Regulation of Autophagy by Nuclear GAPDH and Its Aggregates in Cancer and Neurodegenerative Disorders



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
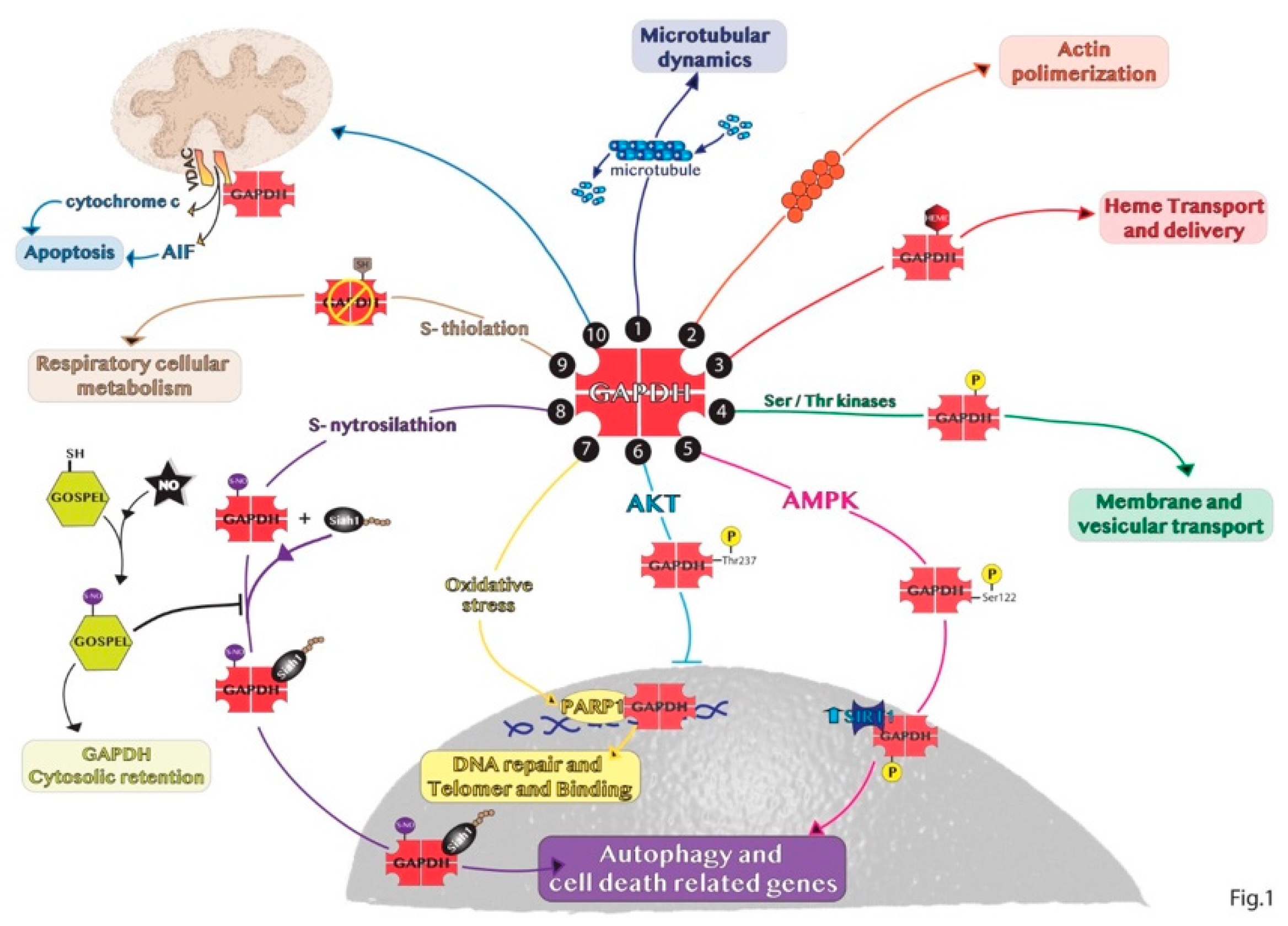
2. Non-Glycolytic Roles of GAPDH
3. Subcellular Localization of GAPDH
3.1. Nuclear Roles of GAPDH
3.2. Non-Nuclear Roles of GAPDH
3.2.1. Cytosolic GAPDH
3.2.2. Mitochondrial GAPDH
3.2.3. Extracellular GAPDH
3.2.4. Other GAPDH Cellular Localizations
4. The Double Role of Autophagy in Cancer
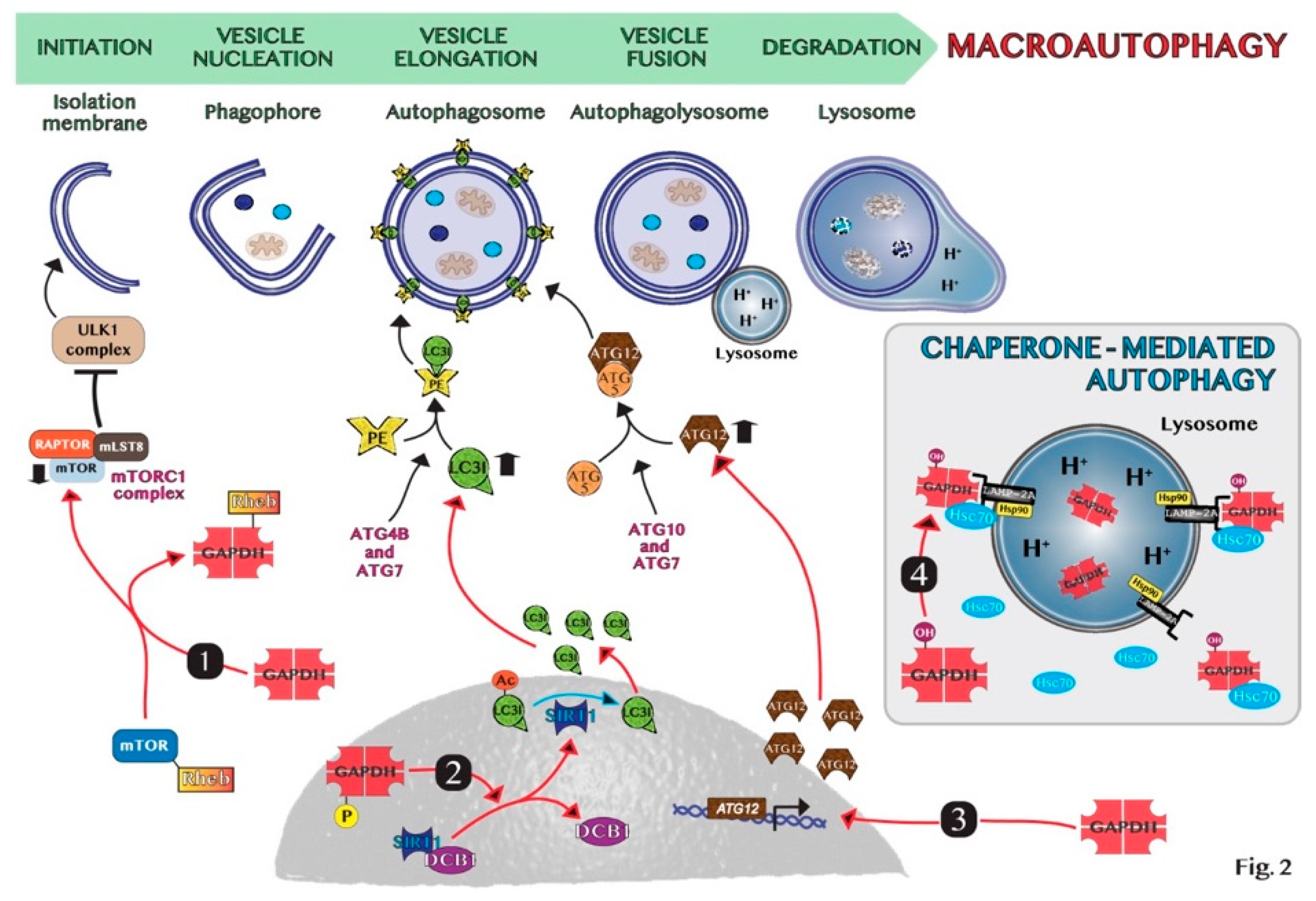
5. GAPDH-Mediated Autophagy
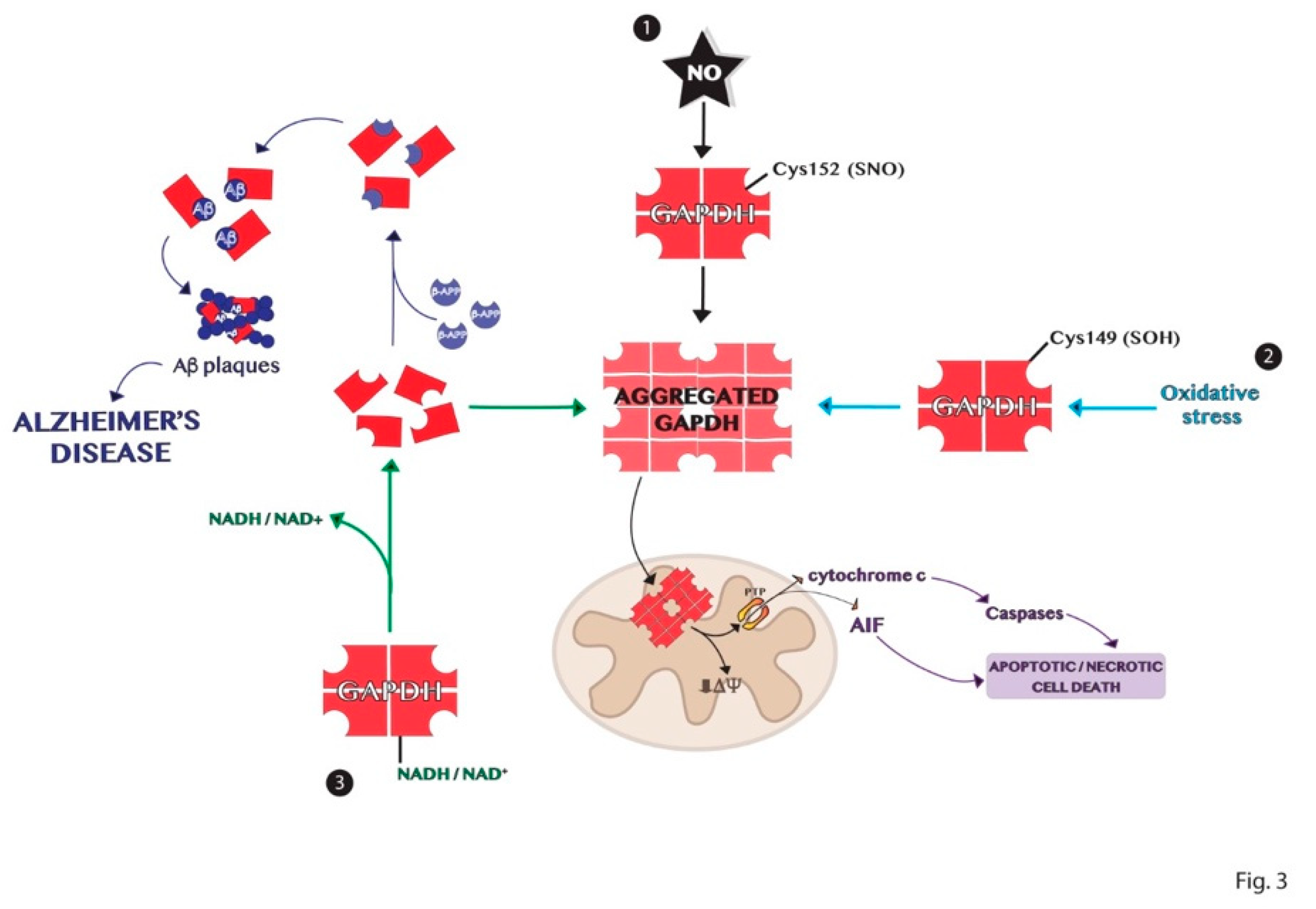
6. Aggregation Mechanisms of GAPDH and Impact on Diseases
GAPDH Aggregates and GAPDH-Mediated Cell Death in Neurodegenerative Diseases
7. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Gao, W.; Kang, J.H.; Liao, Y.; Li, M.; Yin, X.M. Autophagy and cell death. In Essentials of Apoptosis: A Guide for Basic and Clinical Research; Yin, X.M., Dong, Z., Eds.; Humana Press: New York, NY, USA, 2009. [Google Scholar]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Boil. 2018, 20, 1338–1348. [Google Scholar] [CrossRef]
- Puri, P.; Chandra, A. Autophagy Modulation As a Potential Therapeutic Target for Liver Diseases. J. Clin. Exp. Hepatol. 2014, 4, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, H.; Pitt, J.M.; Kroemer, G.; Zitvogel, L. Therapy-induced microenvironmental changes in cancer. J. Mol. Med. 2016, 94, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Rothberg, J.M.; Kumar, V.; Schramm, K.J.; Haller, E.; Proemsey, J.B.; Lloyd, M.C.; Sloane, B.F.; Gillies, R.J.; Kumar, D.V. Chronic Autophagy Is a Cellular Adaptation to Tumor Acidic pH Microenvironments. Cancer Res 2012, 72, 3938–3947. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, C.B.; Robertson, A.G.; Goya, R.; Jones, S.J.; Morin, R.D.; Marra, M.A.; Gorski, S.M. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 2015, 11, 1668–1687. [Google Scholar] [CrossRef]
- Aita, V.M.; Liang, X.H.; Murty, V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.; Levine, B. Cloning and Genomic Organization of Beclin 1, a Candidate Tumor Suppressor Gene on Chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef]
- Laddha, S.V.; Ganesan, S.; Chan, C.S.; White, E. Mutational Landscape of the Essential Autophagy Gene BECN1 in Human Cancers. Mol. Cancer Res. 2014, 12, 485–490. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; Mackay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nat. Cell Boil. 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Yang, A.; RajeshKumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef]
- Comel, A.; Sorrentino, G.; Capaci, V.; Del Sal, G. The cytoplasmic side of p53’s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; Ryan, K.M. DRAM Links Autophagy to p53 and Programmed Cell Death. Autophagy 2007, 3, 72–74. [Google Scholar] [CrossRef]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Pozza, E.D.; Nadal-Serrano, M.; Oliver, J.O.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 19–28. [Google Scholar] [CrossRef]
