Deep Learning/Artificial Intelligence and Blood-Based DNA Epigenomic Prediction of Cerebral Palsy



 ,
, _Guda.png) and
and
Abstract
1. Introduction
2. Results
2.1. Identification of Differential Methylation between CP and Normal Controls
2.2. Newborn Prediction of Cerebral Palsy Using Deep Learning and Other ML Approaches
2.3. Pathway and Network Analyses
2.4. Validation by Pyrosequencing
3. Discussion
3.1. Genes in Axonal Guidance and Actin Cytoskeleton Signaling
3.2. Genes in Insulin Receptor and PI3K/AKT Signaling
3.3. Genes in TGF-β Signaling
3.4. miR-1469 in CP
3.5. Non-Coding RNAs and Small Nuclear RNAs
3.6. Limitations of the Study
3.7. Conclusions
4. Materials and Methods
4.1. Differential Methylation Assay
4.2. Epigenome-Wide Methylation Scan Using Illumina Methylation Arrays
4.3. Validation of Differential Methylation Analysis
4.4. Statistical and Bioinformatic Analysis
4.5. Partial Least Squares Discriminant Analysis (PLS-DA)
4.6. Differentially-Methylated Regions (DMRs) Analysis
4.7. Logistic Regression with AUC (95% CI), Sensitivity and Specificity
4.8. Gene Ontology and Pathway Analysis
4.9. Artificial Intelligence (AI) Analysis Method Data Preprocessing
4.10. Deep Learning (DL)
4.11. Other Machine Learning Algorithms
4.12. Modeling and Evaluation
4.13. CP Prediction Based on AI Analysis
- Epochs (number of passes of the full training set),
- l1 (penalty to converge the weights of the model to 0),
- l2 (penalty to prevent the enlargement of the weights),
- Input dropout ratio (ratio of ignored neurons in the input layer during training),
- Number of hidden layers;
4.14. Overfitting and Computation Time
4.15. Feature Importance
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lundy, C.; Lumsden, D.; Fairhurst, C. Treating complex movement disorders in children with cerebral palsy. Ulst. Med. J. 2009, 78, 157–163. [Google Scholar]
- Moreno-De-Luca, A.; Ledbetter, D.H.; Martin, C.L. Genetic [corrected] insights into the causes and classification of [corrected] cerebral palsies. Lancet Neurol. 2012, 11, 283–292. [Google Scholar] [CrossRef]
- Van Naarden Braun, K.; Doernberg, N.; Schieve, L.; Christensen, D.; Goodman, A.; Yeargin-Allsopp, M. Birth Prevalence of Cerebral Palsy: A Population-Based Study. Pediatrics 2016, 137. [Google Scholar] [CrossRef] [PubMed]
- Benda, W.; McGibbon, N.H.; Grant, K.L. Improvements in muscle symmetry in children with cerebral palsy after equine-assisted therapy (hippotherapy). J. Altern. Complement. Med. 2003, 9, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, L. Children with spastic cerebral palsy, their cognitive functioning, and social participation: A review. Child Neuropsychol. 2010, 16, 209–228. [Google Scholar] [CrossRef]
- Colver, A.; Fairhurst, C.; Pharoah, P.O. Cerebral palsy. Lancet 2014, 383, 1240–1249. [Google Scholar] [CrossRef]
