Alternative Splicing in Angiogenesis



Abstract
1. Alternative Splicing
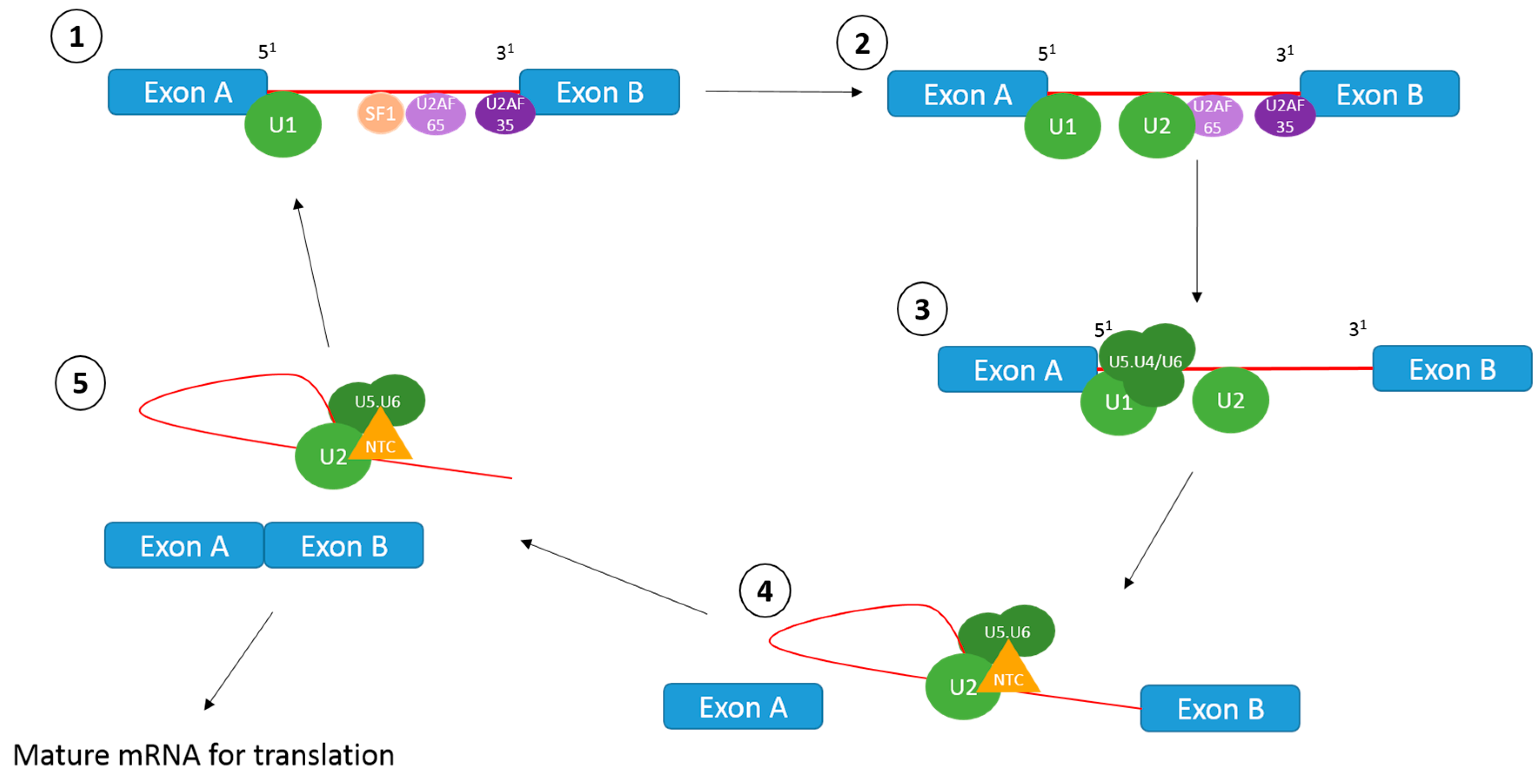
1.1. The Splicing Reaction
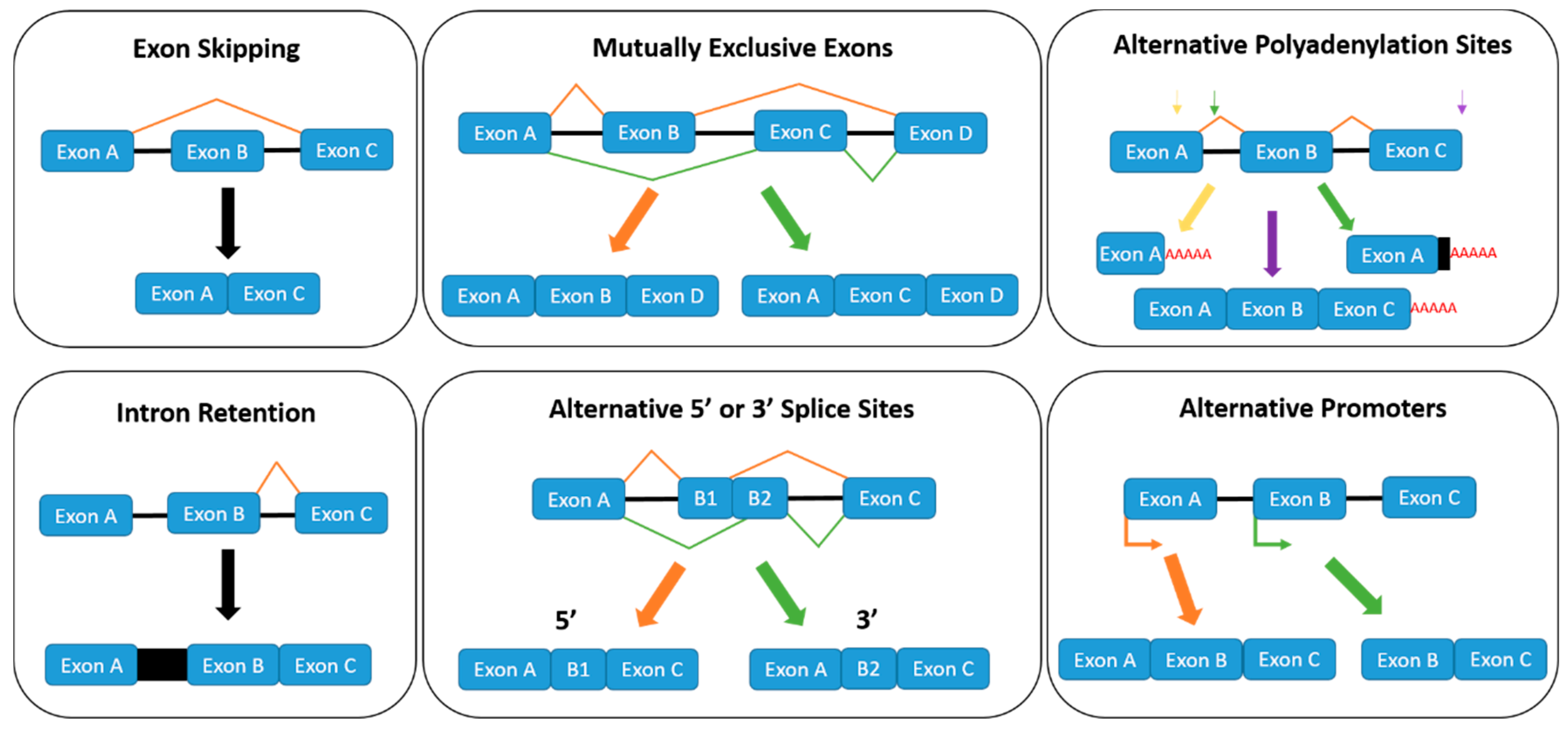
1.2. Regulation of Alternative Splicing
2. Angiogenesis
Angiogenic Vessel Formation
3. Splicing in Angiogenesis
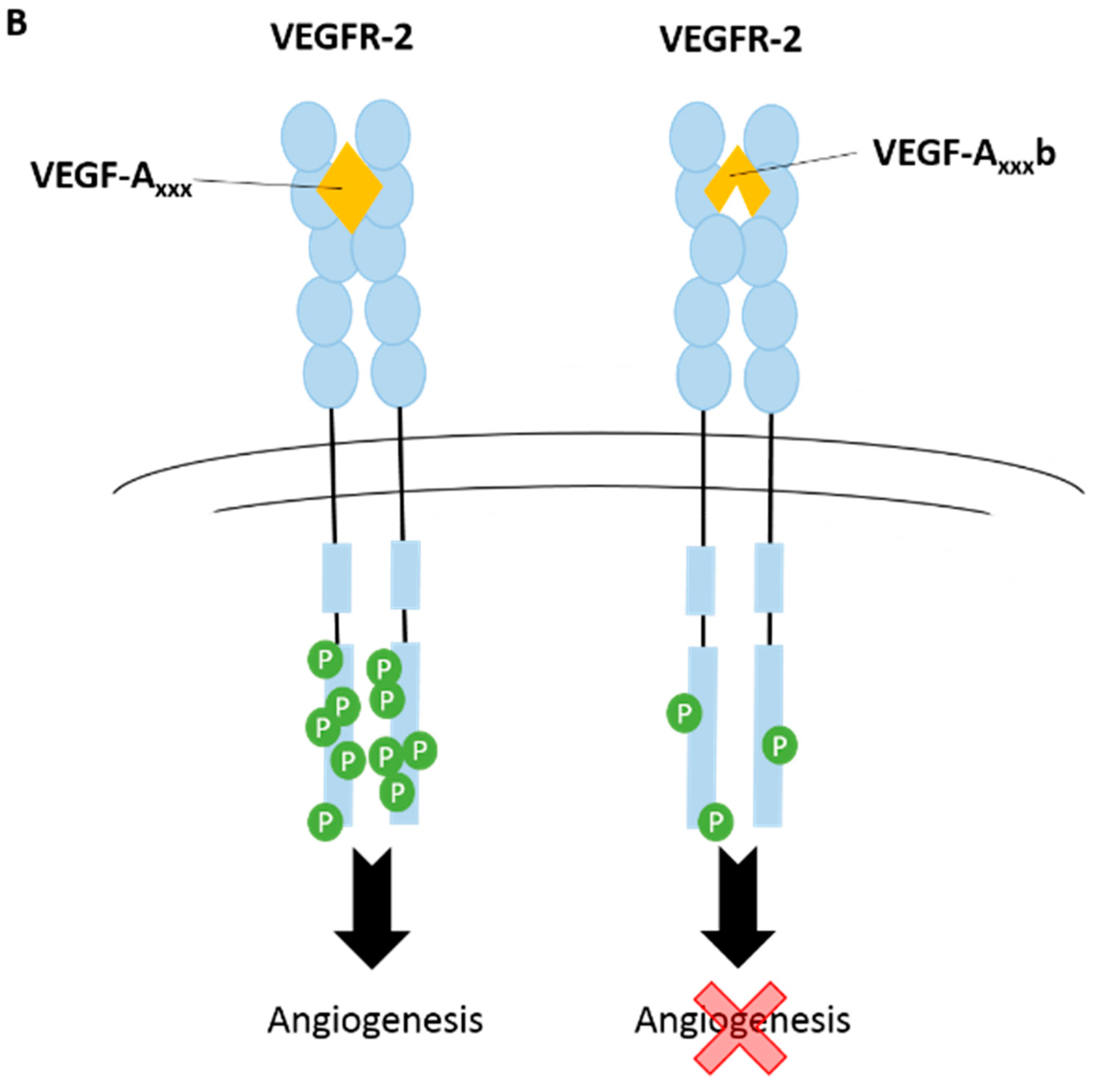
3.1. Vascular Endothelial Growth Factor-A (VEGF-A)
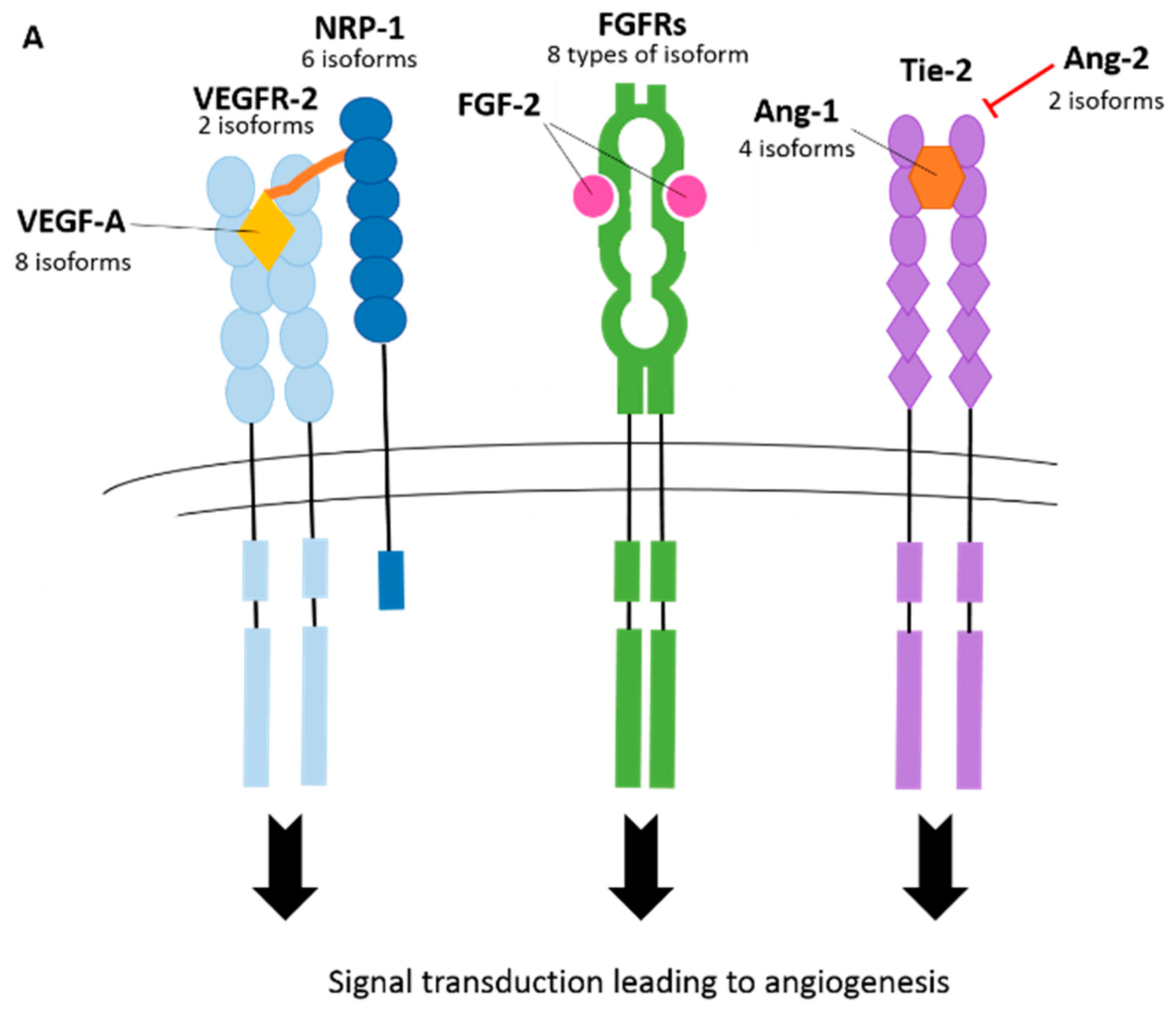
3.2. Vascular Endothelial Growth Factor Receptors (VEGFRs)
3.3. Neuropilins (NRPs)
3.4. Fibroblast Growth Factor Receptors (FGFRs)
3.5. Vasohibins
3.6. Hypoxia Inducible Factor-α (HIF-α)
3.7. Angiopoietin
4. Manipulation of Alternative Splicing as a Potential Therapy for Angiogenic Associated Diseases
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Salzberg, S.L. Open questions: How many genes do we have? BMC Biol. 2018, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef] [PubMed]
- Mthembu, N.N.; Mbita, Z.; Hull, R.; Dlamini, Z. Abnormalities in alternative splicing of angiogenesis-related genes and their role in HIV-related cancers. HIV/AIDS 2017, 9, 77–93. [Google Scholar] [CrossRef]
- Kornblihtt, A.R.; Schor, I.E.; Allo, M.; Dujardin, G.; Petrillo, E.; Munoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nature reviews. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar]
- Lim, K.H.; Ferraris, L.; Filloux, M.E.; Raphael, B.J.; Fairbrother, W.G. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc. Natl. Acad. Sci. USA 2011, 108, 11093–11098. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef]
- Coltri, P.P.; Dos Santos, M.G.P.; da Silva, G.H.G. Splicing and cancer: Challenges and opportunities. RNA 2019, e1527. [Google Scholar] [CrossRef]
- Bowler, E. The Effect of Hypoxia on Alternative Splicing in Prostate Cancer Cell Lines. Ph.D. Thesis, University of the West of England, Bristol, UK, 2017. [Google Scholar]
- Manley, J.L.; Krainer, A.R. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 2010, 24, 1073–1074. [Google Scholar] [CrossRef]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nature reviews. Genetics 2014, 15, 689–701. [Google Scholar]
- Yamazaki, T.; Liu, L.; Lazarev, D.; Al-Zain, A.; Fomin, V.; Yeung, P.L.; Chambers, S.M.; Lu, C.W.; Studer, L.; Manley, J.L. TCF3 alternative splicing controlled by hnRNP H/F regulates E-cadherin expression and hESC pluripotency. Genes Dev. 2018, 32, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Comiskey, D.F., Jr.; Jacob, A.G.; Singh, R.K.; Tapia-Santos, A.S.; Chandler, D.S. Splicing factor SRSF1 negatively regulates alternative splicing of MDM2 under damage. Nucleic Acids Res. 2015, 43, 4202–4218. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Akerman, M.; Clery, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef] [PubMed]
- de Miguel, F.J.; Sharma, R.D.; Pajares, M.J.; Montuenga, L.M.; Rubio, A.; Pio, R. Identification of alternative splicing events regulated by the oncogenic factor SRSF1 in lung cancer. Cancer Res. 2014, 74, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Aslanzadeh, V.; Papa, F.; Zhu, H.; de la Grange, P.; Cambi, F. Global profiling of alternative splicing events and gene expression regulated by hnRNPH/F. PLoS ONE 2012, 7, e51266. [Google Scholar] [CrossRef]
- Kedzierska, H.; Piekielko-Witkowska, A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017, 396, 53–65. [Google Scholar] [CrossRef]
- Howard, J.M.; Sanford, J.R. The RNAissance family: SR proteins as multifaceted regulators of gene expression. RNA 2015, 6, 93–110. [Google Scholar] [CrossRef]
- Serrano, P.; Aubol, B.E.; Keshwani, M.M.; Forli, S.; Ma, C.T.; Dutta, S.K.; Geralt, M.; Wuthrich, K.; Adams, J.A. Directional Phosphorylation and Nuclear Transport of the Splicing Factor SRSF1 Is Regulated by an RNA Recognition Motif. J. Mol. Biol. 2016, 428, 2430–2445. [Google Scholar] [CrossRef]
- Naro, C.; Sette, C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int. J. Cell Biol. 2013, 2013, 151839. [Google Scholar] [CrossRef]
- Ikeda, T.; Yoshitomi, Y.; Saito, H.; Shimasaki, T.; Yamaya, H.; Kobata, T.; Ishigaki, Y.; Tomosugi, N.; Yoshitake, Y.; Yonekura, H. Regulation of soluble Flt-1 (VEGFR-1) production by hnRNP D and protein arginine methylation. Mol. Cell. Biochem. 2016, 413, 155–164. [Google Scholar] [CrossRef]
- Failla, C.M.; Carbo, M.; Morea, V. Positive and Negative Regulation of Angiogenesis by Soluble Vascular Endothelial Growth Factor Receptor-1. Int. J. Mol. Sci. 2018, 19, 1306. [Google Scholar] [CrossRef] [PubMed]
- Otrock, Z.K.; Mahfouz, R.A.; Makarem, J.A.; Shamseddine, A.I. Understanding the biology of angiogenesis: Review of the most important molecular mechanisms. Blood Cells Mol. Dis. 2007, 39, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Djokovic, D.; Calhaz-Jorge, C. Angiogenesis as a therapeutic target in endometriosis. Acta Medica Portuguesa 2014, 27, 489–497. [Google Scholar] [CrossRef][Green Version]
