PTPRK Expression Is Downregulated in Drug Resistant Ovarian Cancer Cell Lines, and Especially in ALDH1A1 Positive CSCs-Like Populations

 , ,
, , 
Abstract
1. Introduction
2. Results
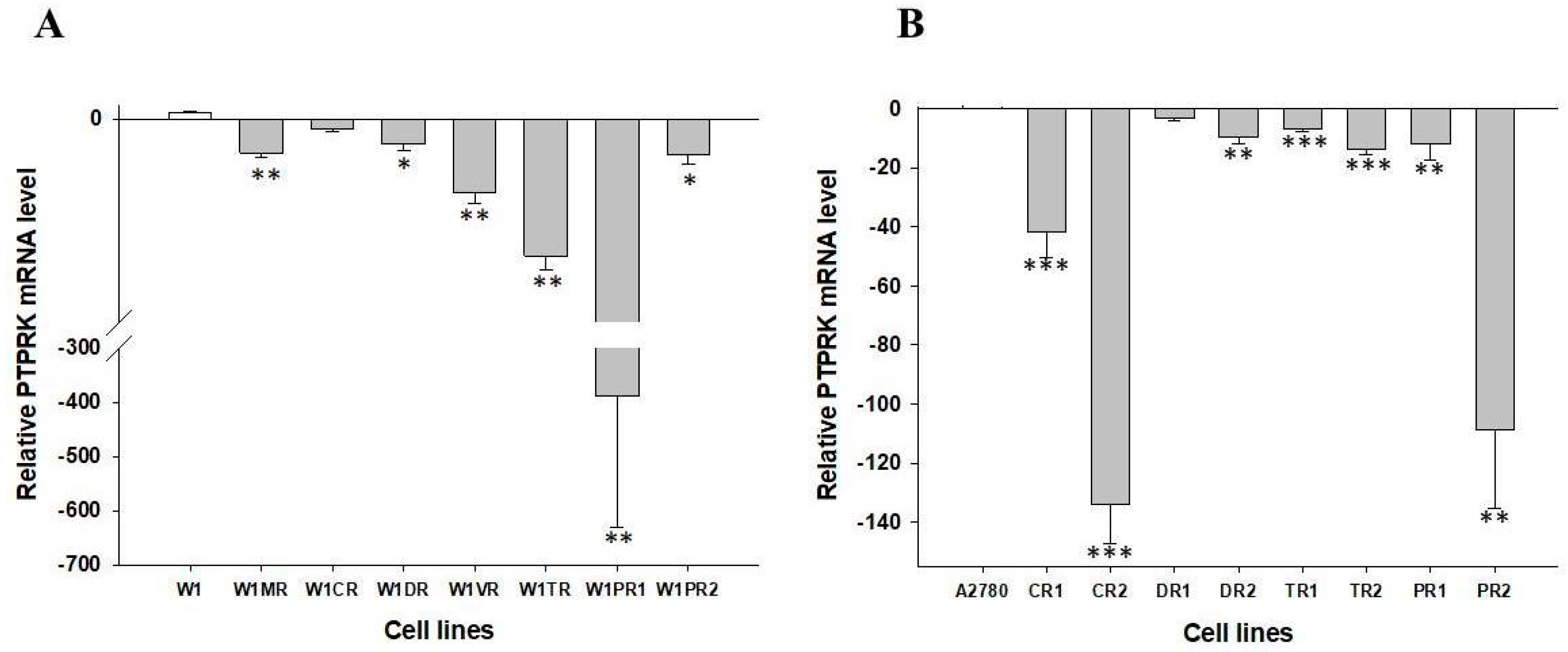
2.1. Analyses of PTPRK Gene Expression in Drug-Resistant Ovarian Cancer Cell Lines
2.2. Immunofluorescence Analysis of PTPRK and pTYR in Drug Sensitive and Resistant Cell Lines
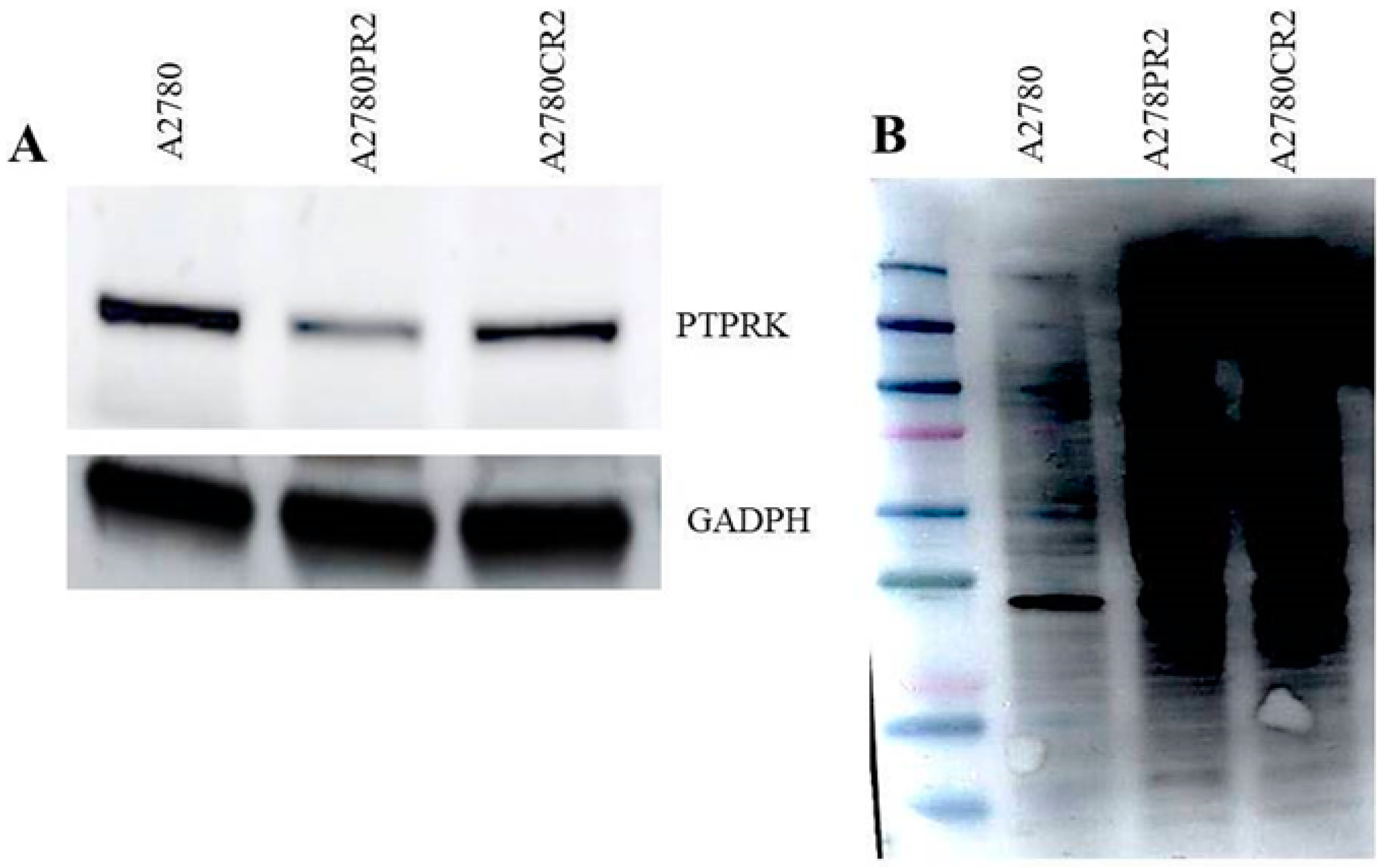
2.3. Western Blot Analysis of PTPRK and pTYR
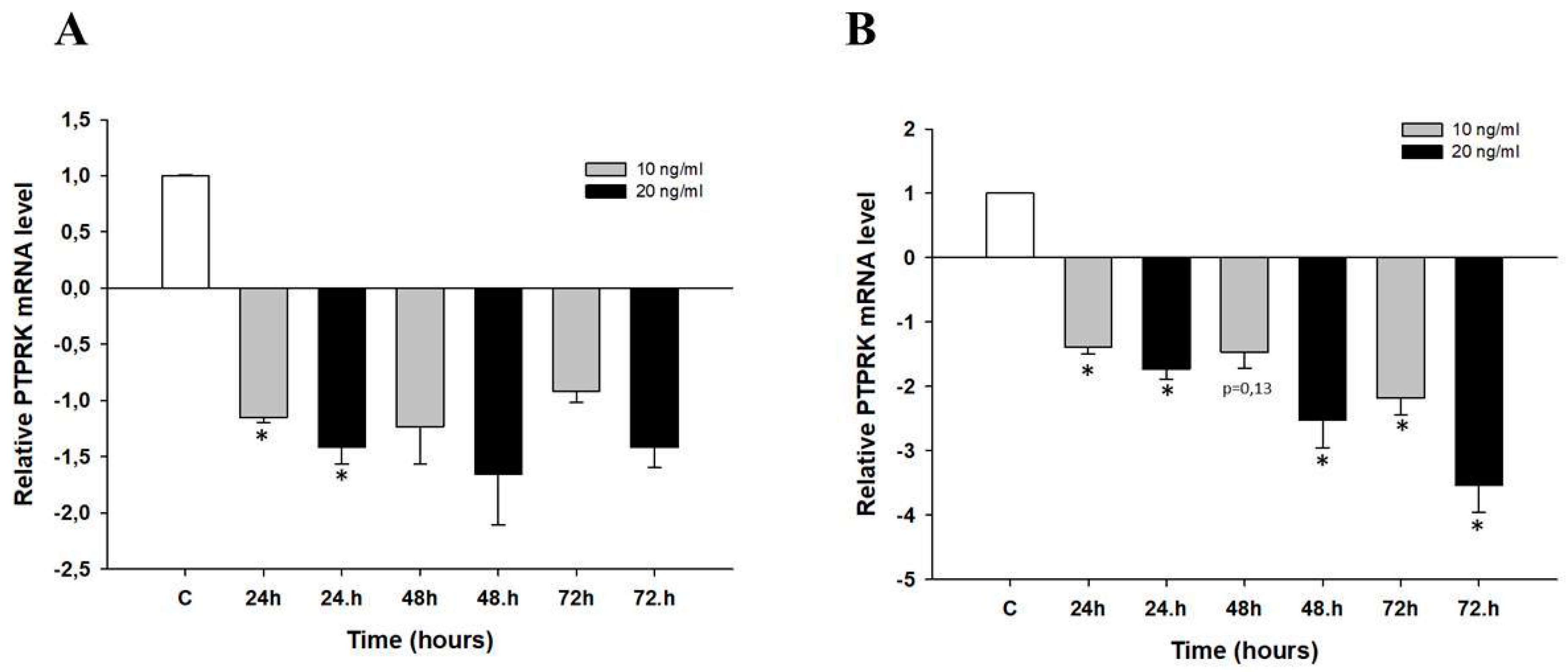
2.4. Early Response to Topotecan (TOP)-Treatment in Ovarian Cancer Cell Lines
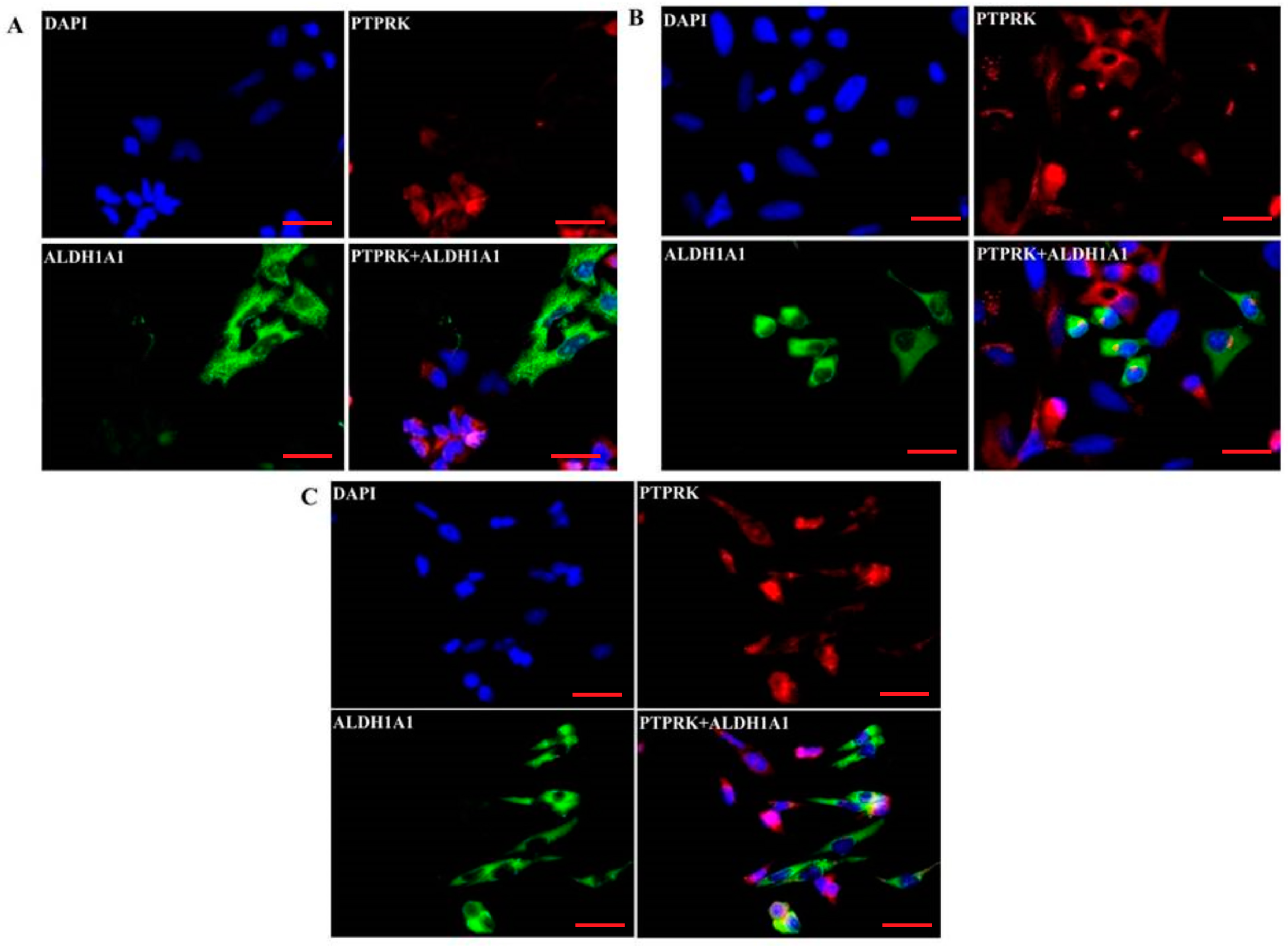
2.5. Co-Expression of PTPRK and ALDH1A1
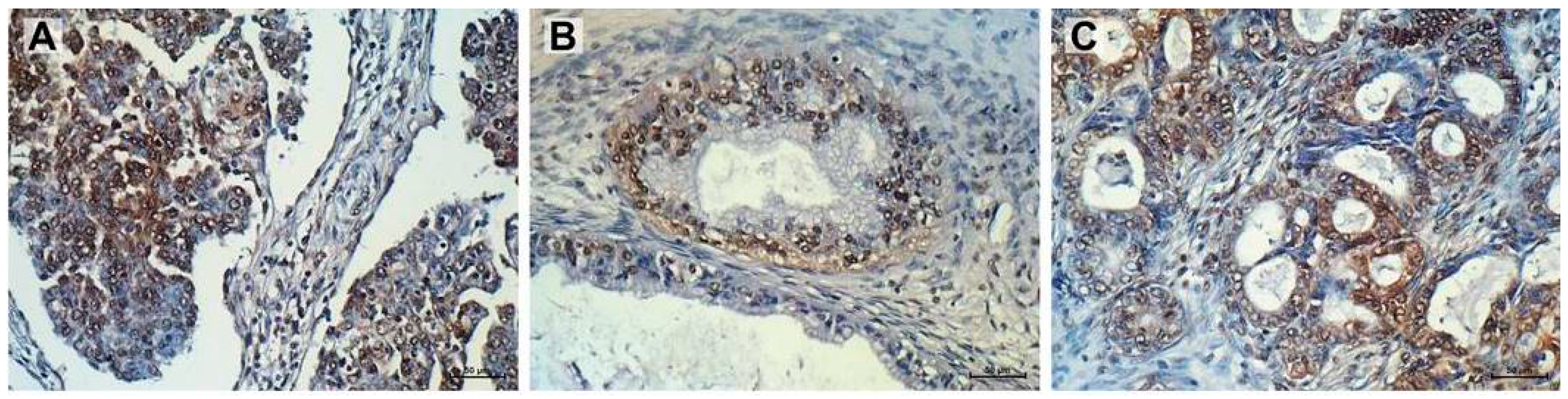
2.6. Immunohistochemistry
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Lines and Cell Culture
4.3. Examination of Gene Expression Using Q-PCR
4.4. Protein Isolation from Cell Culture
4.5. SDS-PAGE and Western Blot Analysis of PTPRK and p-Tyr
4.6. Immunofluorescence Analysis
4.7. Double Immunofluorescence Analysis
4.8. Incubation of Cells with TOP
4.9. Immunohistochemistry
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PTPRK | Protein Tyrosine Phosphatase Receptor Type K |
| CSCs | Cancer Stem Cells |
| CR | Cisplatin Resistance |
| DR | Doxorubicin Resistance |
| MR | Methotrexate Resistance |
| PR | Paclitaxel Resistance |
| TR | Topotecan Resistance |
| VR | Vincristine Resistance |
| ALDH1A1 | Aldehyde Dehydrogenase 1 Family Member A1 |
References
- Webb, P.M.; Jordan, S.J. Epidemiology of epithelial ovarian cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 41, 3–14. [Google Scholar] [CrossRef]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef]