- Zwaigenbaum, L. The intriguing relationship between cerebral palsy and autism. Dev. Med. Child Neurol. 2014, 56, 7–8. [Google Scholar] [CrossRef]
- Romeo, D.M.; Sini, F.; Brogna, C.; Albamonte, E.; Ricci, D.; Mercuri, E. Sex differences in cerebral palsy on neuromotor outcome: A critical review. Dev. Med. Child Neurol. 2016, 58, 809–813. [Google Scholar] [CrossRef]
- Wu, Y.W.; Xing, G.; Fuentes-Afflick, E.; Danielson, B.; Smith, L.H.; Gilbert, W.M. Racial, ethnic, and socioeconomic disparities in the prevalence of cerebral palsy. Pediatrics 2011, 127, e674–e681. [Google Scholar] [CrossRef]
- Shamsoddini, A.; Amirsalari, S.; Hollisaz, M.T.; Rahimnia, A.; Khatibi-Aghda, A. Management of spasticity in children with cerebral palsy. Iran J. Pediatr. 2014, 24, 345–351. [Google Scholar] [PubMed]
- MacLennan, A.H.; Thompson, S.C.; Gecz, J. Cerebral palsy: Causes, pathways, and the role of genetic variants. Am. J. Obs. Gynecol. 2015, 213, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Spittle, A.J.; Morgan, C.; Olsen, J.E.; Novak, I.; Cheong, J.L.Y. Early Diagnosis and Treatment of Cerebral Palsy in Children with a History of Preterm Birth. Clin. Perinatol. 2018, 45, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.; Fahey, M.; Roy, B.; Novak, I. Diagnosing cerebral palsy in full-term infants. J. Paediatr. Child Health 2018, 54, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Fleiss, B.; Gressens, P. Tertiary mechanisms of brain damage: A new hope for treatment of cerebral palsy? Lancet Neurol. 2012, 11, 556–566. [Google Scholar] [CrossRef]
- Schoendorfer, N.C.; Obeid, R.; Moxon-Lester, L.; Sharp, N.; Vitetta, L.; Boyd, R.N.; Davies, P.S. Methylation capacity in children with severe cerebral palsy. Eur. J. Clin. Investig. 2012, 42, 768–776. [Google Scholar] [CrossRef]
- Mohandas, N.; Bass-Stringer, S.; Maksimovic, J.; Crompton, K.; Loke, Y.J.; Walstab, J.; Reid, S.M.; Amor, D.J.; Reddihough, D.; Craig, J.M. Epigenome-wide analysis in newborn blood spots from monozygotic twins discordant for cerebral palsy reveals consistent regional differences in DNA methylation. Clin. Epigenet. 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Jun, S.; Cho, Y.W.; Lee, H.; Kim, G.B.; Seo, J.B.; Kim, N. Deep Learning in Medical Imaging: General Overview. Korean J. Radiol. 2017, 18, 570–584. [Google Scholar] [CrossRef]
- Grapov, D.; Fahrmann, J.; Wanichthanarak, K.; Khoomrung, S. Rise of Deep Learning for Genomic, Proteomic, and Metabolomic Data Integration in Precision Medicine. OMICS 2018, 22, 630–636. [Google Scholar] [CrossRef]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef]
- Angermueller, C.; Parnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef]