- Truby, L.K.; Topkara, V.K. Angiopoietin-2: Marker or mediator of angiogenesis in continuous-flow left ventricular assist device patients? J. Thor. Dis. 2016, 8, 3042–3045. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Choi, J.; Yang, M.J.; Hong, S.P.; Lee, C.K.; Kubota, Y.; Lim, D.S.; Koh, G.Y. A MST1-FOXO1 cascade establishes endothelial tip cell polarity and facilitates sprouting angiogenesis. Nat. Commun. 2019, 10, 838. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Cantu Gutierrez, M.; Dang, L.T.; Khyzha, N.; Chen, Z.; Veitch, S.; Cheng, H.S.; Khor, M.; Antounians, L.; Njock, M.S.; et al. Dynamic regulation of VEGF-inducible genes by an ERK/ERG/p300 transcriptional network. Development 2017, 144, 2428–2444. [Google Scholar] [CrossRef]
- Coma, S.; Allard-Ratick, M.; Akino, T.; van Meeteren, L.A.; Mammoto, A.; Klagsbrun, M. GATA2 and Lmo2 control angiogenesis and lymphangiogenesis via direct transcriptional regulation of neuropilin-2. Angiogenesis 2013, 16, 939–952. [Google Scholar] [CrossRef]
- Chang, S.H.; Hla, T. Post-transcriptional gene regulation by HuR and microRNAs in angiogenesis. Curr. Opin. Hematol. 2014, 21, 235–240. [Google Scholar] [CrossRef]
- Kir, D.; Schnettler, E.; Modi, S.; Ramakrishnan, S. Regulation of angiogenesis by microRNAs in cardiovascular diseases. Angiogenesis 2018, 21, 699–710. [Google Scholar] [CrossRef]
- Jo, D.H.; Kim, J.H.; Kim, K.W.; Suh, Y.G.; Kim, J.H. Allosteric regulation of pathologic angiogenesis: Potential application for angiogenesis-related blindness. Arch. Pharm. Res. 2014, 37, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.G.; Woolard, J.; Amin, E.M.; Konopatskaya, O.; Saleem, M.A.; Churchill, A.J.; Ladomery, M.R.; Harper, S.J.; Bates, D.O. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 2008, 121, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [PubMed]
- Krilleke, D.; DeErkenez, A.; Schubert, W.; Giri, I.; Robinson, G.S.; Ng, Y.S.; Shima, D.T. Molecular mapping and functional characterization of the VEGF164 heparin-binding domain. J. Biol. Chem. 2007, 282, 28045–28056. [Google Scholar] [CrossRef] [PubMed]
- Houck, K.A.; Leung, D.W.; Rowland, A.M.; Winer, J.; Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 1992, 267, 26031–26037. [Google Scholar] [PubMed]
- Houck, K.A.; Ferrara, N.; Winer, J.; Cachianes, G.; Li, B.; Leung, D.W. The vascular endothelial growth factor family: Identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol. Endocrinol. 1991, 5, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Mavrou, A.; Qiu, Y.; Carter, J.G.; Hamdollah-Zadeh, M.; Barratt, S.; Gammons, M.V.; Millar, A.B.; Salmon, A.H.; Oltean, S.; et al. Detection of VEGF-A(xxx)b isoforms in human tissues. PLoS ONE 2013, 8, e68399. [Google Scholar] [CrossRef] [PubMed]
- Pavlakovic, H.; Becker, J.; Albuquerque, R.; Wilting, J.; Ambati, J. Soluble VEGFR-2: An antilymphangiogenic variant of VEGF receptors. Ann. N. Y. Acad. Sci. 2010, 1207, E7–E15. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.L.; Bielenberg, D.R.; Gechtman, Z.; Miao, H.Q.; Takashima, S.; Soker, S.; Klagsbrun, M. Identification of a natural soluble neuropilin-1 that binds vascular endothelial growth factor: In vivo expression and antitumor activity. Proc. Natl. Acad. Sci. USA 2000, 97, 2573–2578. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Spring, S.C.; Terman, B.I. Characterization of a new alternatively spliced neuropilin-1 isoform. Angiogenesis 2003, 6, 39–45. [Google Scholar] [CrossRef]
- Hendricks, C.; Dubail, J.; Brohee, L.; Delforge, Y.; Colige, A.; Deroanne, C. A Novel Physiological Glycosaminoglycan-Deficient Splice Variant of Neuropilin-1 Is Anti-Tumorigenic In Vitro and In Vivo. PLoS ONE 2016, 11, e0165153. [Google Scholar] [CrossRef]
- Gong, S.G. Isoforms of receptors of fibroblast growth factors. J. Cell. Physiol. 2014, 229, 1887–1895. [Google Scholar] [CrossRef]
- Ishiwata, T. Role of fibroblast growth factor receptor-2 splicing in normal and cancer cells. Front. Biosci. 2018, 23, 626–639. [Google Scholar] [CrossRef]
- Johnston, C.L.; Cox, H.C.; Gomm, J.J.; Coombes, R.C. Fibroblast growth factor receptors (FGFRs) localize in different cellular compartments. A splice variant of FGFR-3 localizes to the nucleus. J. Boil. Chem. 1995, 270, 30643–30650. [Google Scholar] [CrossRef]
- Barnard, J.C.; Williams, A.J.; Rabier, B.; Chassande, O.; Samarut, J.; Cheng, S.Y.; Bassett, J.H.; Williams, G.R. Thyroid hormones regulate fibroblast growth factor receptor signaling during chondrogenesis. Endocrinology 2005, 146, 5568–5580. [Google Scholar] [CrossRef]
- Cha, J.Y.; Maddileti, S.; Mitin, N.; Harden, T.K.; Der, C.J. Aberrant receptor internalization and enhanced FRS2-dependent signaling contribute to the transforming activity of the fibroblast growth factor receptor 2 IIIb C3 isoform. J. Biol. Chem. 2009, 284, 6227–6240. [Google Scholar] [CrossRef]
- Twigg, S.R.; Burns, H.D.; Oldridge, M.; Heath, J.K.; Wilkie, A.O. Conserved use of a non-canonical 5′ splice site (/GA) in alternative splicing by fibroblast growth factor receptors 1, 2 and 3. Hum. Mol. Genet. 1998, 7, 685–691. [Google Scholar] [CrossRef]
- Kern, J.; Bauer, M.; Rychli, K.; Wojta, J.; Ritsch, A.; Gastl, G.; Gunsilius, E.; Untergasser, G. Alternative splicing of vasohibin-1 generates an inhibitor of endothelial cell proliferation, migration, and capillary tube formation. Arterioscl. Thromb. Vascular Biol. 2008, 28, 478–484. [Google Scholar] [CrossRef]
- Horie, S.; Suzuki, Y.; Kobayashi, M.; Kadonosono, T.; Kondoh, S.; Kodama, T.; Sato, Y. Distinctive role of vasohibin-1A and its splicing variant vasohibin-1B in tumor angiogenesis. Cancer Gene Ther. 2016, 23, 133–141. [Google Scholar] [CrossRef]