- Tristan, C.; Shahani, N.; Sedlak, T.W.; Sawa, A. The diverse functions of GAPDH: Views from different subcellular compartments. Cell Signal. 2011. [Google Scholar] [CrossRef]
- Sirover, M.A. Subcellular Dynamics of Multifunctional Protein Regulation: Mechanisms of GAPDH Intracellular Translocation. J. Cell. Biochem. 2012, 113, 2193–2200. [Google Scholar] [CrossRef]
- Ikeda, Y.; Yamaji, R.; Irie, K.; Kioka, N.; Murakami, A. Glyceraldehyde-3-phosphate dehydrogenase regulates cyclooxygenase-2 expression by targeting mRNA stability. Arch. Biochem. Biophys. 2012, 528, 141–147. [Google Scholar] [CrossRef]
- Raje, C.I.; Kumar, S.; Harle, A.; Nanda, J.S.; Raje, M. The macrophage cell surface glyceraldehyde-3-phosphate dehydrogenase is a novel transferrin receptor. J. Biol. Chem. 2007. [Google Scholar] [CrossRef]
- Azam, S.; Jouvet, N.; Jilani, A.; Vongsamphanh, R.; Yang, X.; Yang, S.; Ramotar, D. Human Glyceraldehyde-3-phosphate Dehydrogenase Plays a Direct Role in Reactivating Oxidized Forms of the DNA repair enzyme APE1. J. Boil. Chem. 2008, 283, 30632–30641. [Google Scholar] [CrossRef]
- Sirover, M.A. On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: Biochemical mechanisms and regulatory control. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1810, 741–751. [Google Scholar] [CrossRef]
- Tarze, A.; Deniaud, A.; Le Bras, M.; Maillier, E.; Molle, D.; Larochette, N.; Zamzami, N.; Jan, G.; Kroemer, G.; Brenner, C. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 2007. [Google Scholar] [CrossRef]
- Tisdale, E.J.; Talati, N.K.; Artalejo, C.R.; Shisheva, A. GAPDH binds Akt to facilitate cargo transport in the early secretory pathway. Exp. Cell 2016, 349, 310–319. [Google Scholar] [CrossRef]
- Gerszon, J.; Rodacka, A. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase in neurodegenerative processes and the role of low molecular weight compounds in counteracting its aggregation and nuclear translocation. Cancer Rev. 2018, 48, 21–31. [Google Scholar] [CrossRef]
- Nakajima, H.; Amano, W.; Fujita, A.; Fukuhara, A.; Azuma, Y.-T.; Hata, F.; Inui, T.; Takeuchi, T. The Active Site Cysteine of the Proapoptotic Protein Glyceraldehyde-3-phosphate Dehydrogenase Is Essential in Oxidative Stress-induced Aggregation and Cell Death. J. Boil. Chem. 2007, 282, 26562–26574. [Google Scholar] [CrossRef]
- Mazzola, J.L.; Sirover, M.A. Reduction of glyceraldehyde-3-phosphate dehydrogenase activity in Alzheimer’s disease and in Huntington’s disease fibroblasts. J. Neurochem. 2001, 76, 442–449. [Google Scholar] [CrossRef]
- Chuang, D.-M.; Hough, C.; Senatorov, V.V. Glyceraldehyde-3-phosphate dehydrogenase, apoptosis, and neurodegenerative diseases. Annu. Pharmacol. Toxicol. 2005, 45, 269–290. [Google Scholar] [CrossRef]
- Zheng, L.; Roeder, R.G.; Luo, Y. S Phase Activation of the Histone H2B Promoter by OCA-S, a Coactivator Complex that Contains GAPDH as a Key Component. Cell 2003, 114, 255–266. [Google Scholar] [CrossRef]