- National Institute for Health and Care Excellence. Guidance on the Use of Paclitaxel in the Treatment of Ovarian Cancer. Available online: http://www.nice.org.uk/guidance/ta55 (accessed on 22 January 2001).
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy. Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Day, E.K.; Sosale, N.G.; Lazzara, M.J. Cell signaling regulation by protein phosphorylation: A multivariate, heterogeneous, and context-dependent process. Curr. Opin. Biotechnol. 2016, 40, 185–192. [Google Scholar] [CrossRef]
- Frankson, R.; Yu, Z.H.; Bai, Y.; Li, Q.; Zhang, R.Y.; Zhang, Z.Y. Therapeutic Targeting of Oncogenic Tyrosine Phosphatases. Cancer Res. 2017, 77, 5701–5705. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Grandis, J.R. Receptor-type protein tyrosine phosphatases in cancer. Chin. J. Cancer 2015, 34, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Abukhdeir, A.M.; Park, B.H. P21 and p27: Roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med. 2008, 10, e19. [Google Scholar] [CrossRef]
- Ostman, A.; Hellberg, C.; Böhmer, F.D. Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer. 2006, 6, 307–320. [Google Scholar] [CrossRef]
- Craig, S.E.; Brady-Kalnay, S.M. Regulation of development and cancer by the R2B subfamily of RPTPs and the implications of proteolysis. Semin. Cell Dev. Biol. 2015, 37, 108–118. [Google Scholar] [CrossRef]
- Eswaran, J.; Debreczeni, J.E.; Longman, E.; Barr, A.J.; Knapp, S. The crystal structure of human receptor protein tyrosine phosphatase kappa phosphatase domain 1. Protein Sci. 2006, 15, 1500–1505. [Google Scholar] [CrossRef]
- Agarwal, S.; Al-Keilani, M.S.; Alqudah, M.A.Y.; Sibenaller, Z.A.; Ryken, T.C.; Assem, M. Tumor Derived Mutations of Protein Tyrosine Phosphatase Receptor Type K Affect Its Function and Alter Sensitivity to Chemotherapeutics in Glioma. PLoS ONE 2013, 8, e62852. [Google Scholar] [CrossRef]