- Sato, Y.; Sonoda, H. The vasohibin family: A negative regulatory system of angiogenesis genetically programmed in endothelial cells. Arterioscl. Thromb. Vascular Biol. 2007, 27, 37–41. [Google Scholar] [CrossRef]
- Chun, Y.S.; Lee, K.H.; Choi, E.; Bae, S.Y.; Yeo, E.J.; Huang, L.E.; Kim, M.S.; Park, J.W. Phorbol ester stimulates the nonhypoxic induction of a novel hypoxia-inducible factor 1alpha isoform: Implications for tumor promotion. Cancer Res. 2003, 63, 8700–8707. [Google Scholar]
- Lee, K.H.; Park, J.W.; Chun, Y.S. Non-hypoxic transcriptional activation of the aryl hydrocarbon receptor nuclear translocator in concert with a novel hypoxia-inducible factor-1alpha isoform. Nucleic Acids Res. 2004, 32, 5499–5511. [Google Scholar] [CrossRef][Green Version]
- Gothie, E.; Richard, D.E.; Berra, E.; Pages, G.; Pouyssegur, J. Identification of alternative spliced variants of human hypoxia-inducible factor-1α. J. Biol. Chem. 2000, 275, 6922–6927. [Google Scholar] [CrossRef]
- Ando, H.; Natsume, A.; Iwami, K.; Ohka, F.; Kuchimaru, T.; Kizaka-Kondoh, S.; Ito, K.; Saito, K.; Sugita, S.; Hoshino, T.; et al. A hypoxia-inducible factor (HIF)-3alpha splicing variant, HIF-3α4 impairs angiogenesis in hypervascular malignant meningiomas with epigenetically silenced HIF-3α4. Biochem. Biophys. Res. Commun. 2013, 433, 139–144. [Google Scholar] [CrossRef]
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001, 414, 550–554. [Google Scholar] [CrossRef]
- Huang, Y.Q.; Li, J.J.; Karpatkin, S. Identification of a family of alternatively spliced mRNA species of angiopoietin-1. Blood 2000, 95, 1993–1999. [Google Scholar]
- Mezquita, J.; Mezquita, B.; Pau, M.; Mezquita, C. Characterization of a novel form of angiopoietin-2 (Ang-2B) and expression of VEGF and angiopoietin-2 during chicken testicular development and regression. Biochem. Biophys. Res. Commun. 1999, 260, 492–498. [Google Scholar] [CrossRef]
- Kim, I.; Kim, J.H.; Ryu, Y.S.; Jung, S.H.; Nah, J.J.; Koh, G.Y. Characterization and expression of a novel alternatively spliced human angiopoietin-2. J. Biol. Chem. 2000, 275, 18550–18556. [Google Scholar] [CrossRef]
- Gacche, R.N.; Meshram, R.J. Angiogenic factors as potential drug target: Efficacy and limitations of anti-angiogenic therapy. Biochimi. Biophys. Acta 2014, 1846, 161–179. [Google Scholar] [CrossRef]
- Berse, B.; Brown, L.F.; Van de Water, L.; Dvorak, H.F.; Senger, D.R. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol. Biol. Cell 1992, 3, 211–220. [Google Scholar] [CrossRef]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef]
- Panoilia, E.; Schindler, E.; Samantas, E.; Aravantinos, G.; Kalofonos, H.P.; Christodoulou, C.; Patrinos, G.P.; Friberg, L.E.; Sivolapenko, G. A pharmacokinetic binding model for bevacizumab and VEGF165 in colorectal cancer patients. Cancer Chemother. Pharmacol. 2015, 75, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Gitay-Goren, H.; Sharon, R.; Shibuya, M.; Halaban, R.; Levi, B.Z.; Neufeld, G. VEGF121, a vascular endothelial growth factor (VEGF) isoform lacking heparin binding ability, requires cell-surface heparan sulfates for efficient binding to the VEGF receptors of human melanoma cells. J. Biol. Chem. 1995, 270, 11322–11326. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Nagane, M.; Huang, H.S.; Cavenee, W.K. Intracerebral tumor-associated hemorrhage caused by overexpression of the vascular endothelial growth factor isoforms VEGF121 and VEGF165 but not VEGF189. Proc. Natl. Acad. Sci. USA 1997, 94, 12081–12087. [Google Scholar] [CrossRef] [PubMed]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Huang, G.; Zhou, Z.; Wang, H.; Kleinerman, E.S. CAPER-α alternative splicing regulates the expression of vascular endothelial growth factor165 in Ewing sarcoma cells. Cancer 2012, 118, 2106–2116. [Google Scholar] [CrossRef]
- Dowhan, D.H.; Hong, E.P.; Auboeuf, D.; Dennis, A.P.; Wilson, M.M.; Berget, S.M.; O’Malley, B.W. Steroid hormone receptor coactivation and alternative RNA splicing by U2AF65-related proteins CAPERα and CAPERβ. Mol. Cell 2005, 17, 429–439. [Google Scholar] [CrossRef]
- Jafarifar, F.; Yao, P.; Eswarappa, S.M.; Fox, P.L. Repression of VEGFA by CA-rich element-binding microRNAs is modulated by hnRNP L. EMBO J. 2011, 30, 1324–1334. [Google Scholar] [CrossRef]
- Yao, P.; Wu, J.; Lindner, D.; Fox, P.L. Interplay between miR-574-3p and hnRNP L regulates VEGFA mRNA translation and tumorigenesis. Nucleic Acids Res. 2017, 45, 7950–7964. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.S.; Jia, J.; Yao, P.; Majumder, M.; Hatzoglou, M.; Fox, P.L. A stress-responsive RNA switch regulates VEGFA expression. Nature 2009, 457, 915. [Google Scholar] [CrossRef]
- Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar]
- Rennel, E.S.; Varey, A.H.; Churchill, A.J.; Wheatley, E.R.; Stewart, L.; Mather, S.; Bates, D.O.; Harper, S.J. VEGF(121)b, a new member of the VEGF(xxx)b family of VEGF-A splice isoforms, inhibits neovascularisation and tumour growth in vivo. Br. J. Cancer 2009, 101, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Miller-Kasprzak, E.; Jagodzinski, P.P. 5-Aza-2′-deoxycytidine increases the expression of anti-angiogenic vascular endothelial growth factor 189b variant in human lung microvascular endothelial cells. Biomed. Pharmacother. 2008, 62, 158–163. [Google Scholar] [CrossRef]