- Chang, C.; Su, H.; Zhang, D.; Wang, Y.; Shen, Q.; Liu, B.; Huang, R.; Zhou, T.; Peng, C.; Wong, C.C.; et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol. Cell 2015, 60, 930–940. [Google Scholar] [CrossRef]
- Kosova, A.; Khodyreva, S.N.; Lavrik, O.I. Role of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in DNA repair. Biochem. (Moscow) 2017, 82, 643–654. [Google Scholar] [CrossRef]
- Sundararaj, K.P.; Wood, R.E.; Ponnusamy, S.; Salas, A.M.; Szulc, Z.; Bielawska, A.; Obeid, L.M.; Hannun, Y.A.; Ogretmen, B. Rapid Shortening of telomere length in response to ceramide involves the inhibition of telomere binding activity of nuclear glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 2004, 279, 6152–6162. [Google Scholar] [CrossRef]
- Barbini, L.; Rodriguez, J.; Domínguez, F.; Vega, F. Glyceraldehyde-3-phosphate dehydrogenase exerts different biologic activities in apoptotic and proliferating hepatocytes according to its subcellular localization. Mol. Cell. Biochem. 2007, 300, 19–28. [Google Scholar] [CrossRef]
- Sirover, M.A. Pleiotropic effects of moonlighting glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in cancer progression, invasiveness, and metastases. Cancer Metastasis Rev. 2018, 37, 665–676. [Google Scholar] [CrossRef]
- Siswanto, F.M.; Jawi, I.M.; Kartiko, B.H. The role of E3 ubiquitin ligase seven in absentia homolog in the innate immune system: An overview. Vet. World 2018, 11, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Boil. 2005, 7, 665–674. [Google Scholar] [CrossRef]
- Sen, N.; Hara, M.R.; Ahmad, A.S.; Cascio, M.B.; Kamiya, A.; Ehmsen, J.T.; Agrawal, N.; Hester, L.; Doré, S.; Snyder, S.H.; et al. GOSPEL: A Neuroprotective Protein that Binds to GAPDH upon S-Nitrosylation. Neuron 2009, 63, 709. [Google Scholar] [CrossRef]
- Ventura, M.; Mateo, F.; Serratosa, J.; Salaet, I.; Carujo, S.; Bachs, O.; Pujol, M.J. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase is regulated by acetylation. Int. J. Biochem. Cell Boil. 2010, 42, 1672–1680. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Pacchiana, R.; Pozza, E.D.; Cataldo, I.; Bruno, S.; Conti, P.; Cordani, M.; Grimaldi, A.; Butera, G.; Caraglia, M.; et al. UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Radic. Boil. Med. 2017, 113, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Butera, G.; Pacchiana, R.; Mullappilly, N.; Margiotta, M.; Bruno, S.; Conti, P.; Riganti, C.; Donadelli, M. Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Sirover, M.A. Role of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in normal cell function and in cell pathology. J. Cell. Biochem. 1997, 66, 133–140. [Google Scholar] [CrossRef]
- Carlile, G.W.; Chalmers-Redman, R.M.; A Tatton, N.; Pong, A.; E Borden, K.; Tatton, W.G. Reduced apoptosis after nerve growth factor and serum withdrawal: conversion of tetrameric glyceraldehyde-3-phosphate dehydrogenase to a dimer. Mol. Pharmacol. 2000, 57, 2–12. [Google Scholar] [PubMed]
- Butterfield, D.A.; Hardas, S.S.; Lange, M.L.B. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: Many apathways to neurodegenertion. J. Alzheimer’s Dis. 2010. [Google Scholar] [CrossRef]
