Optimisation of Folate-Mediated Liposomal Encapsulated Arsenic Trioxide for Treating HPV-Positive Cervical Cancer Cells In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Liposome Preparation and Characterisation
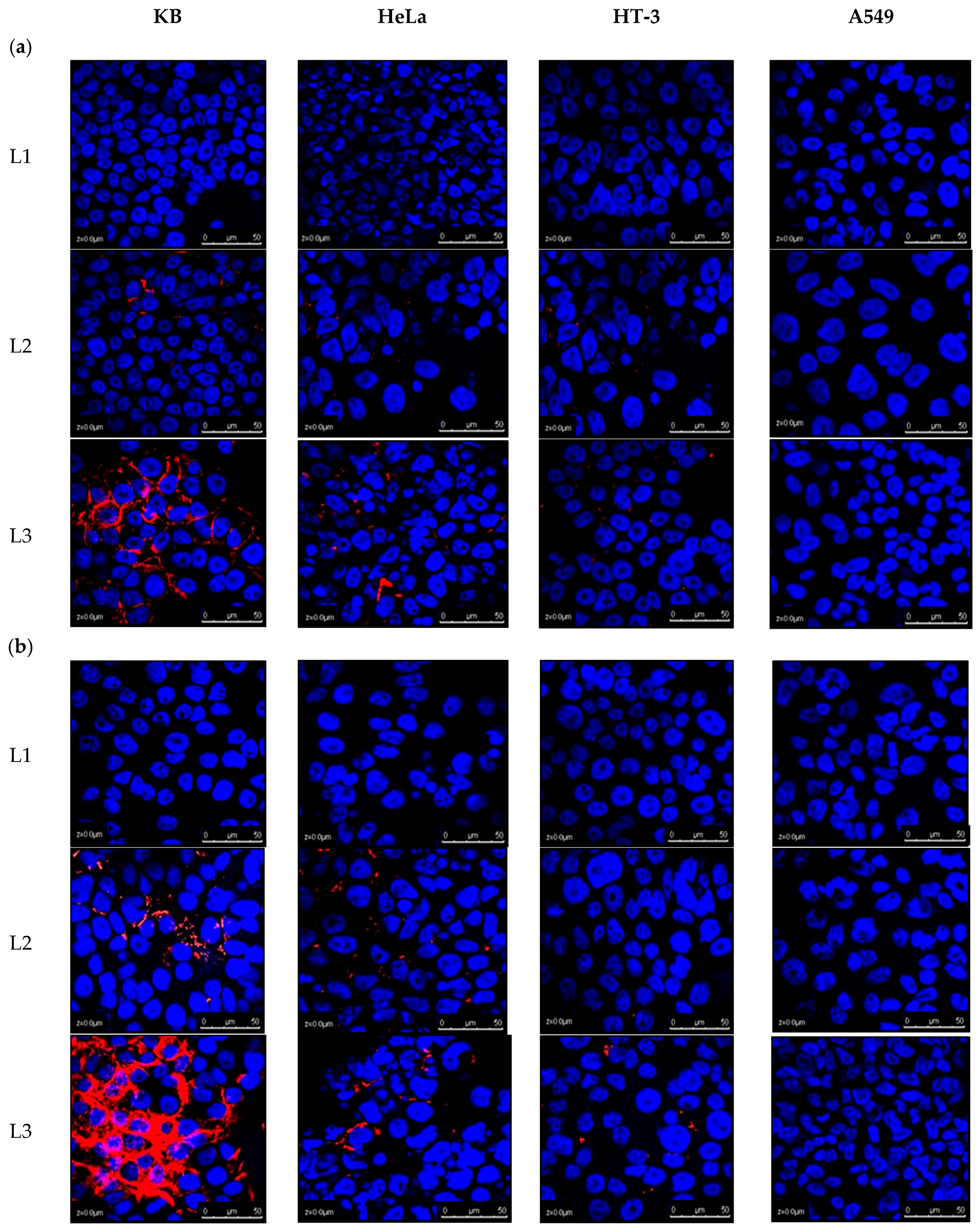
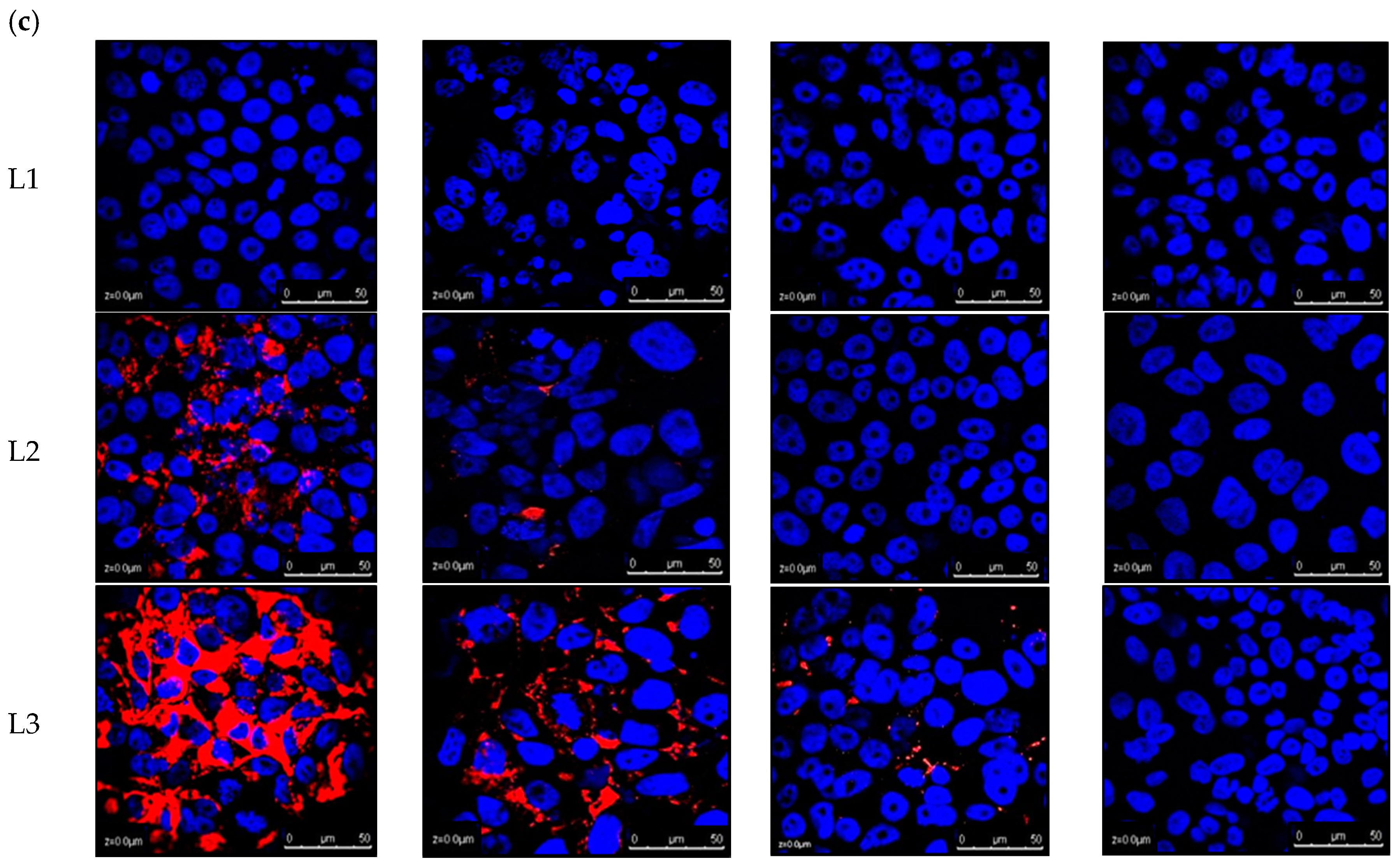
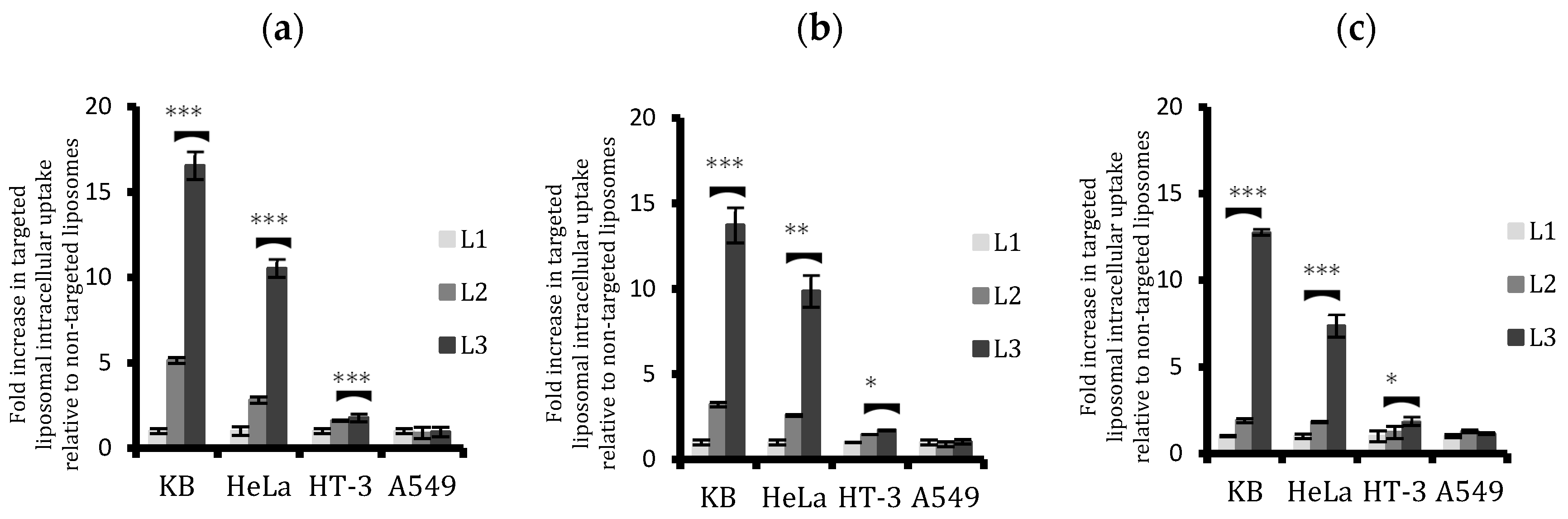
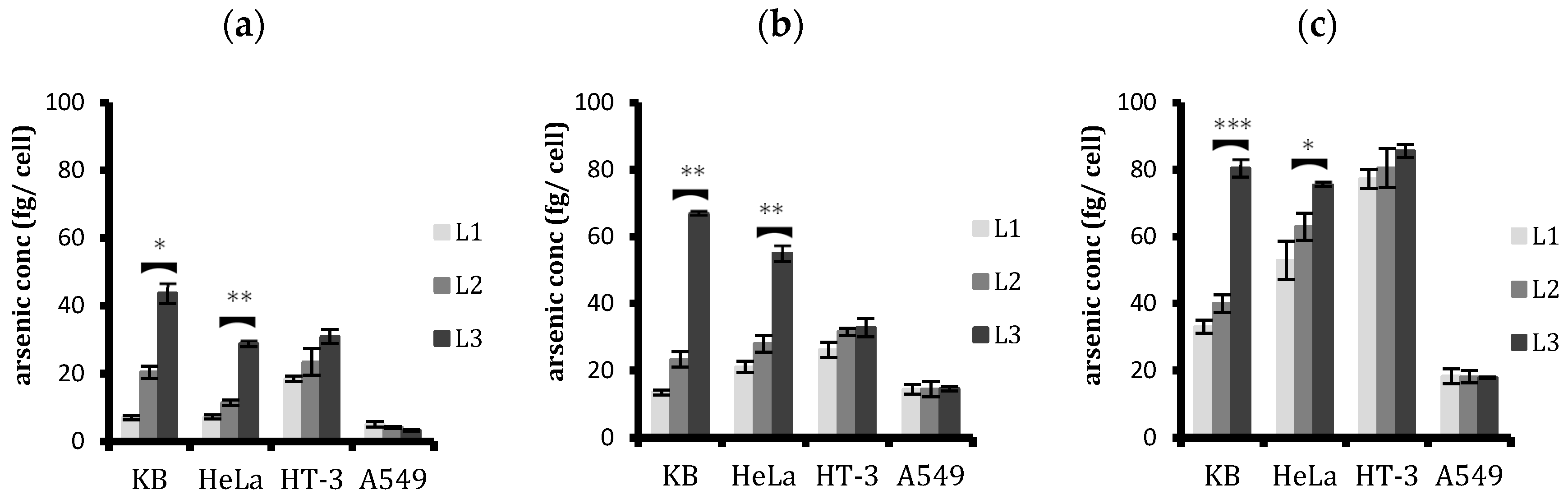
2.2. Analysing Cellular Uptake by Differing Ligand–PEG Spacer Lengths in ATO Liposomes
2.3. Analysing Cytotoxicity of Control Empty Liposomes with Differing Spacer Lengths of Conjugated Ligand
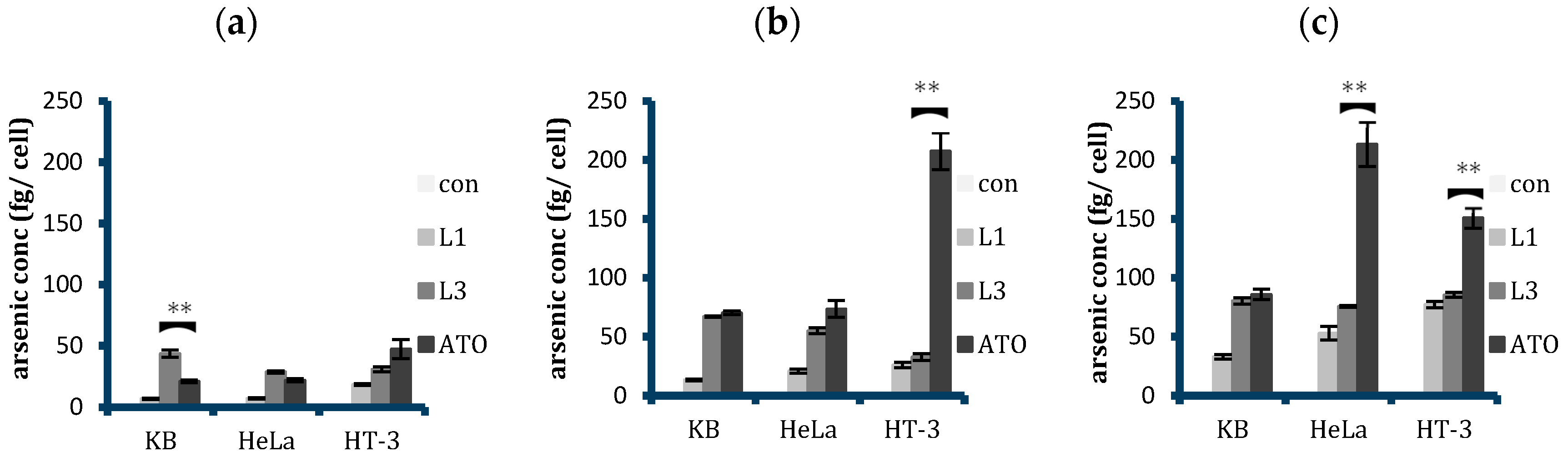
2.4. Quantitative Analysis of Cellular Uptake of Arsenic with Free and Liposomal Arsenic
2.5. Selectivity of Targeted Liposomal ATO in Killing HPV-Infected Cervical Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Liposome Preparation and Characterisation
4.3. Cell Culture
4.4. Qualitative Cellular Uptake Analysis by Confocal Microscopic Visualisation of Liposomal Arsenic
4.5. Flow Cytometric Analysis of Liposomal Arsenic Uptake
4.6. Plate Reader Analysis of Liposomal Uptake by Cells
4.7. Quantitative Analysis of Liposomal Drug Uptake via ICP-MS
4.8. In Vitro Cellular Cytotoxicity Assay
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| % | percentage |
| µg | microgram |
| µl | microliter |
| µM | micromolar |
| A549 | human lung epithelial cancer cell line |
| As | arsenic |
| ATO | arsenic trioxide |
| C | centigrade |
| Chol | cholesterol |
| CLSM | confocal laser scanning microscopy |
| Da | dalton |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DiI | 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine-5,5′-disulfonic acid |
| DLS | dynamic light scattering |
| DMSO | dimethyl sulfoxide |
| DNA | deoxyribonucleic acid |
| DSPE-PEG2000 | methoxypolyethyleneglycol-distearoyl-phosphatidylethanolamine with mPEG MW 2000 Da |
| DSPE-PEG2000-Folate | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-folate(polyethylene glycol)-2000 with mPEG MW2000 Da |
| DSPE-PEG5000-Folate | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-folate(polyethylene glycol)-5000 with mPEG MW5000 Da |
| EPR | enhanced permeability and retention effect |