- Perrin, R.M.; Konopatskaya, O.; Qiu, Y.; Harper, S.; Bates, D.O.; Churchill, A.J. Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor. Diabetologia 2005, 48, 2422–2427. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.G.; Amin, E.M.; Rennel, E.S.; Hoareau-Aveilla, C.; Gammons, M.; Damodoran, G.; Hagiwara, M.; Harper, S.J.; Woolard, J.; Ladomery, M.R.; et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: A novel therapeutic strategy for angiogenesis. J. Biol. Chem. 2010, 285, 5532–5540. [Google Scholar] [CrossRef]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Biselli-Chicote, P.M.; Oliveira, A.R.; Pavarino, E.C.; Goloni-Bertollo, E.M. VEGF gene alternative splicing: Pro- and anti-angiogenic isoforms in cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 363–370. [Google Scholar] [CrossRef]
- Oltean, S.; Gammons, M.; Hulse, R.; Hamdollah-Zadeh, M.; Mavrou, A.; Donaldson, L.; Salmon, A.H.; Harper, S.J.; Ladomery, M.R.; Bates, D.O. SRPK1 inhibition in vivo: Modulation of VEGF splicing and potential treatment for multiple diseases. Biochem. Soc. Trans. 2012, 40, 831–835. [Google Scholar] [CrossRef]
- Kikuchi, R.; Nakamura, K.; MacLauchlan, S.; Ngo, D.T.; Shimizu, I.; Fuster, J.J.; Katanasaka, Y.; Yoshida, S.; Qiu, Y.; Yamaguchi, T.P.; et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med. 2014, 20, 1464–1471. [Google Scholar] [CrossRef]
- Merdzhanova, G.; Gout, S.; Keramidas, M.; Edmond, V.; Coll, J.L.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. The transcription factor E2F1 and the SR protein SC35 control the ratio of pro-angiogenic versus antiangiogenic isoforms of vascular endothelial growth factor-A to inhibit neovascularization in vivo. Oncogene 2010, 29, 5392–5403. [Google Scholar] [CrossRef]
- Witmer, A.N.; Dai, J.; Weich, H.A.; Vrensen, G.F.; Schlingemann, R.O. Expression of vascular endothelial growth factor receptors 1, 2, and 3 in quiescent endothelia. J. Histochem. Cytochem. 2002, 50, 767–777. [Google Scholar] [CrossRef]
- Kabrun, N.; Buhring, H.J.; Choi, K.; Ullrich, A.; Risau, W.; Keller, G. Flk-1 expression defines a population of early embryonic hematopoietic precursors. Development 1997, 124, 2039–2048. [Google Scholar] [PubMed]
- Ishida, A.; Murray, J.; Saito, Y.; Kanthou, C.; Benzakour, O.; Shibuya, M.; Wijelath, E.S. Expression of vascular endothelial growth factor receptors in smooth muscle cells. J. Cell. Physiol. 2001, 188, 359–368. [Google Scholar] [CrossRef]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Fearnley, G.W.; Tomlinson, D.C.; Harrison, M.A.; Ponnambalam, S. The cellular response to vascular endothelial growth factors requires co-ordinated signal transduction, trafficking and proteolysis. Biosci. Rep. 2015, 35, 253. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.D.; Mohammadi, M.; Rahimi, N. A single amino acid substitution in the activation loop defines the decoy characteristic of VEGFR-1/FLT-1. J. Biol. Chem. 2006, 281, 867–875. [Google Scholar] [CrossRef]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C.H. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994, 269, 26988–26995. [Google Scholar]
- Sarabipour, S.; Ballmer-Hofer, K.; Hristova, K. VEGFR-2 conformational switch in response to ligand binding. eLife 2016, 5, e13876. [Google Scholar] [CrossRef]
- Fearnley, G.W.; Smith, G.A.; Abdul-Zani, I.; Yuldasheva, N.; Mughal, N.A.; Homer-Vanniasinkam, S.; Kearney, M.T.; Zachary, I.C.; Tomlinson, D.C.; Harrison, M.A.; et al. VEGF-A isoforms program differential VEGFR2 signal transduction, trafficking and proteolysis. Biol. Open 2016, 5, 571–583. [Google Scholar] [CrossRef]
- Woolard, J.; Wang, W.Y.; Bevan, H.S.; Qiu, Y.; Morbidelli, L.; Pritchard-Jones, R.O.; Cui, T.G.; Sugiono, M.; Waine, E.; Perrin, R.; et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: Mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004, 64, 7822–7835. [Google Scholar] [CrossRef]
- Cebe Suarez, S.; Pieren, M.; Cariolato, L.; Arn, S.; Hoffmann, U.; Bogucki, A.; Manlius, C.; Wood, J.; Ballmer-Hofer, K. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell. Mol. Life Sci. 2006, 63, 2067–2077. [Google Scholar] [CrossRef]
- Kawamura, H.; Li, X.; Harper, S.J.; Bates, D.O.; Claesson-Welsh, L. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 2008, 68, 4683–4692. [Google Scholar] [CrossRef]
- Pan, Q.; Chathery, Y.; Wu, Y.; Rathore, N.; Tong, R.K.; Peale, F.; Bagri, A.; Tessier-Lavigne, M.; Koch, A.W.; Watts, R.J. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J. Biol. Chem. 2007, 282, 24049–24056. [Google Scholar] [CrossRef]
- Fearnley, G.W.; Odell, A.F.; Latham, A.M.; Mughal, N.A.; Bruns, A.F.; Burgoyne, N.J.; Homer-Vanniasinkam, S.; Zachary, I.C.; Hollstein, M.C.; Wheatcroft, S.B.; et al. VEGF-A isoforms differentially regulate ATF-2-dependent VCAM-1 gene expression and endothelial-leukocyte interactions. Mol. Boil. Cell 2014, 25, 2509–2521. [Google Scholar] [CrossRef]
- Shiying, W.; Boyun, S.; Jianye, Y.; Wanjun, Z.; Ping, T.; Jiang, L.; Hongyi, H. The Different Effects of VEGFA121 and VEGFA165 on Regulating Angiogenesis Depend on Phosphorylation Sites of VEGFR2. Inflamm. Bowel Dis. 2017, 23, 603–616. [Google Scholar] [CrossRef]