- Tang, Z.; Yuan, S.; Hu, Y.; Zhang, H.; Wu, W.; Zeng, Z.; Yang, J.; Yun, J.; Xu, R.; Huang, P. Over-expression of GAPDH in human colorectal carcinoma as a preferred target of 3-Bromopyruvate Propyl Ester. J. Bioenerg. Biomembr. 2012, 44, 117–125. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Zhang, F.; Hong, C.-Q.; Giuliano, A.E.; Cui, X.-J.; Zhou, G.-J.; Zhang, G.-J.; Cui, Y.-K. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Boil. Med. 2015, 12, 10–22. [Google Scholar]
- Ganapathy-Kanniappan, S. Evolution of GAPDH as a druggable target of tumor glycolysis? Cancer Ther. Targets 2018, 22, 1–4. [Google Scholar] [CrossRef]
- Liberti, M.V.; Dai, Z.; Wardell, S.E.; Baccile, J.A.; Liu, X.; Gao, X.; Baldi, R.; Mehrmohamadi, M.; Johnson, M.O.; Madhukar, N.S.; et al. A predictive model for selective targeting of the warburg effect through GAPDH inhibition with a natural product. Cell Metab. 2017. [Google Scholar] [CrossRef]
- Huang, Q.; Lan, F.; Zheng, Z.; Xie, F.; Han, J.; Dong, L.; Xie, Y.; Zheng, F. Akt2 Kinase Suppresses Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH)-mediated Apoptosis in Ovarian Cancer Cells via Phosphorylating GAPDH at Threonine 237 and Decreasing Its Nuclear Translocation*. J. Boil. Chem. 2011, 286, 42211–42220. [Google Scholar] [CrossRef]
- A Jacquin, M.; Chiche, J.; Zunino, B.; Bénéteau, M.; Meynet, O.; A Pradelli, L.; Marchetti, S.; Cornille, A.; Carles, M.; Ricci, J.-E. GAPDH binds to active Akt, leading to Bcl-xL increase and escape from caspase-independent cell death. Cell Death Differ. 2013, 20, 1043–1054. [Google Scholar] [CrossRef]
- Tisdale, E.J. Glyceraldehyde-3-phosphate dehydrogenase is required for vesicular transport in the early secretory pathway. J. Biol. Chem. 2001. [Google Scholar] [CrossRef]
- Tisdale, E.J.; Azizi, F.; Artalejo, C.R. Rab2 utilizes glyceraldehyde-3-phosphate dehydrogenase and protein kinase cι to associate with microtubules and to recruit dynein. J. Biol. Chem. 2009. [Google Scholar] [CrossRef]
- Sweeny, E.A.; Singh, A.B.; Chakravarti, R.; Martinez-Guzman, O.; Saini, A.; Haque, M.M.; Garee, G.; Dans, P.D.; Hannibal, L.; Reddi, A.R.; et al. Glyceraldehyde-3-phosphate dehydrogenase is a chaperone that allocates labile heme in cells. J. Boil. Chem. 2018, 293, 14557–14568. [Google Scholar] [CrossRef] [PubMed]
- Kohr, M.J.; Murphy, E.; Steenbergen, C. Glyceraldehyde-3-Phosphate Dehydrogenase Acts as a Mitochondrial Trans-S-Nitrosylase in the Heart. PLOS ONE 2014, 9, e111448. [Google Scholar] [CrossRef] [PubMed]
- Yogalingam, G.; Hwang, S.; Ferreira, J.C.B.; Mochly-Rosen, D. Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) Phosphorylation by Protein Kinase Cδ (PKCδ) Inhibits Mitochondria Elimination by Lysosomal-like Structures following Ischemia and Reoxygenation-induced Injury*. J. Boil. Chem. 2013, 288, 18947–18960. [Google Scholar] [CrossRef]
- Chauhan, A.S.; Rawat, P.; Malhotra, H.; Sheokand, N.; Kumar, M.; Patidar, A.; Chaudhary, S.; Jakhar, P.; Raje, C.I.; Raje, M. Secreted multifunctional Glyceraldehyde-3-phosphate dehydrogenase sequesters lactoferrin and iron into cells via a non-canonical pathway. Sci. Rep. 2015, 5, 18465. [Google Scholar] [CrossRef]