- Sun, P.H.; Ye, L.; Mason, M.D.; Jiang, W.G. Protein tyrosine phosphatase kappa (PTPRK) is a negative regulator of adhesion and invasion of breast cancer cells, and associates with poor prognosis of breast cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 1129–1139. [Google Scholar] [CrossRef]
- Chen, Y.W.; Guo, T.; Shen, L.; Wong, K.Y.; Tao, Q.; Choi, W.W.; Au-Yeung, R.K.; Chan, Y.P.; Wong, M.L.; Tang, J.C.; et al. Receptor-type tyrosine-protein phosphatase κ directly targets STAT3 activation for tumor suppression in nasal NK/T-cell lymphoma. Blood 2015, 125, 1589–1600. [Google Scholar] [CrossRef]
- Lupia, M.; Cavallaro, U. Ovarian cancer stem cells: Still an elusive entity? Mol. Cancer 2017, 16, 64. [Google Scholar] [CrossRef]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef]
- Januchowski, R.; Wojtowicz, K.; Zabel, M. The role of aldehyde dehydrogenase (ALDH) in cancer drug resistance. Biomed. Pharmacother. 2013, 67, 669–680. [Google Scholar] [CrossRef]
- Croker, A.K.; Rodriguez-Torres, M.; Xia, Y.; Pardhan, S.; Leong, H.S.; Lewis, J.D.; Allan, A.L. Differential Functional Roles of ALDH1A1 and ALDH1A3 in Mediating Metastatic Behavior and Therapy Resistance of Human Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 2039. [Google Scholar] [CrossRef]
- Januchowski, R.; Wojtowicz, K.; Sterzyńska, K.; Sosińska, P.; Andrzejewska, M.; Zawierucha, P.; Nowicki, M.; Zabel, M. Inhibition of ALDH1A1 activity decreases expression of drug transporters and reduces chemotherapy resistance in ovarian cancer cell lines. Int. J. Biochem. Cell Biol. 2016, 78, 248–259. [Google Scholar] [CrossRef]