- Albuquerque, R.J.; Hayashi, T.; Cho, W.G.; Kleinman, M.E.; Dridi, S.; Takeda, A.; Baffi, J.Z.; Yamada, K.; Kaneko, H.; Green, M.G. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat. Med. 2009, 15, 1023. [Google Scholar] [CrossRef]
- Ferrara, N.; Davis-Smyth, T. The biology of vascular endothelial growth factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef]
- Gille, H.; Kowalski, J.; Li, B.; LeCouter, J.; Moffat, B.; Zioncheck, T.F.; Pelletier, N.; Ferrara, N. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J. Biol. Chem. 2001, 276, 3222–3230. [Google Scholar] [CrossRef]
- Abou Faycal, C.; Gazzeri, S.; Eymin, B. A VEGF-A/SOX2/SRSF2 network controls VEGFR1 pre-mRNA alternative splicing in lung carcinoma cells. Sci. Rep. 2019, 9, 336. [Google Scholar] [CrossRef]
- Heydarian, M.; McCaffrey, T.; Florea, L.; Yang, Z.; Ross, M.; Zhou, W.; Maynard, S. Novel splice variants of sFlt1 are upregulated in preeclampsia. Placenta 2009, 30, 250–255. [Google Scholar] [CrossRef]
- Wu, F.T.; Stefanini, M.O.; Mac Gabhann, F.; Kontos, C.D.; Annex, B.H.; Popel, A.S. A systems biology perspective on sVEGFR1: Its biological function, pathogenic role and therapeutic use. J. Cell. Mol. Med. 2010, 14, 528–552. [Google Scholar] [CrossRef]
- Ikeda, T.; Sun, L.; Tsuruoka, N.; Ishigaki, Y.; Yoshitomi, Y.; Yoshitake, Y.; Yonekura, H. Hypoxia down-regulates sFlt-1 (sVEGFR-1) expression in human microvascular endothelial cells by a mechanism involving mRNA alternative processing. Biochem. J. 2011, 436, 399–407. [Google Scholar] [CrossRef]
- Fellows, A.; Mierke, D.F.; Nichols, R.C. AUF1-RGG peptides up-regulate the VEGF antagonist, soluble VEGF receptor-1 (sFlt-1). Cytokine 2013, 64, 337–342. [Google Scholar] [CrossRef]
- Boeckel, J.N.; Guarani, V.; Koyanagi, M.; Roexe, T.; Lengeling, A.; Schermuly, R.T.; Gellert, P.; Braun, T.; Zeiher, A.; Dimmeler, S. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 3276–3281. [Google Scholar] [CrossRef]
- Kangsamaksin, T.; Murtomaki, A.; Kofler, N.M.; Cuervo, H.; Chaudhri, R.A.; Tattersall, I.W.; Rosenstiel, P.E.; Shawber, C.J.; Kitajewski, J. NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov. 2015, 5, 182–197. [Google Scholar] [CrossRef]
- Raimondi, C.; Brash, J.T.; Fantin, A.; Ruhrberg, C. NRP1 function and targeting in neurovascular development and eye disease. Prog. Retin. Eye Res. 2016, 52, 64–83. [Google Scholar] [CrossRef]
- Yao, X.; Bouyain, S. Splicing and proteolytic processing in VEGF signaling: Now it is the coreceptor’s turn. Structure 2015, 23, 610–611. [Google Scholar] [CrossRef][Green Version]
- Herzog, B.; Pellet-Many, C.; Britton, G.; Hartzoulakis, B.; Zachary, I.C. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol. Boil. Cell 2011, 22, 2766–2776. [Google Scholar] [CrossRef]
- Parker, M.W.; Xu, P.; Li, X.; Vander Kooi, C.W. Structural basis for selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1. J. Biol. Chem. 2012, 287, 11082–11089. [Google Scholar] [CrossRef]
- Vander Kooi, C.W.; Jusino, M.A.; Perman, B.; Neau, D.B.; Bellamy, H.D.; Leahy, D.J. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. USA 2007, 104, 6152–6157. [Google Scholar] [CrossRef]
- Rossignol, M.; Gagnon, M.L.; Klagsbrun, M. Genomic organization of human neuropilin-1 and neuropilin-2 genes: Identification and distribution of splice variants and soluble isoforms. Genomics 2000, 70, 211–222. [Google Scholar] [CrossRef]
- Cackowski, F.C.; Xu, L.; Hu, B.; Cheng, S.Y. Identification of two novel alternatively spliced Neuropilin-1 isoforms. Genomics 2004, 84, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; David, G.; Esko, J.D. Repetitive Ser-Gly sequences enhance heparan sulfate assembly in proteoglycans. J. Biol. Chem. 1995, 270, 27127–27135. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Takashima, S.; Asano, Y.; Kato, H.; Liao, Y.; Yamazaki, S.; Tsukamoto, O.; Seguchi, O.; Yamamoto, H.; Fukushima, T.; et al. Glycosaminoglycan modification of neuropilin-1 modulates VEGFR2 signaling. EMBO J. 2006, 25, 3045–3055. [Google Scholar] [CrossRef]
- Frankel, P.; Pellet-Many, C.; Lehtolainen, P.; D’Abaco, G.M.; Tickner, M.L.; Cheng, L.; Zachary, I.C. Chondroitin sulphate-modified neuropilin 1 is expressed in human tumour cells and modulates 3D invasion in the U87MG human glioblastoma cell line through a p130Cas-mediated pathway. EMBO Rep. 2008, 9, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Yaqoob, U.; Cao, S.; Shergill, U.; Jagavelu, K.; Geng, Z.; Yin, M.; de Assuncao, T.M.; Cao, Y.; Szabolcs, A.; Thorgeirsson, S.; et al. Neuropilin-1 stimulates tumor growth by increasing fibronectin fibril assembly in the tumor microenvironment. Cancer Res. 2012, 72, 4047–4059. [Google Scholar] [CrossRef]
- Xu, Y.; Yuan, L.; Mak, J.; Pardanaud, L.; Caunt, M.; Kasman, I.; Larrivee, B.; Del Toro, R.; Suchting, S.; Medvinsky, A.; et al. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J. Biol. Chem. 2010, 188, 115–130. [Google Scholar]
- Yuan, L.; Moyon, D.; Pardanaud, L.; Breant, C.; Karkkainen, M.J.; Alitalo, K.; Eichmann, A. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development 2002, 129, 4797–4806. [Google Scholar]