| FA | folic acid |
| FACS | fluorescence activated cell sorting |
| FBS | fetal bovine serum |
| FITC | fluorescein isothiocyanate |
| fg | femtogram |
| FR | folate receptor |
| Ga | gallium |
| h | hour(s) |
| HeLa | human cervical epithelial cells of adenocarcinoma |
| HPV | human papilloma virus |
| HT-3 | human cervical cancer cell line (HPV-negative) |
| ICP-MS | inductively coupled plasma-mass spectrometer |
| ICP-OES | inductively coupled plasma-optical emission spectrometer |
| KB | human nasopharyngeal cell line contaminated with HeLa cells |
| kDa | kilodalton |
| Lipo | liposome |
| mAb | monoclonal antibody |
| min | minute(s) |
| mM | millimolar |
| mol | molarity |
| MPS | mononuclear phagocytic system |
| MTT | 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide |
| NaH2PO4 | sodium dihydrogen phosphate |
| Na2HPO4 | disodium hydrogen phosphate |
| Ni | nickle |
| Ni(OAc)2 | nickle acetate |
| nm | nanometre |
| P | phospholipids |
| PBS | phosphate buffered saline |
| PC | phosphatidyl choline |
| PEG | polyethylene glycol |
| PMT | photomultiplier tube |
| SD | standard deviation |
| UK | United Kingdom |
| USA | United States of America |
| v/v | volume by volume |
References
- Parkin, D.M.; Bray, F. The burden of HPV-related cancers. Vaccine 2006, 24, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Bosch, F.X.; Lorincz, A.; Munoz, N.; Meijer, C.; Shah, K.V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.F.; Peto, J.; Meijer, C.J.L.M.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Nair, S.; Pillai, M.R. Human papillomavirus and disease mechanisms: Relevance to oral and cervical cancers. Oral Dis. 2005, 11, 350–359. [Google Scholar] [CrossRef] [PubMed]
- NeufCoeur, P.E.; Arafa, M.; Delvenne, P.; Saussez, S. Involvement of human papillomavirus in upper aero-digestive tracts cancers. Bull. Cancer 2009, 96, 941–950. [Google Scholar] [PubMed]
- Shukla, S.; Bharti, A.C.; Mahata, S.; Hussain, S.; Kumar, R.; Hedau, S.; Das, B.C. Infection of human papillomaviruses in cancers of different human organ sites. Indian J. Med. Res. 2009, 130, 222–233. [Google Scholar]