- Januchowski, R.; Sterzyńska, K.; Zawierucha, P.; Ruciński, M.; Świerczewska, M.; Partyka, M.; Bednarek-Rajewska, K.; Brązert, M.; Nowicki, M.; Zabel, M.; et al. Microarray-based detection and expression analysis of new genes associated with drug resistance in ovarian cancer cell lines. Oncotarget 2017, 8, 49944–49958. [Google Scholar] [CrossRef]
- Sterzyńska, K.; Klejewski, A.; Wojtowicz, K.; Świerczewska, M.; Nowacka, M.; Kaźmierczak, D.; Andrzejewska, M.; Rusek, D.; Brązert, M.; Brązert, J.; et al. Mutual Expression of ALDH1A1, LOX, and Collagens in Ovarian Cancer Cell Lines as Combined CSCs- and ECM-Related Models of Drug Resistance Development. Int. J. Mol. Sci. 2018, 20, 54. [Google Scholar] [CrossRef] [PubMed]
- Freimund, A.E.; Beach, J.A.; Christie, E.L.; Bowtell, D.D.L. Mechanisms of Drug Resistance in High-Grade Serous Ovarian Cancer. Hematol. Oncol. Clin. North Am. 2018, 32, 983–996. [Google Scholar] [CrossRef]
- Wojtowicz, K.; Januchowski, R.; Nowicki, M.; Zabel, M. Inhibition of protein glycosylation reverses the MDR phenotype of cancer cell lines. Biomed. Pharmacother. 2015, 74, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updat. 2012, 15, 39–49. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Lee, J.T.; Chang, F.; Bertrand, F.E.; Navolanic, P.M.; Terrian, D.M.; Franklin, R.A.; D’Assoro, A.B.; et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzyme Regul. 2006, 46, 249–279. [Google Scholar] [CrossRef]
- Januchowski, R.; Wojtowicz, K.; Sujka-Kordowska, P.; Andrzejewska, M.; Zabel, M. MDR gene expression analysis of six drug-resistant ovarian cancer cell lines. Biomed. Res. Int. 2013, 2013, 241763. [Google Scholar] [CrossRef]