- Caunt, M.; Mak, J.; Liang, W.C.; Stawicki, S.; Pan, Q.; Tong, R.K.; Kowalski, J.; Ho, C.; Reslan, H.B.; Ross, J.; et al. Blocking neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell 2008, 13, 331–342. [Google Scholar] [CrossRef]
- Parker, M.W.; Linkugel, A.D.; Goel, H.L.; Wu, T.; Mercurio, A.M.; Vander Kooi, C.W. Structural basis for VEGF-C binding to neuropilin-2 and sequestration by a soluble splice form. Structure 2015, 23, 677–687. [Google Scholar] [CrossRef]
- Sleeman, M.; Fraser, J.; McDonald, M.; Yuan, S.; White, D.; Grandison, P.; Kumble, K.; Watson, J.D.; Murison, J.G. Identification of a new fibroblast growth factor receptor, FGFR5. Gene 2001, 271, 171–182. [Google Scholar] [CrossRef]
- Leelayuwat, C.; Abraham, L.J.; Pinelli, M.; Townend, D.C.; Wilks, A.F.; Dawkins, R.L. The primate MHC contains sequences related to the fibroblast growth factor receptor gene family. Tissue Antigens 1996, 48, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, D.; Rosato, B.; Nanni, M.; Belleudi, F.; Torrisi, M.R. Expression of the FGFR2c mesenchymal splicing variant in human keratinocytes inhibits differentiation and promotes invasion. Mol. Carcinogenesis 2018, 57, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Michelle, L.; Barbier, J.; Chabot, B. hnRnP and hnRnP-Like Proteins in Splicing control. RNA Binding Proteins 2012, 1–25. [Google Scholar]
- Hovhannisyan, R.H.; Carstens, R.P. Heterogeneous ribonucleoprotein M is a splicing regulatory protein that can enhance or silence splicing of alternatively spliced exons. J. Biol. Chem. 2007, 282, 36265–36274. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.C.; Lin, W.C.; Lin, Y.J.; Lin, J.C. The impact of RNA binding motif protein 4-regulated splicing cascade on the progression and metabolism of colorectal cancer cells. Oncotarget 2015, 6, 38046–38060. [Google Scholar] [CrossRef]
- Yan, G.; Fukabori, Y.; McBride, G.; Nikolaropolous, S.; McKeehan, W. Exon switching and activation of stromal and embryonic fibroblast growth factor (FGF)-FGF receptor genes in prostate epithelial cells accompany stromal independence and malignancy. Mol. Cell. Biol. 1993, 13, 4513–4522. [Google Scholar] [CrossRef]
- Savagner, P.; Valles, A.; Jouanneau, J.; Yamada, K.; Thiery, J. Alternative splicing in fibroblast growth factor receptor 2 is associated with induced epithelial-mesenchymal transition in rat bladder carcinoma cells. Mol. Biol. Cell 1994, 5, 851–862. [Google Scholar] [CrossRef]
- Oltean, S.; Sorg, B.S.; Albrecht, T.; Bonano, V.I.; Brazas, R.M.; Dewhirst, M.W.; Garcia-Blanco, M.A. Alternative inclusion of fibroblast growth factor receptor 2 exon IIIc in Dunning prostate tumors reveals unexpected epithelial mesenchymal plasticity. Proc. Natl. Acad. Sci. USA 2006, 103, 14116–14121. [Google Scholar] [CrossRef]
- Narita, K.; Fujii, T.; Ishiwata, T.; Yamamoto, T.; Kawamoto, Y.; Kawahara, K.; Nakazawa, N.; Naito, Z. Keratinocyte growth factor induces vascular endothelial growth factor-A expression in colorectal cancer cells. Int. J. Oncol. 2009, 34, 355–360. [Google Scholar]
- Cho, K.; Ishiwata, T.; Uchida, E.; Nakazawa, N.; Korc, M.; Naito, Z.; Tajiri, T. Enhanced expression of keratinocyte growth factor and its receptor correlates with venous invasion in pancreatic cancer. Am. J. Pathol. 2007, 170, 1964–1974. [Google Scholar] [CrossRef]
- Lang, L.; Teng, Y. Fibroblast Growth Factor Receptor 4 Targeting in Cancer: New Insights into Mechanisms and Therapeutic Strategies. Cells 2019, 8, 31. [Google Scholar] [CrossRef]
- Kostrzewa, M.; Muller, U. Genomic structure and complete sequence of the human FGFR4 gene. Mamm. Genome 1998, 9, 131–135. [Google Scholar] [CrossRef]
- Roghani, M.; Moscatelli, D. Prostate cells express two isoforms of fibroblast growth factor receptor 1 with different affinities for fibroblast growth factor-2. Prostate 2007, 67, 115–124. [Google Scholar] [CrossRef]
- Groth, C.; Lardelli, M. The structure and function of vertebrate fibroblast growth factor receptor 1. Int. J. Dev. Boil. 2002, 46, 393–400. [Google Scholar]
- Zhao, M.; Zhuo, M.L.; Zheng, X.; Su, X.; Meric-Bernstam, F. FGFR1beta is a driver isoform of FGFR1 alternative splicing in breast cancer cells. Oncotarget 2019, 10, 30–44. [Google Scholar] [CrossRef]
- Bruno, I.G.; Jin, W.; Cote, G.J. Correction of aberrant FGFR1 alternative RNA splicing through targeting of intronic regulatory elements. Hum. Mol. Genet. 2004, 13, 2409–2420. [Google Scholar] [CrossRef]
- Jin, W.; Cote, G.J. Enhancer-dependent splicing of FGFR1 alpha-exon is repressed by RNA interference-mediated down-regulation of SRp55. Cancer Res. 2004, 64, 8901–8905. [Google Scholar] [CrossRef]
- Burgar, H.R.; Burns, H.D.; Elsden, J.L.; Lalioti, M.D.; Heath, J.K. Association of the signaling adaptor FRS2 with fibroblast growth factor receptor 1 (Fgfr1) is mediated by alternative splicing of the juxtamembrane domain. J. Biol. Chem. 2002, 277, 4018–4023. [Google Scholar] [CrossRef]