- Steenbergen, R.D.; de Wilde, J.; Wilting, S.M.; Brink, A.A.; Snijders, P.J.; Meijer, C.J. HPV-mediated transformation of the anogenital tract. J. Clin. Virol. 2005, 32, 25–33. [Google Scholar] [CrossRef]
- Akhtar, A.; Wang, S.X.; Ghali, L.; Bell, C.; Wen, X. Effective Delivery of Arsenic Trioxide to HPV-Positive Cervical Cancer Cells Using Optimised Liposomes: A Size and Charge Study. Int. J. Mol. Sci. 2018, 19, 1081. [Google Scholar] [CrossRef]
- Um, S.J.; Lee, S.Y.; Kim, E.J.; Myoung, J.; Namkoong, S.E.; Park, J.S. Down-regulation of human papillomavirus E6/E7 oncogene by arsenic trioxide in cervical carcinoma cells. Cancer Lett. 2002, 181, 11–22. [Google Scholar] [CrossRef]
- Wen, X.; Li, D.; Zhang, Y.; Liu, S.; Ghali, L.; Iles, R.K. Arsenic trioxide induces cervical cancer apoptosis, but specifically targets human papillomavirus-infected cell populations. Anticancer Drugs 2012, 23, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Chen, Z. Acute promyelocytic leukemia: From highly fatal to highly curable. Blood 2008, 111, 2505–2515. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, Z.; Lallemand-Breitenbach, V.; de Thé, H. How acute promyelocytic leukaemia revived arsenic. Nat. Rev. Cancer 2002, 2, 705–7113. [Google Scholar] [CrossRef]
- Emadi, A.; Gore, S.D. Arsenic trioxide—An old drug rediscovered. Blood Rev. 2010, 24, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Larochette, N.; Decaudin, D.; Jacotot, E.; Brenner, C.; Marzo, I.; Susin, S.A.; Zamzami, N.; Xie, Z.; Reed, J.; Kroemer, G. Arsenite induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Exp. Cell Res. 1999, 249, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; de Thé, H. Arsenic trioxide, a novel mitochondriotoxic anticancer agent? J. Natl. Cancer Inst. 1999, 9, 743–745. [Google Scholar] [CrossRef]
- Platanias, L.C. Biological responses to arsenic compounds. J. Biol. Chem. 2009, 284, 18583–18587. [Google Scholar] [CrossRef] [PubMed]