- Januchowski, R.; Sterzyńska, K.; Zaorska, K.; Sosińska, P.; Klejewski, A.; Brązert, M.; Nowicki, M.; Zabel, M. Analysis of MDR genes expression and cross-resistance in eight drug resistant ovarian cancer cell lines. J. Ovarian Res. 2016, 9, 65. [Google Scholar] [CrossRef]
- Januchowski, R.; Świerczewska, M.; Sterzyńska, K.; Wojtowicz, K.; Nowicki, M.; Zabel, M. Increased expression of several collagen genes is associated with drug resistance in ovarian cancer cell lines. J. Cancer 2016, 25, 1295–1310. [Google Scholar] [CrossRef]
- Klejewski, A.; Sterzyńska, K.; Wojtowicz, K.; Świerczewska, M.; Partyka, M.; Brązert, M.; Nowicki, M.; Zabel, M.; Januchowski, R. The significance of lumican expression in ovarian cancer drug-resistant cell lines. Oncotarget 2017, 8, 74466–74478. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Zawierucha, P.; Ruciński, M.; Nowicki, M.; Zabel, M. Extracellular matrix proteins expression profiling in chemoresistant variants of the A2780 ovarian cancer cell line. Biomed. Res. Int. 2014, 2014, 365867. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Zawierucha, P.; Ruciński, M.; Zabel, M. Microarray-based detection and expression analysis of extracellular matrix proteins in drug resistant ovarian cancer cell lines. Oncol. Rep. 2014, 32, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Sterzyńska, K.; Klejewski, A.; Wojtowicz, K.; Świerczewska, M.; Andrzejewska, M.; Rusek, D.; Sobkowski, M.; Kędzia, W.; Brązert, J.; Nowicki, M.; et al. The Role of Matrix Gla Protein (MGP) Expression in Paclitaxel and Topotecan Resistant Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2018, 19, 2901. [Google Scholar] [CrossRef] [PubMed]