- Watanabe, K.; Hasegawa, Y.; Yamashita, H.; Shimizu, K.; Ding, Y.; Abe, M.; Ohta, H.; Imagawa, K.; Hojo, K.; Maki, H.; et al. Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J. Clin. Investig. 2004, 114, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Matsumoto, N.; Kuramoto, Y.; Sakata, A.; Motonagare, R.; Aikawa, A.; Imoto, M.; Toda, A.; Honda, S.; Shimeno, H.; et al. Vasohibin induces prolyl hydroxylase-mediated degradation of hypoxia-inducible factor-1alpha in human umbilical vein endothelial cells. FEBS Lett. 2012, 586, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y. The vasohibin family: A novel family for angiogenesis regulation. J. Biochem. 2013, 153, 5–11. [Google Scholar] [CrossRef]
- Takahashi, Y.; Koyanagi, T.; Suzuki, Y.; Saga, Y.; Kanomata, N.; Moriya, T.; Suzuki, M.; Sato, Y. Vasohibin-2 expressed in human serous ovarian adenocarcinoma accelerates tumor growth by promoting angiogenesis. Mol. Cancer Res. 2012, 10, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Margariti, A.; Hu, Y.; Sato, Y.; Zeng, L.; Ivetic, A.; Habi, O.; Mason, J.C.; Wang, X.; Xu, Q. Protein kinase C{delta} deficiency accelerates neointimal lesions of mouse injured artery involving delayed reendothelialization and vasohibin-1 accumulation. Arterioscl. Thromb. Vasc. Biol. 2010, 30, 2467–2474. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Wu, D.; Bu, X.; Qiao, Y. Hypoxia promotes apoptosis of neuronal cells through hypoxia-inducible factor-1α-microRNA-204-B-cell lymphoma-2 pathway. Exp. Biol. Med. 2016, 241, 177–183. [Google Scholar] [CrossRef]
- Kim, S.H.; Hwang, D.; Park, H.; Yang, E.G.; Chung, H.S.; Kim, S.Y. The action of HIF-3alpha variants on HIF-2α-HIF-1β heterodimer formation is directly probed in live cells. Exp. Cell Res. 2015, 336, 329–337. [Google Scholar] [CrossRef]
- Green, L.; Cookson, A.; Bruce, I.N.; Donn, R.P.; Ray, D.W. Identification of multiple, oxygen-stable HIF1 alpha isoforms, and augmented expression of adrenomedullin in rheumatoid arthritis. Clin. Exp. Rheumatol. 2013, 31, 672–682. [Google Scholar]
- Lukashev, D.; Sitkovsky, M. Preferential expression of the novel alternative isoform I.3 of hypoxia-inducible factor 1alpha in activated human T lymphocytes. Hum. Immunol. 2008, 69, 421–425. [Google Scholar]
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3α locus. J. Boil. Chem. 2002, 277, 32405–32408. [Google Scholar] [CrossRef]
- Maynard, M.A.; Evans, A.J.; Shi, W.; Kim, W.Y.; Liu, F.F.; Ohh, M. Dominant-negative HIF-3 alpha 4 suppresses VHL-null renal cell carcinoma progression. Cell Cycle 2007, 6, 2810–2816. [Google Scholar] [CrossRef][Green Version]
- Thomas, M.; Augustin, H.G. The role of the Angiopoietins in vascular morphogenesis. Angiogenesis 2009, 12, 125–137. [Google Scholar] [CrossRef]
- Fiedler, U.; Scharpfenecker, M.; Koidl, S.; Hegen, A.; Grunow, V.; Schmidt, J.M.; Kriz, W.; Thurston, G.; Augustin, H.G. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood 2004, 103, 4150–4156. [Google Scholar] [CrossRef]
- Pergolizzi, M.; Bizzozero, L.; Riccitelli, E.; Pascal, D.; Samarelli, A.V.; Bussolino, F.; Arese, M. Modulation of Angiopoietin 2 release from endothelial cells and angiogenesis by the synaptic protein Neuroligin 2. Biochem. Biophys. Res. Commun. 2018, 501, 165–171. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef]
- Pichiule, P.; Chavez, J.C.; LaManna, J.C. Hypoxic regulation of angiopoietin-2 expression in endothelial cells. J. Biol. Chem. 2004, 279, 12171–12180. [Google Scholar] [CrossRef]
- Mezquita, J.; Mezquita, P.; Montserrat, P.; Mezquita, B.; Francone, V.; Vilagrasa, X.; Mezquita, C. Genomic structure and alternative splicing of chicken angiopoietin-2. Biochem. Biophys. Res. Commun. 2000, 275, 643–651. [Google Scholar] [CrossRef]
- Michael, I.P.; Orebrand, M.; Lima, M.; Pereira, B.; Volpert, O.; Quaggin, S.E.; Jeansson, M. Angiopoietin-1 deficiency increases tumor metastasis in mice. BMC Cancer 2017, 17, 539. [Google Scholar] [CrossRef]
- Yang, P.; Chen, N.; Yang, D.; Crane, J.; Huang, B.; Dong, R.; Yi, X.; Guo, J.; Cai, J.; Wang, Z. Cervical cancer cell-derived angiopoietins promote tumor progression. Tumour Boil. 2017, 39, 1010428317711658. [Google Scholar] [CrossRef]
- Dong, Z.; Chen, J.; Yang, X.; Zheng, W.; Wang, L.; Fang, M.; Wu, M.; Yao, M.; Yao, D. Ang-2 promotes lung cancer metastasis by increasing epithelial-mesenchymal transition. Oncotarget 2018, 9, 12705–12717. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Angiopoietin-2 is implicated in the regulation of tumor angiogenesis. Am. J. Pathol. 2001, 158, 563–570. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Therap. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Verma, A. Recent Advances in Antisense Oligonucleotide Therapy in Genetic Neuromuscular Diseases. Ann. Indian Acad. Neurol. 2018, 21, 3–8. [Google Scholar] [PubMed]
- Gammons, M.V.; Fedorov, O.; Ivison, D.; Du, C.; Clark, T.; Hopkins, C.; Hagiwara, M.; Dick, A.D.; Cox, R.; Harper, S.J.; et al. Topical antiangiogenic SRPK1 inhibitors reduce choroidal neovascularization in rodent models of exudative AMD. Investig. Ophthalmol. Visual Sci. 2013, 54, 6052–6062. [Google Scholar] [CrossRef]