- Seidler, K.A.; Seidler, N.W. Role of extracellular GAPDH in Streptococcus pyogenes Virulence. Mo. Med. 2013, 110, 236–240. [Google Scholar] [PubMed]
- Cueille, N.; Blanc, C.T.; Riederer, I.M.; Riederer, B.M. Microtubule-Associated Protein 1B Binds Glyceraldehyde-3-phosphate Dehydrogenase. J. Proteome 2007, 6, 2640–2647. [Google Scholar] [CrossRef] [PubMed]
- Muronetz, V.; Wang, Z.; Keith, T.; Knull, H.; Srivastava, D. Binding Constants and Stoichiometries of Glyceraldehyde 3-Phosphate Dehydrogenase-Tubulin Complexes. Arch. Biochem. Biophys. 1994, 313, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, H.; Bereiter-Hahn, J. Glyceraldehyde-3-phosphate dehydrogenase associates with actin filaments in serum deprived nih 3t3 cells only. Cell Boil. Int. 2002, 26, 155–164. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Cell Dev. Boil. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nat. Cell Boil. 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, metabolism, and cancer HHS public access. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Ranieri, R.; Ciaglia, E.; Amodio, G.; Picardi, P.; Proto, M.C.; Gazzerro, P.; Laezza, C.; Remondelli, P.; Bifulco, M.; Pisanti, S. N6-isopentenyladenosine dual targeting of AMPK and Rab7 prenylation inhibits melanoma growth through the impairment of autophagic flux. Cell Death Differ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Green, D.R.; Ricci, J.-E. Novel roles for GAPDH in cell death and carcinogenesis. Cell Death Differ. 2009, 16, 1573–1581. [Google Scholar] [CrossRef]
- Henry, E.; Fung, N.; Liu, J.; Drakakaki, G.; Coaker, G. Beyond Glycolysis: GAPDHs Are Multi-functional Enzymes Involved in Regulation of ROS, Autophagy, and Plant Immune Responses. PLOS Genet. 2015, 11, e1005199. [Google Scholar] [CrossRef]
- Han, S.; Wang, Y.; Zheng, X.; Jia, Q.; Zhao, J.; Bai, F.; Hong, Y.; Liu, Y. Cytoplastic Glyceraldehyde-3-Phosphate Dehydrogenases Interact with ATG3 to Negatively Regulate Autophagy and Immunity in Nicotiana benthamiana. Cancer Cell 2015, 27, 1316–1331. [Google Scholar]
- Guha, P.; Harraz, M.M.; Snyder, S.H. Cocaine elicits autophagic cytotoxicity via a nitric oxide-GAPDH signaling cascade. Proc. Natl. Acad. Sci. USA 2015, 1–6. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Dando, I.; Torrens-Mas, M.; Butturini, E.; Pacchiana, R.; Oppici, E.; Cavallini, C.; Gasperini, S.; Tamassia, N.; et al. Mutant p53 blocks SESN1/AMPK/PGC-1α/UCP2 axis increasing mitochondrial O2ˉ· production in cancer cells. Br. J. Cancer 2018, 119, 994–1008. [Google Scholar] [CrossRef]