- Sterzyńska, K.; Klejewski, A.; Świerczewska, M.; Nowicki, M.; Brązert, J.; Januchowski, R. Myotilin, a New Topotecan Resistant Protein in Ovarian Cancer Cell Lines. J. Cancer 2018, 9, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- Świerczewska, M.; Klejewski, A.; Brązert, M.; Kaźmierczak, D.; Iżycki, D.; Nowicki, M.; Zabel, M.; Januchowski, R. New and Old Genes Associated with Primary and Established Responses to Paclitaxel Treatment in Ovarian Cancer Cell Lines. Molecules 2018, 23, 891. [Google Scholar] [CrossRef]
- Klejewski, A.; Świerczewska, M.; Zaorska, K.; Brązert, M.; Nowicki, M.; Zabel, M.; Januchowski, R. New and Old Genes Associated with Topotecan Resistance Development in Ovarian Cancer Cell Lines. Anticancer Res. 2017, 37, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Świerczewska, M.; Klejewski, A.; Wojtowicz, K.; Brązert, M.; Iżycki, D.; Nowicki, M.; Zabel, M.; Januchowski, R. New and Old Genes Associated with Primary and Established Responses to Cisplatin and Topotecan Treatment in Ovarian Cancer Cell Lines. Molecules 2017, 22, 1717. [Google Scholar] [CrossRef]
- McArdle, L.; Rafferty, M.; Maelandsmo, G.M.; Bergin, O.; Farr, C.J.; Dervan, P.A.; O’Loughlin, S.; Herlyn, M.; Easty, D.J. Protein tyrosine phosphatase genes downregulated in melanoma. J. Invest. Dermatol. 2001, 117, 1255–1260. [Google Scholar] [CrossRef]
- Nakamura, M.; Kishi, M.; Sakaki, T.; Hashimoto, H.; Nakase, H.; Shimada, K.; Ishida, E.; Konishi, N. Novel tumor suppressor loci on 6q22-23 in primary central nervous system lymphomas. Cancer Res. 2003, 63, 737–741. [Google Scholar] [PubMed]