- Gammons, M.V.; Lucas, R.; Dean, R.; Coupland, S.E.; Oltean, S.; Bates, D.O. Targeting SRPK1 to control VEGF-mediated tumour angiogenesis in metastatic melanoma. Br. J. Cancer 2014, 111, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Batson, J.; Toop, H.D.; Redondo, C.; Babaei-Jadidi, R.; Chaikuad, A.; Wearmouth, S.F.; Gibbons, B.; Allen, C.; Tallant, C.; Zhang, J.; et al. Development of Potent, Selective SRPK1 Inhibitors as Potential Topical Therapeutics for Neovascular Eye Disease. ACS Chem. Biol. 2017, 12, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, J.M.; Wu, G.; Zeng, C.; Zhu, J.; Meng, F.; Patel, S.; Wang, W.; Ficarro, S.B.; Leggett, A.L.; Powell, C.E.; et al. SRPKIN-1: A Covalent SRPK1/2 Inhibitor that Potently Converts VEGF from Pro-angiogenic to Anti-angiogenic Isoform. Cell Chem. Biol. 2018, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]




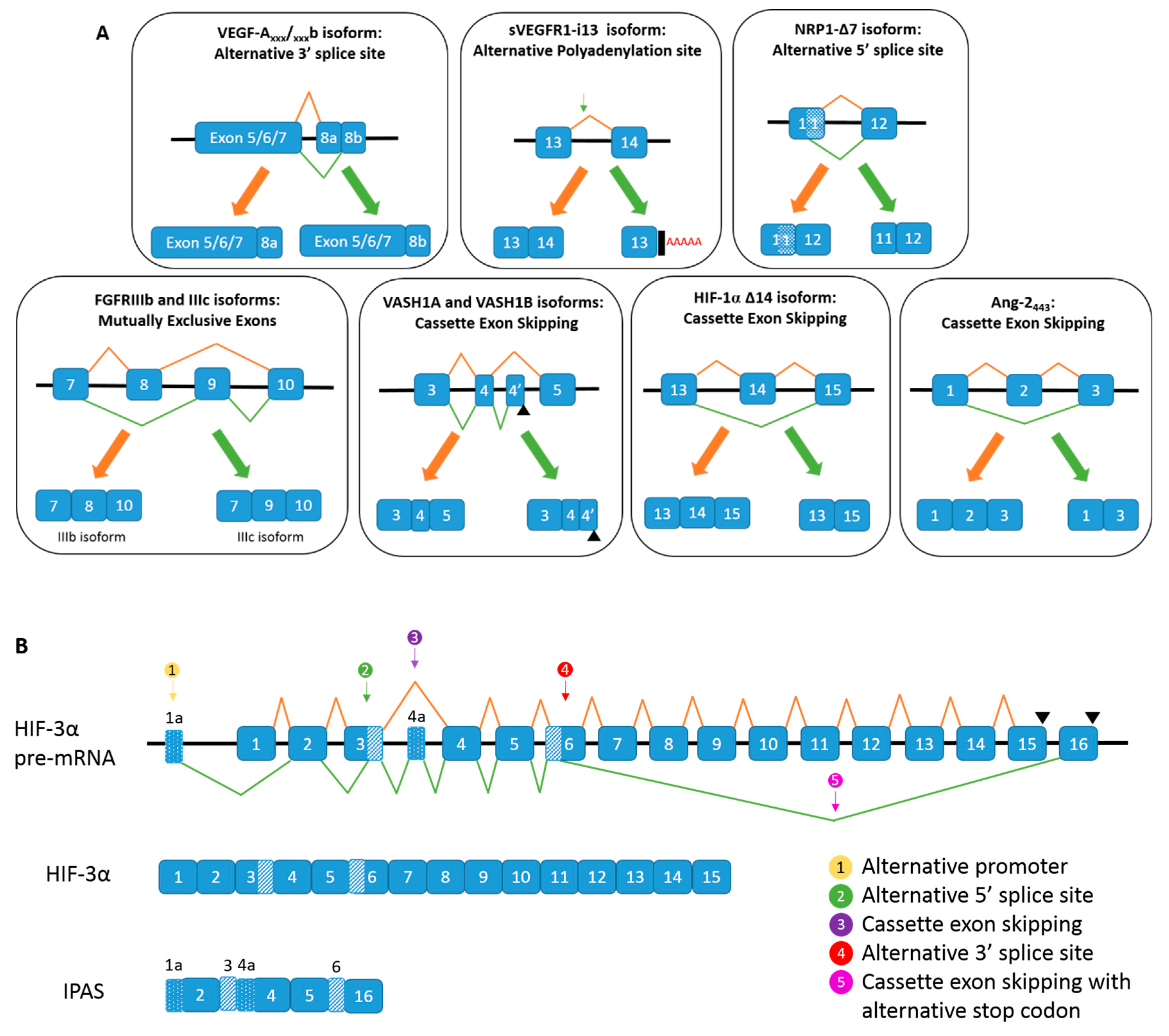
 . (B) Alternative spicing of HIF-3α, and the generation of IPAS mRNA.
. (B) Alternative spicing of HIF-3α, and the generation of IPAS mRNA.
. (B) Alternative spicing of HIF-3α, and the generation of IPAS mRNA.
. (B) Alternative spicing of HIF-3α, and the generation of IPAS mRNA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Splice Variants | Function |
|---|---|---|
| VEGF-A | VEGF-A111 | Proangiogenic. Diffusible [33,34] |
| VEGF-A121 | Proangiogenic. Diffusible. Implicated in tumorigenesis [33,35,36] | |
| VEGF-A145 | Proangiogenic. Bind to cell surface and extracellular matrix [33,34] | |
| VEGF-A165 | Proangiogenic. Moderately diffusible. Implicated in tumorigenesis [33,36] | |
| VEGF-A189 | Proangiogenic. Implicated in tumorigenesis [33,36,37] | |
| VEGF-A206 | Proangiogenic. Strongly bind to cell surface and ECM [33,37] | |
| VEGF-A183 | Proangiogenic [33] | |
| VEGF-Axxxb | Anti-angiogenic. Downregulated in cancer, diabetic retinopathy, Denys Drash syndrome, retinal vein occlusion. Upregulated in systemic sclerosis and asthma [33,38] | |
| VEGFR1 | sVEGFR1 | Potent anti-angiogenic [39] |
| VEGFR2 | sVEGFR2 | Decreases lymphangiogenesis. Downregulated in neuroblastoma [39] |
| esVEGFR2 | Decreases lymphangiogenesis [39] | |
| NRP-1 | s11NRP1, s12NRP1, sIIINRP1, sIVNRP1 | Soluble isoform. Antagonists of NRP1 signalling. Anti-angiogenic and anti-tumorigenic [40] |
| NRP1-ΔE16 | No functional difference to full length NRP1 [41] | |
| NRP1Δ7 | Affects glycosylation status of NRP1. Anti-tumorigenic in prostate cancer and breast cancer cells [42] | |
| FGFRs | IIIb | EMT. Found in epithelial tissues. Evidence as a tumour suppressor and as a tumour promoter [43,44] |
| IIIc | EMT. Found in mesenchymal tissues. Tumourigenic [43] | |
| FGFRα | Contains autoinhibitory IgI domain which results in a lower affinity for FGFs and decreased signalling compared to FGFRβ [45,46] | |
| FGFRβ | Higher affinity for FGFs and enhanced signalling. Increases proliferation and linked to tumourigenesis [43,45,46] | |
| Soluble receptors | Can be found in locations in the cell other than the cell membrane. Precise function unknown [43,45] | |
| C1, C2 and C3 | C3 has the most transforming activity, C2 has moderate transforming activity and C1 has the least transforming activity. C3 implicated in oncogenesis [47] | |
| Deletion of the VT motif | Prevention of binding of some effector molecules. Suggested to be unable to activate the downstream Ras/MAPK signalling pathway [48] | |
| Vasohibin-1 | VASH1A and VASH-1B | Both are anti-angiogenic. VASH-1A promotes normalisation of abnormal tumour blood vessels. VASH-1B prunes vasculature [49,50] |
| Vasohibin-2 | 355aa | Predominantly expressed in HUVECs. Function unknown [51] |
| 290aa | Anti-angiogenic activity [52] | |
| 311aa, 156aa, 117aa, 104aa | Function unknown | |
| HIF-1α | HIF-1αΔ11 | Promotes tumorigenesis through enhancement of HIF activity [52] |
| HIF-1αΔ12 and HIF-1αΔ11&12 | Inhibits dimerisation of HIF-1α and HIF-1β. Act as dominant regulators of HIF-1 transcription [53] | |
| HIF-1αΔ14 | Less potent activator of HIF-1 transcription than canonical form of HIF-1α [54] | |
| HIF-1α417 | Amplifies HIF-1β-mediated transcription of EPO gene [53] | |
| HIF-1TAG | Function unknown | |
| HIF-1α Alt1 | Function unknown | |
| HIF-3α | IPAS | Dimerises with HIF-α subunits but cannot initiate transcription of HIF target genes, such as VEGF. Dampens angiogenesis [53,55,56] |
| HIF-3α4 | Forms complex with HIF-1α and prevents HIF transcription. Hampers angiogenesis and proliferation [55] | |
| Ang-1 | 1.5 kb, 1.3 kb | Bind to Tie-2 and induce its autophosphorylation [57] |
| 0.9 kb, 0.7 kb | Bind to Tie-2 but do not induce its autophosphorylation [57] | |
| Ang-2 | Ang-2B | Not precise function but indication of inactivation of the vasculature and vascular remodelling [58] |
| Ang-2443 | Antagonist of Tie-2 signalling activation. Suggestive role in the regulation of inflammatory processes [59] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowler, E.; Oltean, S. Alternative Splicing in Angiogenesis. Int. J. Mol. Sci. 2019, 20, 2067. https://doi.org/10.3390/ijms20092067
Bowler E, Oltean S. Alternative Splicing in Angiogenesis. International Journal of Molecular Sciences. 2019; 20(9):2067. https://doi.org/10.3390/ijms20092067
Chicago/Turabian StyleBowler, Elizabeth, and Sebastian Oltean. 2019. "Alternative Splicing in Angiogenesis" International Journal of Molecular Sciences 20, no. 9: 2067. https://doi.org/10.3390/ijms20092067
APA StyleBowler, E., & Oltean, S. (2019). Alternative Splicing in Angiogenesis. International Journal of Molecular Sciences, 20(9), 2067. https://doi.org/10.3390/ijms20092067