- Kwon, H.J.; Rhim, J.H.; Jang, I.-S.; Kim, G.-E.; Park, S.C.; Yeo, E.-J. Activation of AMP-activated protein kinase stimulates the nuclear localization of glyceraldehyde 3-phosphate dehydrogenase in human diploid fibroblasts. Exp. Mol. Med. 2010, 42, 254–269. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M.; Lippincott-Schwartz, J. Activation of Chaperone-mediated Autophagy during Oxidative Stress. Mol. Boil. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Mol. Cell. Boil. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.N.; Ha, S.H.; Kim, J.; Koh, A.; Lee, C.S.; Jeon, H.; Kim, D.-H.; Suh, P.-G.; Ryu, S.H.; Kim, J.H.; et al. Glycolytic Flux Signals to mTOR through Glyceraldehyde-3-Phosphate Dehydrogenase-Mediated Regulation of Rheb. Mol. Cell. Boil. 2009, 29, 3991–4001. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Acad. Sci. 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; et al. Deacetylation of Nuclear LC3 Drives Autophagy Initiation under Starvation. Mol. Cell 2015, 57, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Ricci, J.-E.; Tait, S.; Milasta, S.; Maurer, U.; Bouchier-Hayes, L.; Fitzgerald, P.; Guio-Carrion, A.; Waterhouse, N.J.; Li, C.W.; et al. GAPDH and Autophagy Preserve Survival after Apoptotic Cytochrome c Release in the Absence of Caspase Activation. Cell 2007, 129, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Finkel, T. GAPDH and the search for alternative energy. Nat. Cell Boil. 2007, 9, 869–870. [Google Scholar] [CrossRef]
- Bertin, S.; Samson, M.; Pons, C.; Gavelli, A.; Baqué, P.; Brossette, N.; Pagnotta, S.; Pierrefite-Carle, V.; Guigonis, J.-M.; Ricci, J.-E. Comparative Proteomics Study Reveals That Bacterial CpG Motifs Induce Tumor Cell Autophagy in Vitro and in Vivo*S. Mol. Cell. Proteom. 2008, 7, 2311–2322. [Google Scholar] [CrossRef]
- Soltany-Rezaee-Rad, M.; Mottaghi-Dastjerdi, N.; Setayesh, N.; Roshandel, G.; Ebrahimifard, F.; Sepehrizadeh, Z. Overexpression of FOXO3, MYD88, and GAPDH Identified by Suppression Subtractive Hybridization in Esophageal Cancer Is Associated with Autophagy. Gastroenterol. Pr. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Guan, J.; Sun, J.; Sun, F.; Lou, B.; Zhang, D.; Mashayekhi, V.; Sadeghi, N.; Storm, G.; Mastrobattista, E.; He, Z. Hypoxia-induced tumor cell resistance is overcome by synergistic GAPDH-siRNA and chemotherapy co-delivered by long-circulating and cationic-interior liposomes. Nanoscale 2017, 9, 9190–9201. [Google Scholar] [CrossRef]
- Dodson, M.; Liang, Q.; Johnson, M.S.; Redmann, M.; Fineberg, N.; Darley-Usmar, V.M.; Zhang, J. Inhibition of glycolysis attenuates 4-hydroxynonenal-dependent autophagy and exacerbates apoptosis in differentiated SH-SY5Y neuroblastoma cells. Autophagy 2013, 9, 1996–2008. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Barinova, K.V.; Stroylova, Y.Y.; Semenyuk, P.I.; Schmalhausen, E.V.; Barinova, K.V. Glyceraldehyde-3-phosphate dehydrogenase: Aggregation mechanisms and impact on amyloid neurodegenerative diseases. Int. J. Boil. Macromol. 2017, 100, 55–66. [Google Scholar] [CrossRef]
- I Arutyunova, E.; Danshina, P.V.; Domnina, L.V.; Pleten, A.P.; I Muronetz, V. Oxidation of glyceraldehyde-3-phosphate dehydrogenase enhances its binding to nucleic acids. Biochem. Biophys. Commun. 2003, 307, 547–552. [Google Scholar] [CrossRef]