- van Eyk, C.L.; Corbett, M.A.; Gardner, A.; van Bon, B.W.; Broadbent, J.L.; Harper, K.; MacLennan, A.H.; Gecz, J. Analysis of 182 cerebral palsy transcriptomes points to dysregulation of trophic signalling pathways and overlap with autism. Transl. Psychiatry 2018, 8, 88. [Google Scholar] [CrossRef]
- Botzki, A.; Kreft, Ł.; Soete, A.; Hulpiau, P.; De Bleser, P.; Saeys, Y. ConTra v3: A tool to identify transcription factor binding sites across species, update 2017. Nucleic Acids Res. 2017, 45, W490–W494. [Google Scholar]
- Novak, I.; Morgan, C.; Adde, L.; Blackman, J.; Boyd, R.N.; Brunstrom-Hernandez, J.; Cioni, G.; Damiano, D.; Darrah, J.; Eliasson, A.C.; et al. Early, Accurate Diagnosis and Early Intervention in Cerebral Palsy: Advances in Diagnosis and Treatment. JAMA Pediatr. 2017, 171, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Onishi, K.; Hollis, E.; Zou, Y. Axon guidance and injury-lessons from Wnts and Wnt signaling. Curr. Opin. Neurobiol. 2014, 27, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Boitard, M.; Bocchi, R.; Egervari, K.; Petrenko, V.; Viale, B.; Gremaud, S.; Zgraggen, E.; Salmon, P.; Kiss, J.Z. Wnt signaling regulates multipolar-to-bipolar transition of migrating neurons in the cerebral cortex. Cell Rep. 2015, 10, 1349–1361. [Google Scholar] [CrossRef]
- Tsutsui, Y.; Nagahama, M.; Mizutani, A. Neuronal migration disorders in cerebral palsy. Neuropathology 1999, 19, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Houlihan, C.M.; Stevenson, R.D. Bone density in cerebral palsy. Phys. Med. Rehabil. Clin. N. Am. 2009, 20, 493–508. [Google Scholar] [CrossRef]
- Fontaine, R.; Mesples, B.; Lelievre, V.; Gressens, P. 125 TGF-Beta-1 Mediates IL-9/Mast Cells Interactions in a Mouse Model of Periventricular Leukomalacia. Pediatr. Res. 2005, 58, 376. [Google Scholar] [CrossRef][Green Version]
- Kawaguchi, N.; Sundberg, C.; Kveiborg, M.; Moghadaszadeh, B.; Asmar, M.; Dietrich, N.; Thodeti, C.K.; Nielsen, F.C.; Moller, P.; Mercurio, A.M.; et al. ADAM12 induces actin cytoskeleton and extracellular matrix reorganization during early adipocyte differentiation by regulating beta1 integrin function. J. Cell Sci. 2003, 116, 3893–3904. [Google Scholar] [CrossRef]
- Kruer, M.C.; Jepperson, T.; Dutta, S.; Steiner, R.D.; Cottenie, E.; Sanford, L.; Merkens, M.; Russman, B.S.; Blasco, P.A.; Fan, G.; et al. Mutations in gamma adducin are associated with inherited cerebral palsy. Ann. Neurol. 2013, 74, 805–814. [Google Scholar] [CrossRef]
- Sunmonu, N.A.; Li, K.; Li, J.Y. Numerous isoforms of Fgf8 reflect its multiple roles in the developing brain. J. Cell Physiol. 2011, 226, 1722–1726. [Google Scholar] [CrossRef]
- Peterson, M.D.; Gordon, P.M.; Hurvitz, E.A.; Burant, C.F. Secondary muscle pathology and metabolic dysregulation in adults with cerebral palsy. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1085–E1093. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, C.; Kahn, C.R. Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arter. Thromb. Vasc. Biol. 2012, 32, 2052–2059. [Google Scholar] [CrossRef]