- Stevenson, W.S.; Best, O.G.; Przybylla, A.; Chen, Q.; Singh, N.; Koleth, M.; Pierce, S.; Kennedy, T.; Tong, W.; Kuang, S.Q.; et al. DNA methylation of membrane-bound tyrosine phosphatase genes in acute lymphoblastic leukaemia. Leukemia 2014, 28, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Assem, M.; Sibenaller, Z.; Agarwal, S.; Al-Keilani, M.S.; Alqudah, M.A.; Ryken, T.C. Enhancing diagnosis, prognosis, and therapeutic outcome prediction of gliomas using genomics. OMICS 2012, 16, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Stolarczyk, E.I.; Reiling, C.J.; Paumi, C.M. Regulation of ABC transporter function via phosphorylation by protein kinases. Curr. Pharm. Biotechnol. 2011, 12, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xu, K.; Linn, D.E.; Yang, X.; Guo, Z.; Shimelis, H.; Nakanishi, T.; Ross, D.D.; Chen, H.; Fazli, L.; et al. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J. Biol. Chem. 2008, 283, 3349–3356. [Google Scholar] [CrossRef] [PubMed]
- Weinstein-Oppenheimer, C.R.; Henríquez-Roldán, C.F.; Davis, J.M.; Navolanic, P.M.; Saleh, O.A.; Steelman, L.S.; Franklin, R.A.; Robinson, P.J.; McMahon, M.; McCubrey, J.A. Role of the Raf signal transduction cascade in the in vitro resistance to the anticancer drug doxorubicin. Clin. Cancer Res. 2001, 7, 2898–2907. [Google Scholar] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.A.M.A.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef]
- Januchowski, R.; Wojtowicz, K.; Andrzejewska, M.; Zabel, M. Expression of MDR1 and MDR3 gene products in paclitaxel-, doxorubicin- and vincristine-resistant cell lines. Biomed. Pharmacother. 2014, 68, 111–117. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Sequence (5′–3′ Direction) | Ensembl Transcript (ENST) Number (Available Online: http://www.ensembl.org) | Product Size (bp) |
|---|---|---|---|
| PTPRK | CCCAGGACCTCCACTAATCA ATTCCCAGTCCACAGCAATC | 00000368226 | 110 bp |
| GADPH | GAAGGTGAAGGTCGGAGTCA GACAAGCTTCCCGTTCTCAG | 00000229239 | 199 bp |