- Kubo, T.; Nakajima, H.; Nakatsuji, M.; Itakura, M.; Kaneshige, A.; Azuma, Y.-T.; Inui, T.; Takeuchi, T. Active site cysteine-null glyceraldehyde-3-phosphate dehydrogenase (GAPDH) rescues nitric oxide-induced cell death. Nitric Oxide 2016, 53, 13–21. [Google Scholar] [CrossRef]
- Wang, Q.; Woltjer, R.L.; Cimino, P.J.; Pan, C.; Montine, K.S.; Zhang, J. Proteomic analysis of neurofibrillary tangles in Alzheimer disease identifies GAPDH as a detergent-insoluble paired helical filament tau binding protein. FASEB J. 2005, 19, 869–871. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Tajima, H.; Kuwae, T.; Takeshima, T.; Nakano, T.; Tanaka, M.; Sunaga, K.; Fukuhara, Y.; Nakashima, K.; Ohama, E.; et al. Pro-apoptotic protein glyceraldehyde-3-phosphate dehydrogenase promotes the formation of Lewy body-like inclusions. Eur. J. Neurosci. 2005, 21, 317–326. [Google Scholar] [CrossRef]
- Bae, B.-I.; Hara, M.R.; Cascio, M.B.; Wellington, C.L.; Hayden, M.R.; Ross, C.A.; Ha, H.C.; Li, X.-J.; Snyder, S.H.; Sawa, A. Mutant Huntingtin: Nuclear translocation and cytotoxicity mediated by GAPDH. Proc. Acad. Sci. 2006, 103, 3405–3409. [Google Scholar] [CrossRef]
- Verdier, Y.; Foldi, I.; Sergeant, N.; Fülöp, L.; Penke, Z.; Janáky, T.; Szücs, M.; Penke, B. Characterization of the interaction between Aβ 1–42 and glyceraldehyde phosphodehydrogenase. J. Pept. Sci. 2008, 14, 755–762. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s Disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–767. [Google Scholar] [CrossRef]
- Mazzola, J.L.; Sirover, M.A. Alteration of nuclear glyceraldehyde-3-phosphate dehydrogenase structure in Huntington’s disease fibroblasts. Mol. Brain 2002, 100, 95–101. [Google Scholar] [CrossRef]
- Cumming, R.C.; Schubert, D. Amyloid-β induces disulfide bonding and aggregation of GAPDH in Alzheimer’s disease. FASEB J. 2005, 19, 2060–2062. [Google Scholar] [CrossRef]
- Koshy, B.; Matilla, T.; Burright, E.N.; Merry, D.E.; Fischbeck, K.H.; Orr, H.T.; Zoghbi, H.Y. Spinocerebellar ataxia type-1 and spinobulbar muscular atrophy gene products interact with glyceraldehyde-3-phosphate dehydrogenase. Hum. Mol. Genet. 1996, 5, 1311–1318. [Google Scholar] [CrossRef]
- Burke, J.R.; Enghild, J.J.; Martin, M.E.; Jou, Y.-S.; Myers, R.M.; Roses, A.D.; Vance, J.M.; Strittmatter, W.J. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat. Med. 1996, 2, 347–350. [Google Scholar] [CrossRef]
- Shiozawa, M.; Fukutani, Y.; Arai, N.; Cairns, N.J.; Mizutani, T.; Isaki, K.; Lantos, P.L.; Wada, Y. Glyceraldehyde 3-phosphate dehydrogenase and endothelin-1 immunoreactivity is associated with cerebral white matter damage in dentatorubral-pallidoluysian atrophy. Neuropathology 2003, 23, 36–43. [Google Scholar] [CrossRef]
- Nakajima, H.; Itakura, M.; Kubo, T.; Kaneshige, A.; Harada, N.; Izawa, T.; Azuma, Y.-T.; Kuwamura, M.; Yamaji, R.; Takeuchi, T. Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) Aggregation Causes Mitochondrial Dysfunction during Oxidative Stress-induced Cell Death*. J. Boil. Chem. 2017, 292, 4727–4742. [Google Scholar] [CrossRef]