- Mullonkal, C.J.; Toledo-Pereyra, L.H. Akt in ischemia and reperfusion. J. Investig. Surg. 2007, 20, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Babcock, M.A.; Kostova, F.V.; Ferriero, D.M.; Johnston, M.V.; Brunstrom, J.E.; Hagberg, H.; Maria, B.L. Injury to the preterm brain and cerebral palsy: Clinical aspects, molecular mechanisms, unanswered questions, and future research directions. J. Child Neurol. 2009, 24, 1064–1084. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, W.-C.; Séjourné, J.; Clipperton-Allen, A.E.; Page, D.T. Pten Mutations Alter Brain Growth Trajectory and Allocation of Cell Types through Elevated β-Catenin Signaling. J. Neurosci. 2015, 35, 10252–10267. [Google Scholar] [CrossRef]
- Ismail, A.; Ning, K.; Al-Hayani, A.; Sharrack, B.; Azzouz, M. PTEN: A molecular target for neurodegeneratIve disorders. Transl. Neurosci. 2012, 3, 132–142. [Google Scholar] [CrossRef]
- Charles, M.S.; Drunalini Perera, P.N.; Doycheva, D.M.; Tang, J. Granulocyte-colony stimulating factor activates JAK2/PI3K/PDE3B pathway to inhibit corticosterone synthesis in a neonatal hypoxic-ischemic brain injury rat model. Exp. Neurol. 2015, 272, 152–159. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Habarou, F.; Brassier, A.; Rio, M.; Chretien, D.; Monnot, S.; Barbier, V.; Barouki, R.; Bonnefont, J.P.; Boddaert, N.; Chadefaux-Vekemans, B.; et al. Pyruvate carboxylase deficiency: An underestimated cause of lactic acidosis. Mol. Genet. Metab. Rep. 2015, 2, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Lissens, W.; Vreken, P.; Barth, P.G.; Wijburg, F.A.; Ruitenbeek, W.; Wanders, R.J.; Seneca, S.; Liebaers, I.; De Meirleir, L. Cerebral palsy and pyruvate dehydrogenase deficiency: Identification of two new mutations in the E1alpha gene. Eur. J. Pediatr. 1999, 158, 853–857. [Google Scholar] [CrossRef]
- Dobolyi, A.; Vincze, C.; Pal, G.; Lovas, G. The neuroprotective functions of transforming growth factor beta proteins. Int. J. Mol. Sci. 2012, 13, 8219–8258. [Google Scholar] [CrossRef] [PubMed]
- Kulak-Bejda, A.; Kulak, P.; Bejda, G.; Krajewska-Kulak, E.; Kulak, W. Stem cells therapy in cerebral palsy: A systematic review. Brain Dev. 2016, 38, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Macias, M.J.; Martin-Malpartida, P.; Massague, J. Structural determinants of Smad function in TGF-beta signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef]
- Krishnan, V.; Ito, Y. RUNX3 loss turns on the dark side of TGF-beta signaling. Oncoscience 2017, 4, 156–157. [Google Scholar] [PubMed]
- Park, B.Y.; Saint-Jeannet, J.P. Expression analysis of Runx3 and other Runx family members during Xenopus development. Gene Expr. Patterns 2010, 10, 159–166. [Google Scholar] [CrossRef][Green Version]
