sox2 | Putative tumour stem cell marker in SCC | [29] |
| FRMD4A | Putative tumour stem cell marker in SCC; interacts with Hippo pathway | [39] |
| YAP1 | Key downstream effector of the Hippo pathway, influencing the behaviour of stem cells in the normal epithelium and in SCC | [58] |
| Lrig1 (loss) | Putative tumour stem cell marker in SCC and tumour suppressor. | [41] |
| Grhl3 (loss) | Conserved developmental transcription factor, essential for epidermal differentiation, barrier formation and SCC initiation | [51] |
| TARC/CCL17 | Cytokine driving hyperproliferation in Grhl3 deletion model | [51] |
| IL-1α | Cytokine driving papilloma formation in MEK1 overexpression model | [40] |
| IL-17R | Cytokine implicated in the mobilization of Lrig1-positive stem cells in wound healing experiments, and in tumorigenesis | [41] |
| Survivin | Overexpression of Survivin promotes the development of SCC in multiple tissue types and results in poor prognosis | [32] |
| miR-204 (loss) | Loss of miR-204 drives STAT3 pro-inflammatory response in sun-damaged skin leading to the development of SCC. | [55] |
| MEK1 | Overexpression promotes hyperproliferation and skin inflammation, leading to papilloma formation | [40] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldie, S.J.; Chincarini, G.; Darido, C. Targeted Therapy Against the Cell of Origin in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 2201. https://doi.org/10.3390/ijms20092201
Goldie SJ, Chincarini G, Darido C. Targeted Therapy Against the Cell of Origin in Cutaneous Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2019; 20(9):2201. https://doi.org/10.3390/ijms20092201
Chicago/Turabian StyleGoldie, Stephen J., Ginevra Chincarini, and Charbel Darido. 2019. "Targeted Therapy Against the Cell of Origin in Cutaneous Squamous Cell Carcinoma" International Journal of Molecular Sciences 20, no. 9: 2201. https://doi.org/10.3390/ijms20092201
APA StyleGoldie, S. J., Chincarini, G., & Darido, C. (2019). Targeted Therapy Against the Cell of Origin in Cutaneous Squamous Cell Carcinoma. International Journal of Molecular Sciences, 20(9), 2201. https://doi.org/10.3390/ijms20092201