| β-actin | TCTGGCACCACACCTTCTAC GATAGCACAGCCTGGATAGC | 00000331789 | 169 bp |
| HRPT1 | CTGAGGATTTGGAAAGGGTG AATCCAGCAGGTCAGCAAAG | 00000298556 | 156 bp |
| β2M | CGCTACTCTCTCTTTCTGGC ATGTCGGATGGATGAAACCC | 00000558401 | 133 bp |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Świerczewska, M.; Sterzyńska, K.; Wojtowicz, K.; Kaźmierczak, D.; Iżycki, D.; Nowicki, M.; Zabel, M.; Januchowski, R. PTPRK Expression Is Downregulated in Drug Resistant Ovarian Cancer Cell Lines, and Especially in ALDH1A1 Positive CSCs-Like Populations. Int. J. Mol. Sci. 2019, 20, 2053. https://doi.org/10.3390/ijms20082053
Świerczewska M, Sterzyńska K, Wojtowicz K, Kaźmierczak D, Iżycki D, Nowicki M, Zabel M, Januchowski R. PTPRK Expression Is Downregulated in Drug Resistant Ovarian Cancer Cell Lines, and Especially in ALDH1A1 Positive CSCs-Like Populations. International Journal of Molecular Sciences. 2019; 20(8):2053. https://doi.org/10.3390/ijms20082053
Chicago/Turabian StyleŚwierczewska, Monika, Karolina Sterzyńska, Karolina Wojtowicz, Dominika Kaźmierczak, Dariusz Iżycki, Michał Nowicki, Maciej Zabel, and Radosław Januchowski. 2019. "PTPRK Expression Is Downregulated in Drug Resistant Ovarian Cancer Cell Lines, and Especially in ALDH1A1 Positive CSCs-Like Populations" International Journal of Molecular Sciences 20, no. 8: 2053. https://doi.org/10.3390/ijms20082053
APA StyleŚwierczewska, M., Sterzyńska, K., Wojtowicz, K., Kaźmierczak, D., Iżycki, D., Nowicki, M., Zabel, M., & Januchowski, R. (2019). PTPRK Expression Is Downregulated in Drug Resistant Ovarian Cancer Cell Lines, and Especially in ALDH1A1 Positive CSCs-Like Populations. International Journal of Molecular Sciences, 20(8), 2053. https://doi.org/10.3390/ijms20082053