- Greenberg, D.S.; Soreq, H. MicroRNA therapeutics in neurological disease. Curr. Pharm. Des. 2014, 20, 6022–6027. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kwon, E.J.; Tsai, L.H. MicroRNAs in learning, memory, and neurological diseases. Learn Mem. 2012, 19, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Diaz, M.; Miranda-Roman, M.A.; Soto, D.; Quintero-Aguilo, M.; Ortiz-Zuazaga, H.; Marcos-Martinez, M.J.; Vivas-Mejia, P.E. MicroRNA-27a distinguishes glioblastoma multiforme from diffuse and anaplastic astrocytomas and has prognostic value. Am. J. Cancer Res. 2015, 5, 201–218. [Google Scholar] [PubMed]
- Freischmidt, A.; Muller, K.; Zondler, L.; Weydt, P.; Volk, A.E.; Bozic, A.L.; Walter, M.; Bonin, M.; Mayer, B.; von Arnim, C.A.; et al. Serum microRNAs in patients with genetic amyotrophic lateral sclerosis and pre-manifest mutation carriers. Brain 2014, 137, 2938–2950. [Google Scholar] [CrossRef]
- Kan, A.A.; van Erp, S.; Derijck, A.A.; de Wit, M.; Hessel, E.V.; O’Duibhir, E.; de Jager, W.; Van Rijen, P.C.; Gosselaar, P.H.; de Graan, P.N.; et al. Genome-wide microRNA profiling of human temporal lobe epilepsy identifies modulators of the immune response. Cell. Mol. Life Sci. 2012, 69, 3127–3145. [Google Scholar] [CrossRef] [PubMed]
- de la Morena, M.T.; Eitson, J.L.; Dozmorov, I.M.; Belkaya, S.; Hoover, A.R.; Anguiano, E.; Pascual, M.V.; van Oers, N.S. Signature MicroRNA expression patterns identified in humans with 22q11.2 deletion/DiGeorge syndrome. Clin. Immunol. 2013, 147, 11–22. [Google Scholar] [CrossRef]
- Santosh, P.S.; Arora, N.; Sarma, P.; Pal-Bhadra, M.; Bhadra, U. Interaction map and selection of microRNA targets in Parkinson’s disease-related genes. J. Biomed. Biotechnol. 2009, 2009, 363145. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S. The State of Long Non-Coding RNA Biology. Noncoding Rna 2018, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Valadkhan, S.; Gunawardane, L.S. Role of small nuclear RNAs in eukaryotic gene expression. Essays Biochem. 2013, 54, 79–90. [Google Scholar] [CrossRef]
- Radhakrishna, U.; Albayrak, S.; Alpay-Savasan, Z.; Zeb, A.; Turkoglu, O.; Sobolewski, P.; Bahado-Singh, R.O. Genome-Wide DNA Methylation Analysis and Epigenetic Variations Associated with Congenital Aortic Valve Stenosis (AVS). PLoS ONE 2016, 11, e0154010. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Zaffra, R.; Albayarak, S.; Chelliah, A.; Bolinjkar, R.; Turkoglu, O.; Radhakrishna, U. Epigenetic markers for newborn congenital heart defect (CHD). J Matern Fetal Neonatal Med 2016, 29, 1881–1887. [Google Scholar] [CrossRef]