- Dilda, P.J.; Hogg, P.J. Arsenical-based cancer drugs. Cancer Treat. Rev. 2007, 33, 542–564. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Pan, S.; Dong, X.; Qiao, H.; Jiang, H.; Krissansen, G.W.; Sun, X. Opposing effects of arsenic trioxide on hepatocellular carcinomas in mice. Cancer Sci. 2006, 97, 675–681. [Google Scholar] [CrossRef]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Cheng, Z.; Al Zaki, A.; Hui, J.Z.; Muzykantov, V.R.; Tsourkas, A. Multifunctional nanoparticles: Cost versus benefit of adding targeting and imaging capabilities. Science 2012, 338, 903–910. [Google Scholar] [CrossRef]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef]
- Koshkaryev, A.; Sawant, R.; Deshpande, M.; Torchilin, V. Immunoconjugates and long circulating systems: Origins, current state of the art and future directions. Adv. Drug Deliv. Rev. 2013, 65, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Xiao, Z.; Kamaly, N.; Farokhzad, O.C. Self-assembled targeted nanoparticles: Evolution of technologies and bench to bedside translation. Acc. Chem. Res. 2011, 44, 1123–1134. [Google Scholar] [CrossRef]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.J.; Low, P.S. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochimica et Biophysica Acta (BBA) Biomembranes 1995, 1233, 134–144. [Google Scholar] [CrossRef]
- Ni, S.; Stephenson, S.M.; Lee, R.J. Folate receptor targeted delivery of liposomal daunorubicin into tumor cells. Anticancer Res. 2002, 22, 2131–2135. [Google Scholar] [PubMed]
- Pan, X.Q.; Wang, H.; Shukla, S.; Sekido, M.; Adams, D.M.; Tjarks, W.; Barth, R.F.; Lee, R.J. Boron-containing folate receptor-targeted liposomes as potential delivery agents for neutron capture therapy. Bioconju. Chem. 2002, 13, 435–442. [Google Scholar] [CrossRef]
- Sudimack, J.; Lee, R.J. Targeted drug delivery via the folate receptor. Adv. Drug Deliv. Rev. 2000, 41, 147–162. [Google Scholar] [CrossRef]
- Werner, M.E.; Karve, S.; Sukumar, R.; Cummings, N.D.; Copp, J.A.; Chen, R.C.; Zhang, T.; Wang, A.Z. Folate-targeted nanoparticle delivery of chemo-and radiotherapeutics for the treatment of ovarian cancer peritoneal metastasis. Biomaterials 2011, 32, 8548–8554. [Google Scholar] [CrossRef]
- Zhao, X.B.; Lee, R.J. Tumor-selective targeted delivery of genes and antisense oligodeoxyribonucleotides via the folate receptor. Adv. Drug Deliv. Rev. 2004, 56, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Allen, T.M. Ligand-targeted liposomal anticancer drugs. Prog. Lipid Res. 2003, 42, 439–462. [Google Scholar] [CrossRef]