- Hara, M.R.; Thomas, B.; Cascio, M.B.; Bae, B.-I.; Hester, L.D.; Dawson, V.L.; Dawson, T.M.; Sawa, A.; Snyder, S.H. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc. Acad. Sci. 2006, 103, 3887–3889. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Nikotina, A.D.; Semenyuk, P.I.; Evstafyeva, D.B.; Mikhaylova, E.R.; Muronetz, V.I.; Shevtsov, M.A.; Tolkacheva, A.V.; Dobrodumov, A.V.; Shavarda, A.L.; et al. Small molecules preventing GAPDH aggregation are therapeutically applicable in cell and rat models of oxidative stress. Radic. Boil. Med. 2016, 92, 29–38. [Google Scholar] [CrossRef]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef]
- Kedi, X.; Ming, Y.; Yongping, W.; Yi, Y.; Xiaoxiang, Z. Free cholesterol overloading induced smooth muscle cells death and activated both ER- and mitochondrial-dependent death pathway. Atherosclerosis 2009, 207, 123–130. [Google Scholar] [CrossRef]
- Persichini, T.; Mariotto, S.; Suzuki, H.; Butturini, E.; Mastrantonio, R.; Cantoni, O.; Colasanti, M. Cross-Talk Between NO Synthase Isoforms in Neuro-Inflammation: Possible Implications in HIV-Associated Neurocognitive Disorders. Med. Chem. 2016, 23, 2706–2714. [Google Scholar] [CrossRef]
- Hwang, S.; Disatnik, M.-H.; Mochly-Rosen, D. Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington’s disease. EMBO Mol. Med. 2015, 7, 1307–1326. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.D. Glyceraldehyde-3-phosphate dehydrogenase as a target for small-molecule disease-modifying therapies in human neurodegenerative disorders. J. Cancer Neurosci. 2004, 29, 337–345. [Google Scholar]
- Nakajima, H.; Amano, W.; Kubo, T.; Fukuhara, A.; Ihara, H.; Azuma, Y.-T.; Tajima, H.; Inui, T.; Sawa, A.; Takeuchi, T. Glyceraldehyde-3-phosphate Dehydrogenase Aggregate Formation Participates in Oxidative Stress-induced Cell Death*. J. Boil. Chem. 2009, 284, 34331–34341. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butera, G.; Mullappilly, N.; Masetto, F.; Palmieri, M.; Scupoli, M.T.; Pacchiana, R.; Donadelli, M. Regulation of Autophagy by Nuclear GAPDH and Its Aggregates in Cancer and Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 2062. https://doi.org/10.3390/ijms20092062
Butera G, Mullappilly N, Masetto F, Palmieri M, Scupoli MT, Pacchiana R, Donadelli M. Regulation of Autophagy by Nuclear GAPDH and Its Aggregates in Cancer and Neurodegenerative Disorders. International Journal of Molecular Sciences. 2019; 20(9):2062. https://doi.org/10.3390/ijms20092062
Chicago/Turabian StyleButera, Giovanna, Nidula Mullappilly, Francesca Masetto, Marta Palmieri, Maria Teresa Scupoli, Raffaella Pacchiana, and Massimo Donadelli. 2019. "Regulation of Autophagy by Nuclear GAPDH and Its Aggregates in Cancer and Neurodegenerative Disorders" International Journal of Molecular Sciences 20, no. 9: 2062. https://doi.org/10.3390/ijms20092062
APA StyleButera, G., Mullappilly, N., Masetto, F., Palmieri, M., Scupoli, M. T., Pacchiana, R., & Donadelli, M. (2019). Regulation of Autophagy by Nuclear GAPDH and Its Aggregates in Cancer and Neurodegenerative Disorders. International Journal of Molecular Sciences, 20(9), 2062. https://doi.org/10.3390/ijms20092062