- Liu, Y.; Aryee, M.J.; Padyukov, L.; Fallin, M.D.; Hesselberg, E.; Runarsson, A.; Reinius, L.; Acevedo, N.; Taub, M.; Ronninger, M.; et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013, 31, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, L.; Chen, L.; Ren, W.; Mei, A.; Chen, X.; Deng, Y. Two novel mutations of the NCSTN gene in Chinese familial acne inverse. J. Eur. Acad. Derm. Venereol. 2013, 27, 1571–1574. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm-Benartzi, C.S.; Koestler, D.C.; Karagas, M.R.; Flanagan, J.M.; Christensen, B.C.; Kelsey, K.T.; Marsit, C.J.; Houseman, E.A.; Brown, R. Review of processing and analysis methods for DNA methylation array data. Br. J. Cancer 2013, 109, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Daca-Roszak, P.; Pfeifer, A.; Zebracka-Gala, J.; Rusinek, D.; Szybinska, A.; Jarzab, B.; Witt, M.; Zietkiewicz, E. Impact of SNPs on methylation readouts by Illumina Infinium HumanMethylation450 BeadChip Array: Implications for comparative population studies. BMC Genom. 2015, 16, 1003. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Jannot, A.S.; Ehret, G.; Perneger, T. P < 5 × 10(-8) has emerged as a standard of statistical significance for genome-wide association studies. J. Clin. Epidemiol. 2015, 68, 460–465. [Google Scholar] [PubMed]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, T.R.; Shihab, H.A.; Hemani, G.; Min, J.L.; Woodward, G.; Lyttleton, O.; Zheng, J.; Duggirala, A.; McArdle, W.L.; Ho, K.; et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016, 17, 61. [Google Scholar] [CrossRef]
- Alakwaa, F.M.; Chaudhary, K.; Garmire, L.X. Deep Learning Accurately Predicts Estrogen Receptor Status in Breast Cancer Metabolomics Data. J. Proteome Res. 2018, 17, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Candel, A.; Parmar, V.; LeDell, E.; Arora, A. Deep Learning with H2O. Available online: http://h2o.ai/resources/ (accessed on 27 April 2019).
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]




{kind=link}
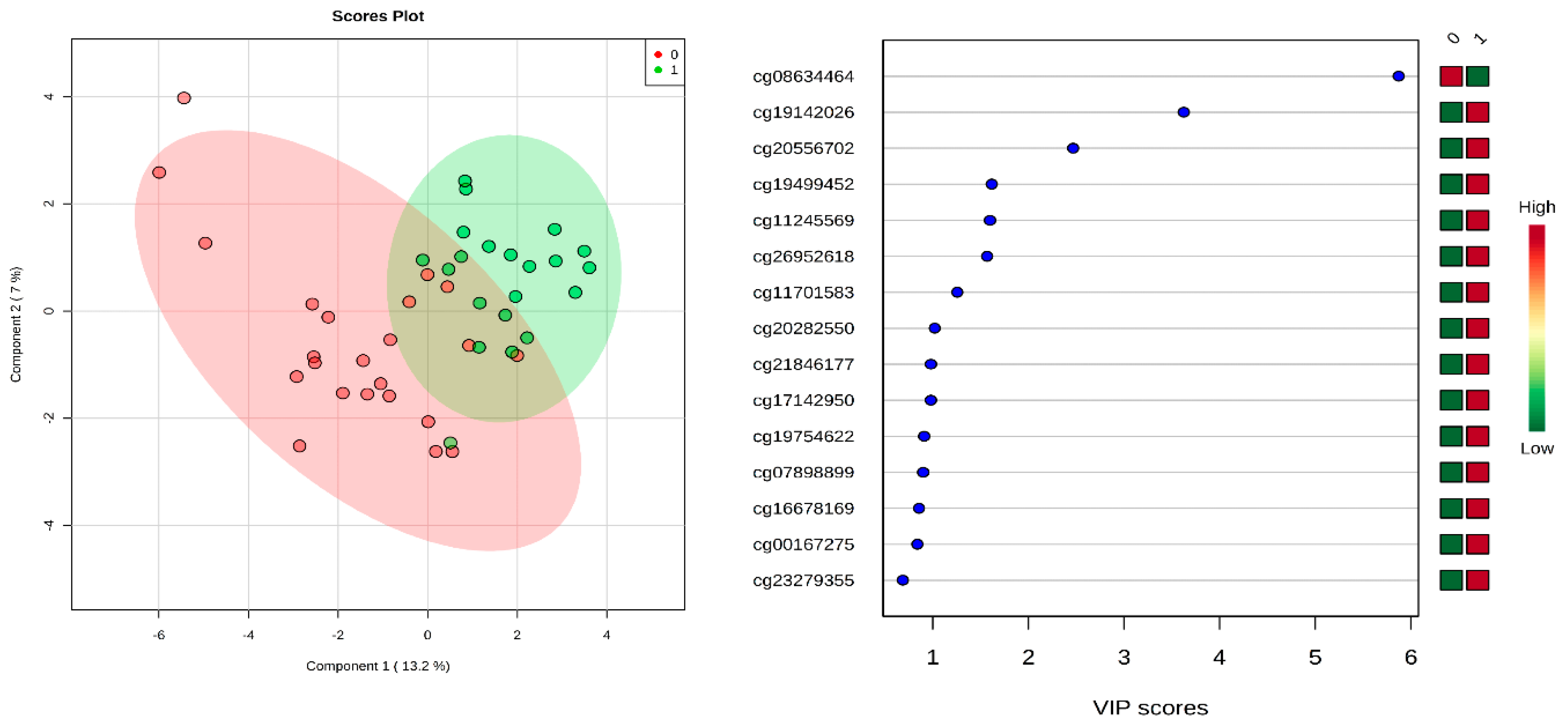{kind=link}
{kind=link}
| Target ID | Chr | Gene | FDR p-Val | Fold Change | % Methylation | AUC | CI | ||
|---|---|---|---|---|---|---|---|---|---|
| Cases | Control | Lower | Upper | ||||||
| cg13187827 | 6 | C6orf27 | 4.56 × 10-28 | 0.47 | 12.88 | 27.47 | 0.94 | 0.86 | 1 |
| cg01561596 | 13 | UFM1 | 0.00296 | 0.43 | 1.57 | 3.67 | 0.91 | 0.82 | 1 |
| cg03586379 | 3 | SLC25A36 | 1.02 × 10−5 | 0.41 | 2.33 | 5.64 | 0.91 | 0.82 | 1 |
| cg08052428 | 9 | RALGDS | 1.53 × 10−8 | 0.48 | 4.66 | 9.63 | 0.90 | 0.8 | 1 |
| cg07898899 | 1 | S100A13 | 3.72 × 10−20 | 0.42 | 7.11 | 16.87 | 0.89 | 0.79 | 0.99 |
| cg17142950 | 1 | SAMD13 | 1.33 × 10−30 | 0.44 | 12.21 | 27.61 | 0.88 | 0.77 | 0.98 |
| cg20376421 | 12 | MYL6B | 4.40 × 10−7 | 0.49 | 4.14 | 8.41 | 0.88 | 0.78 | 0.99 |
| cg10230427 | 6 | BAG2 | 6.70 × 10−12 | 0.41 | 4.22 | 10.24 | 0.87 | 0.76 | 0.98 |
| cg14347670 | 6 | CCND3 | 5.68 × 10−8 | 0.4 | 2.81 | 7.07 | 0.87 | 0.75 | 0.98 |