- Wang, M.; Thanou, M. Targeting nanoparticles to cancer. Pharmacol. Res. 2010, 62, 90–99. [Google Scholar] [CrossRef]
- Lee, R.J.; Low, P.S. Delivery of liposomes into cultured KB cells via folate receptor-mediated endocytosis. J. Biol. Chem. 1994, 269, 3198–3204. [Google Scholar]
- Gabizon, A.; Horowitz, A.T.; Goren, D.; Tzemach, D.; Mandelbaum-Shavit, F.; Qazen, M.M.; Zalipsky, S. Targeting folate receptor with folate linked to extremities of poly (ethylene glycol)-grafted liposomes: In vitro studies. Bioconjug. Chem. 1999, 10, 289–298. [Google Scholar] [CrossRef]
- Canal, F.; Vicent, M.J.; Pasut, G.; Schiavon, O. Relevance of folic acid/polymer ratio in targeted PEG-“epirubicin conjugates”. J. Control. Release 2010, 146, 388–399. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, L.; Dong, Y.; Zhang, X.; Lin, J.; Chen, Z. Folate-mediated poly (3-hydroxybutyrate-co-3-hydroxyoctanoate) nanoparticles for targeting drug delivery. Eur. J. Pharm. Biopharm. 2010, 76, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jia, J.; Lai, Y.; Ma, Y.; Weng, J.; Sun, L. Conjugating folic acid to gold nanoparticles through glutathione for targeting and detecting cancer cells. Bioorg. Med. Chem. 2010, 18, 5528–5534. [Google Scholar] [CrossRef]
- Peng, Z.H.; Sima, M.; Salama, M.E.; Kopečková, P.; Kopeček, J. Spacer length impacts the efficacy of targeted docetaxel conjugates in prostate-specific membrane antigen expressing prostate cancer. J. Drug Target. 2013, 21, 968–980. [Google Scholar] [CrossRef]
- Gu, F.; Zhang, L.; Teply, B.A.; Mann, N.; Wang, A.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc. Natl. Acad. Sci. USA 2008, 105, 2586–2591. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Kim, B.Y.S.; Rutka, J.T.; Chan, W.C.W. Nanoparticle-mediated cellular response is size-dependent. Nat. Nanotechnol. 2008, 3, 145–150. [Google Scholar] [CrossRef]
- Talekar, M.; Kendall, J.; Denny, W.; Garg, S. Targeting of nanoparticles in cancer: Drug delivery and diagnostics. Anticancer Drugs 2011, 22, 949–962. [Google Scholar] [CrossRef]
- Yu, B.O.; Tai, H.C.; Xue, W.; Lee, L.J.; Lee, R.J. Receptor-targeted nanocarriers for therapeutic delivery to cancer. Mol. Membr. Biol. 2010, 27, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, S.M.; Low, P.S.; Lee, R.J. Folate receptor-mediated targeting of liposomal drugs to cancer cells. Methods Enzymol. 2004, 387, 33–50. [Google Scholar] [PubMed]
- Reddy, J.A.; Allagadda, V.M.; Leamon, C.P. Targeting therapeutic and imaging agents to folate receptor positive tumors. Curr. Pharm. Biotechnol. 2005, 6, 131–150. [Google Scholar] [CrossRef]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Ãberg, C.; Mahon, E.; Dawson, K.A. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef]
- Holland, J.W.; Hui, C.; Cullis, P.R.; Madden, T.D. Poly(ethylene glycol)−lipid conjugates regulate the calcium-induced fusion of liposomes composed of phosphatidylethanolamine and phosphatidylserine. Biochemistry 1996, 35, 2618–2624. [Google Scholar] [CrossRef]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Weissleder, R.; Kelly, K.; Sun, E.Y.; Shtatland, T.; Josephson, L. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nat. Biotechnol. 2005, 23, 1418–1423. [Google Scholar] [CrossRef]
- Elias, D.R.; Poloukhtine, A.; Popik, V.; Tsourkas, A. Effect of ligand density, receptor density, and nanoparticle size on cell targeting. Nanomedicine 2013, 9, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.A.; Abburi, C.; Hofland, H.; Howard, S.J.; Vlahov, I.; Wils, P.; Leamon, C.P. Folate-targeted, cationic liposome-mediated gene transfer into disseminated peritoneal tumors. Gene Ther. 2002, 9, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Saul, J.M.; Annapragada, A.; Natarajan, J.V.; Bellamkonda, R.V. Controlled targeting of liposomal doxorubicin via the folate receptor in vitro. J. Control. Release 2003, 92, 49–67. [Google Scholar] [CrossRef]
- Pan, X.Q.; Wang, H.; Lee, R.J. Boron delivery to a murine lung carcinoma using folate receptor-targeted liposomes. Anticancer Res. 2002, 22, 1629–1633. [Google Scholar]