| cg20640432 | 19 | CREB3L3 | 0.00015 | 0.5 | 2.91 | 5.86 | 0.87 | 0.75 | 0.98 |
| cg00472801 | 6 | KHDRBS2 | 8.40 × 10−7 | 0.5 | 4.08 | 8.23 | 0.86 | 0.74 | 0.97 |
| cg03307401 | 19 | KLK13 | 0.00017 | 0.36 | 1.45 | 4.09 | 0.86 | 0.74 | 0.97 |
| cg11961138 | 17 | IGFBP4 | 2.48 × 10−21 | 0.39 | 6.14 | 15.87 | 0.86 | 0.74 | 0.97 |
| cg12204727 | 15 | COMMD4 | 0.02176 | 0.5 | 1.63 | 3.27 | 0.86 | 0.75 | 0.97 |
| cg12206423 | 13 | SLITRK5 | 0.00012 | 0.49 | 2.91 | 5.9 | 0.86 | 0.74 | 0.97 |
| cg17852224 | 22 | MAPK8IP2 | 1.45 × 10−11 | 0.47 | 5.51 | 11.83 | 0.86 | 0.74 | 0.97 |
| cg20871904 | 4 | YTHDC1 | 3.95 × 10−5 | 0.47 | 2.75 | 5.92 | 0.86 | 0.74 | 0.97 |
| cg26707202 | 4 | SMAD1 | 1.68 × 10−6 | 0.42 | 2.66 | 6.35 | 0.86 | 0.74 | 0.97 |
| cg01067849 | 6 | WRNIP1 | 0.00058 | 0.42 | 1.76 | 4.23 | 0.85 | 0.73 | 0.97 |
| cg02782426 | 3 | ENTPD3 | 1.94 × 10−7 | 0.47 | 3.9 | 8.26 | 0.85 | 0.74 | 0.97 |
| cg03433549 | 12 | PA2G4 | 0.00456 | 0.47 | 1.86 | 3.91 | 0.85 | 0.73 | 0.97 |
| cg08931196 | 11 | RNF26 | 0.03450 | 0.47 | 1.33 | 2.81 | 0.85 | 0.73 | 0.97 |
| cg15277906 | 8 | GDF6 | 0.00073 | 0.5 | 2.5 | 5.05 | 0.85 | 0.73 | 0.97 |
| cg20810398 | 1 | EXOSC10 | 0.04950 | 0.48 | 1.27 | 2.64 | 0.85 | 0.73 | 0.97 |
| cg22624212 | 21 | WDR4 | 0.00137 | 0.43 | 1.75 | 4.04 | 0.85 | 0.73 | 0.97 |
| SVM | GLM | PAM | RF | LDA | DL | |
|---|---|---|---|---|---|---|
| AUC 95% CI | 0.9875 (0.6875–1) | 0.9765 (0.6765–1) | 0.8468 (0.6468–1) | 0.9087 (0.6087–1) | 0.9675 (0.6675–1) | 0.9760 (0.6760–1) |
| Sensitivity | 0.9200 | 0.8500 | 0.7500 | 0.7500 | 0.8000 | 0.9500 |
| Specificity | 0.9200 | 0.8500 | 0.9000 | 0.9000 | 0.9000 | 0.9440 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Deep Learning/Artificial Intelligence and Blood-Based DNA Epigenomic Prediction of Cerebral Palsy. Int. J. Mol. Sci. 2019, 20, 2075. https://doi.org/10.3390/ijms20092075
Bahado-Singh RO, Vishweswaraiah S, Aydas B, Mishra NK, Guda C, Radhakrishna U. Deep Learning/Artificial Intelligence and Blood-Based DNA Epigenomic Prediction of Cerebral Palsy. International Journal of Molecular Sciences. 2019; 20(9):2075. https://doi.org/10.3390/ijms20092075
Chicago/Turabian StyleBahado-Singh, Ray O., Sangeetha Vishweswaraiah, Buket Aydas, Nitish Kumar Mishra, Chittibabu Guda, and Uppala Radhakrishna. 2019. "Deep Learning/Artificial Intelligence and Blood-Based DNA Epigenomic Prediction of Cerebral Palsy" International Journal of Molecular Sciences 20, no. 9: 2075. https://doi.org/10.3390/ijms20092075
APA StyleBahado-Singh, R. O., Vishweswaraiah, S., Aydas, B., Mishra, N. K., Guda, C., & Radhakrishna, U. (2019). Deep Learning/Artificial Intelligence and Blood-Based DNA Epigenomic Prediction of Cerebral Palsy. International Journal of Molecular Sciences, 20(9), 2075. https://doi.org/10.3390/ijms20092075