- Ohguchi, Y.; Kawano, K.; Hattori, Y.; Maitani, Y. Selective delivery of folate-PEG-linked, nanoemulsion-loaded aclacinomycin A to KB nasopharyngeal cells and xenograft: Effect of chain length and amount of folate-PEG linker. J. Drug Target. 2008, 16, 660–667. [Google Scholar] [CrossRef]
- Chen, H.; Pazicni, S.; Krett, N.L.; Ahn, R.W.; Penner-Hahn, J.E.; Rosen, S.T.; O’Halloran, T.V. Coencapsulation of Arsenic-and Platinum-based Drugs for Targeted Cancer Treatment. Angew. Chem. Int. Ed. 2009, 48, 9295–9299. [Google Scholar] [CrossRef]
- Suen, W.L.L.; Chau, Y. Size-dependent internalisation of folate-decorated nanoparticles via the pathways of clathrin and caveolae-mediated endocytosis in ARPE-19 cells. J. Pharm. Pharmacol. 2014, 66, 564–573. [Google Scholar] [CrossRef]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin-and caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef]
- Turek, J.J.; Leamon, C.P.; Low, P.S. Endocytosis of folate-protein conjugates: Ultrastructural localization in KB cells. J. Cell Sci. 1993, 106, 423–430. [Google Scholar]
- Kawano, K.; Maitani, Y. Effects of polyethylene glycol spacer length and ligand density on folate receptor targeting of liposomal Doxorubicin in vitro. J. Drug Deliv. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; MacDonald, R.C.; Li, S.; Krett, N.L.; Rosen, S.T.; O’Halloran, T.V. Lipid encapsulation of arsenic trioxide attenuates cytotoxicity and allows for controlled anticancer drug release. J. Am. Chem. Soc. 2006, 128, 13348–13349. [Google Scholar] [CrossRef] [PubMed]
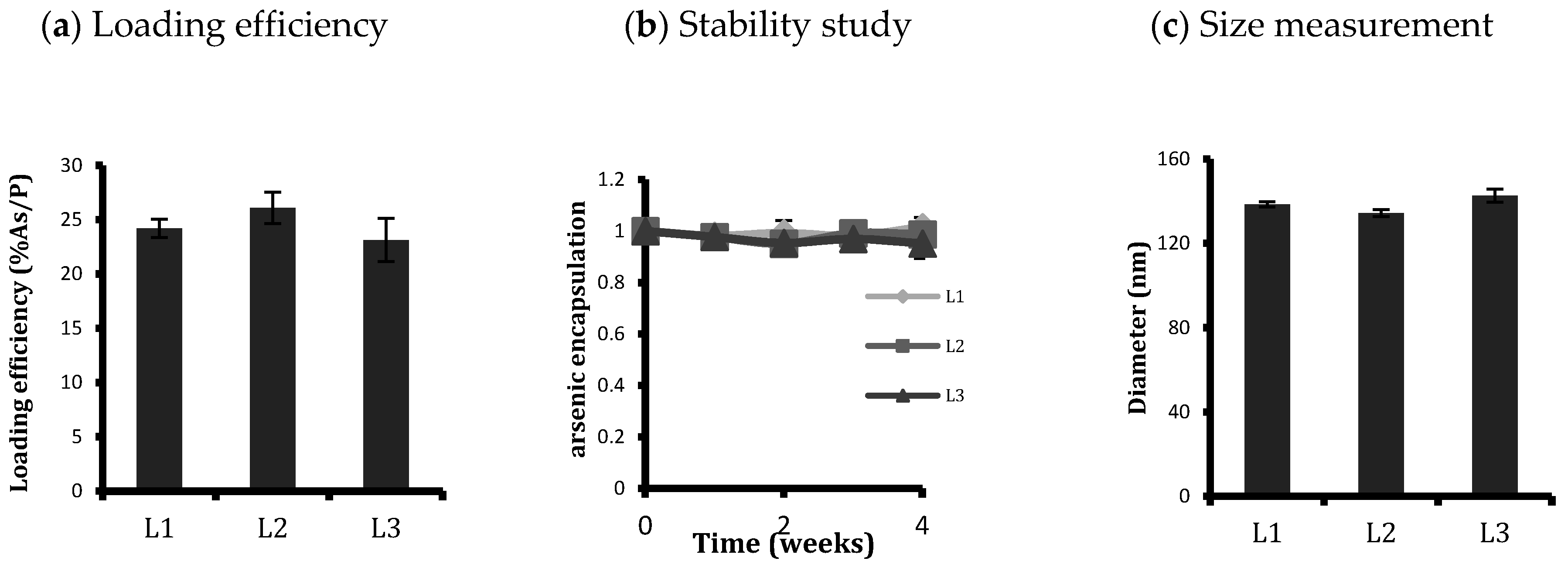

 represents the concentrations of phospholipid at dilutions used for treatment of liposomal ATO.
represents the concentrations of phospholipid at dilutions used for treatment of liposomal ATO.
represents the concentrations of phospholipid at dilutions used for treatment of liposomal ATO.
represents the concentrations of phospholipid at dilutions used for treatment of liposomal ATO.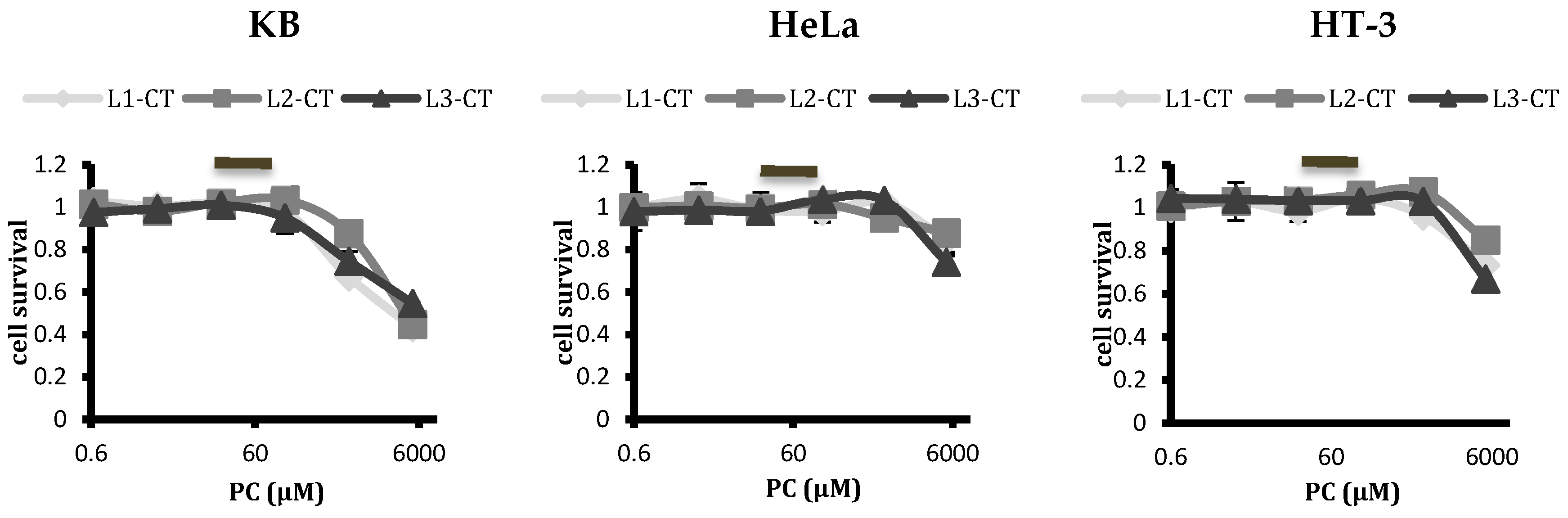
 points to 5 µM ATO concentrations that were typically used for treatment.
points to 5 µM ATO concentrations that were typically used for treatment.
points to 5 µM ATO concentrations that were typically used for treatment.
points to 5 µM ATO concentrations that were typically used for treatment.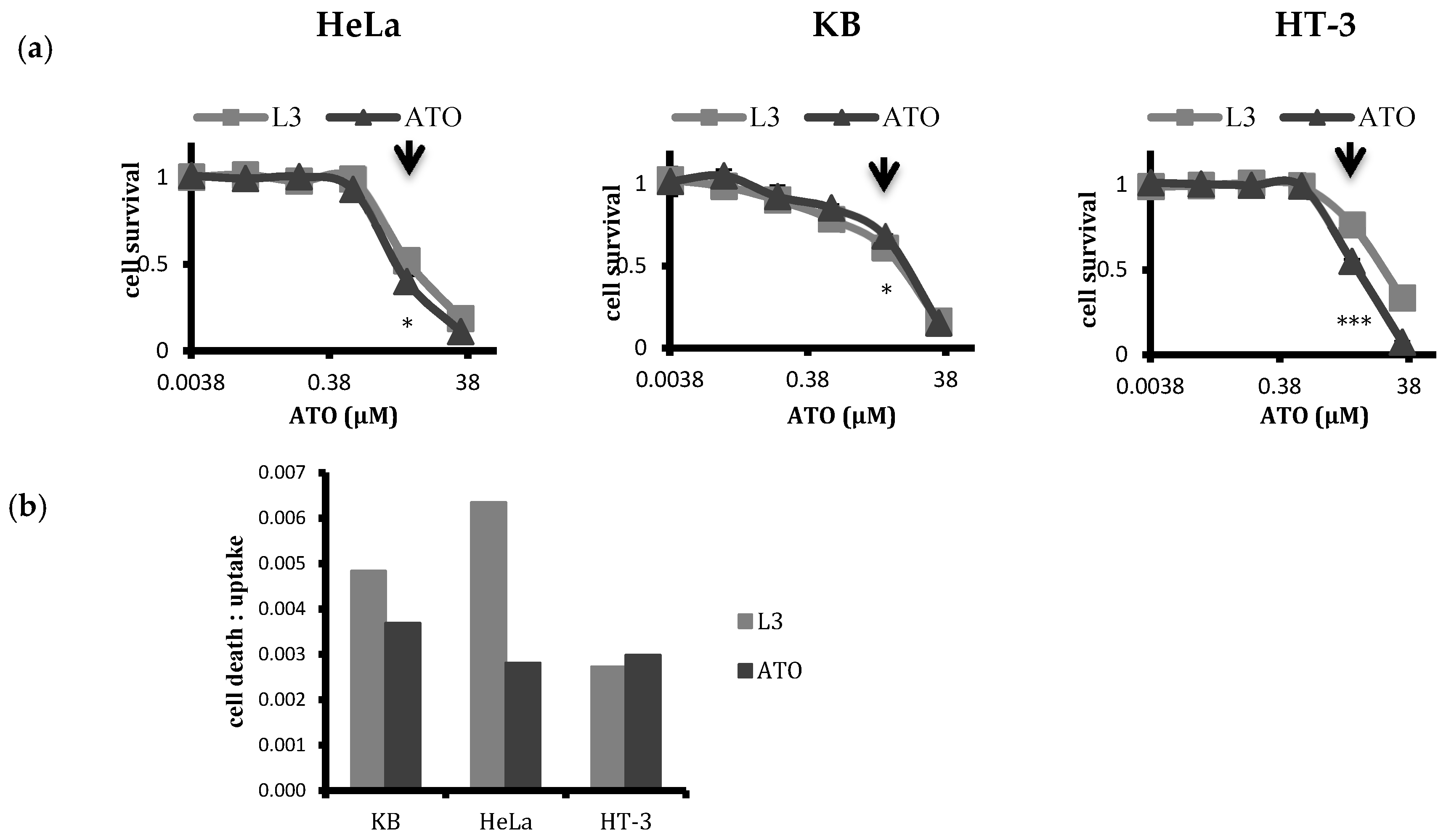
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhtar, A.; Ghali, L.; Wang, S.X.; Bell, C.; Li, D.; Wen, X. Optimisation of Folate-Mediated Liposomal Encapsulated Arsenic Trioxide for Treating HPV-Positive Cervical Cancer Cells In Vitro. Int. J. Mol. Sci. 2019, 20, 2156. https://doi.org/10.3390/ijms20092156
Akhtar A, Ghali L, Wang SX, Bell C, Li D, Wen X. Optimisation of Folate-Mediated Liposomal Encapsulated Arsenic Trioxide for Treating HPV-Positive Cervical Cancer Cells In Vitro. International Journal of Molecular Sciences. 2019; 20(9):2156. https://doi.org/10.3390/ijms20092156
Chicago/Turabian StyleAkhtar, Anam, Lucy Ghali, Scarlet Xiaoyan Wang, Celia Bell, Dong Li, and Xuesong Wen. 2019. "Optimisation of Folate-Mediated Liposomal Encapsulated Arsenic Trioxide for Treating HPV-Positive Cervical Cancer Cells In Vitro" International Journal of Molecular Sciences 20, no. 9: 2156. https://doi.org/10.3390/ijms20092156
APA StyleAkhtar, A., Ghali, L., Wang, S. X., Bell, C., Li, D., & Wen, X. (2019). Optimisation of Folate-Mediated Liposomal Encapsulated Arsenic Trioxide for Treating HPV-Positive Cervical Cancer Cells In Vitro. International Journal of Molecular Sciences, 20(9), 2156. https://doi.org/10.3390/ijms20092156