Pigment Nephropathy: Novel Insights into Inflammasome-Mediated Pathogenesis

 , and
, and
Abstract
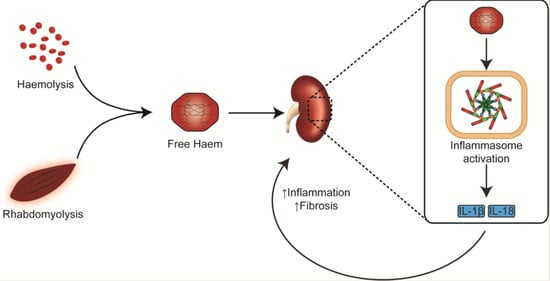
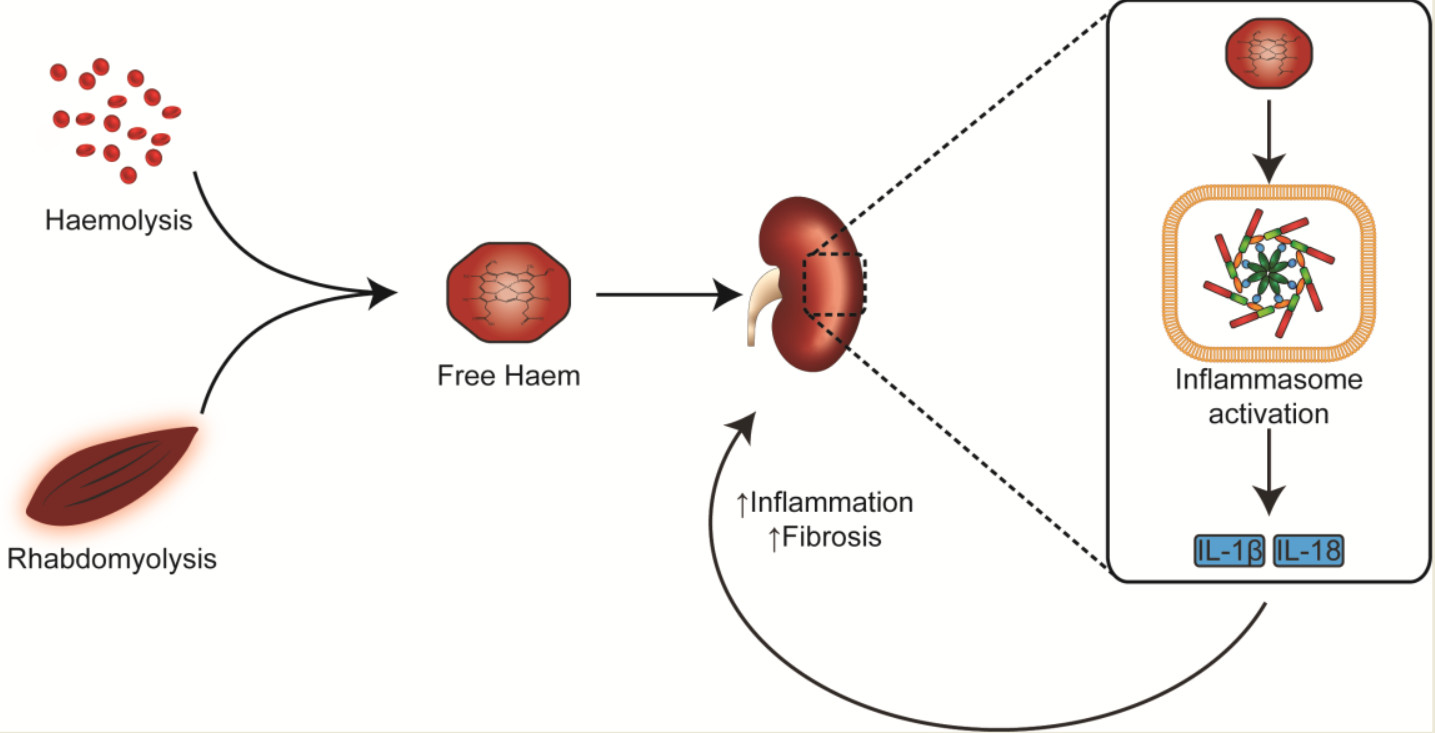{kind=link}
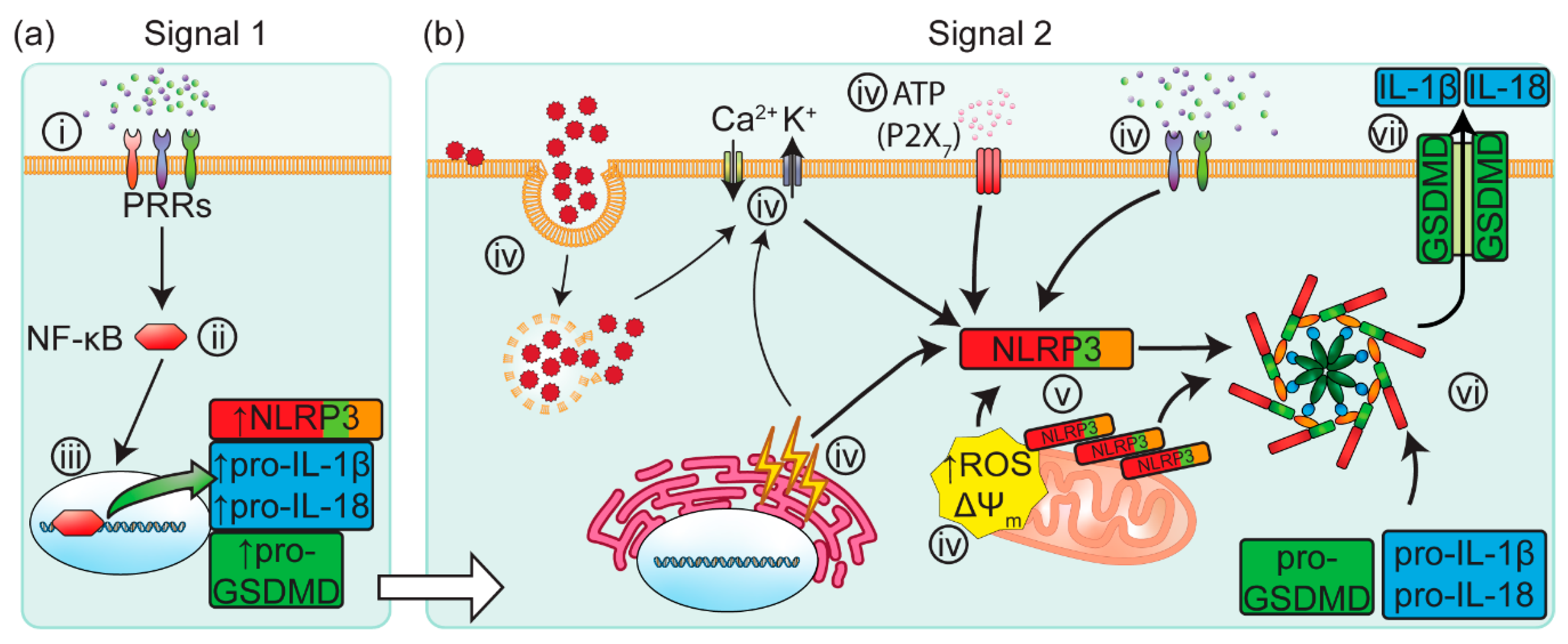{kind=link}
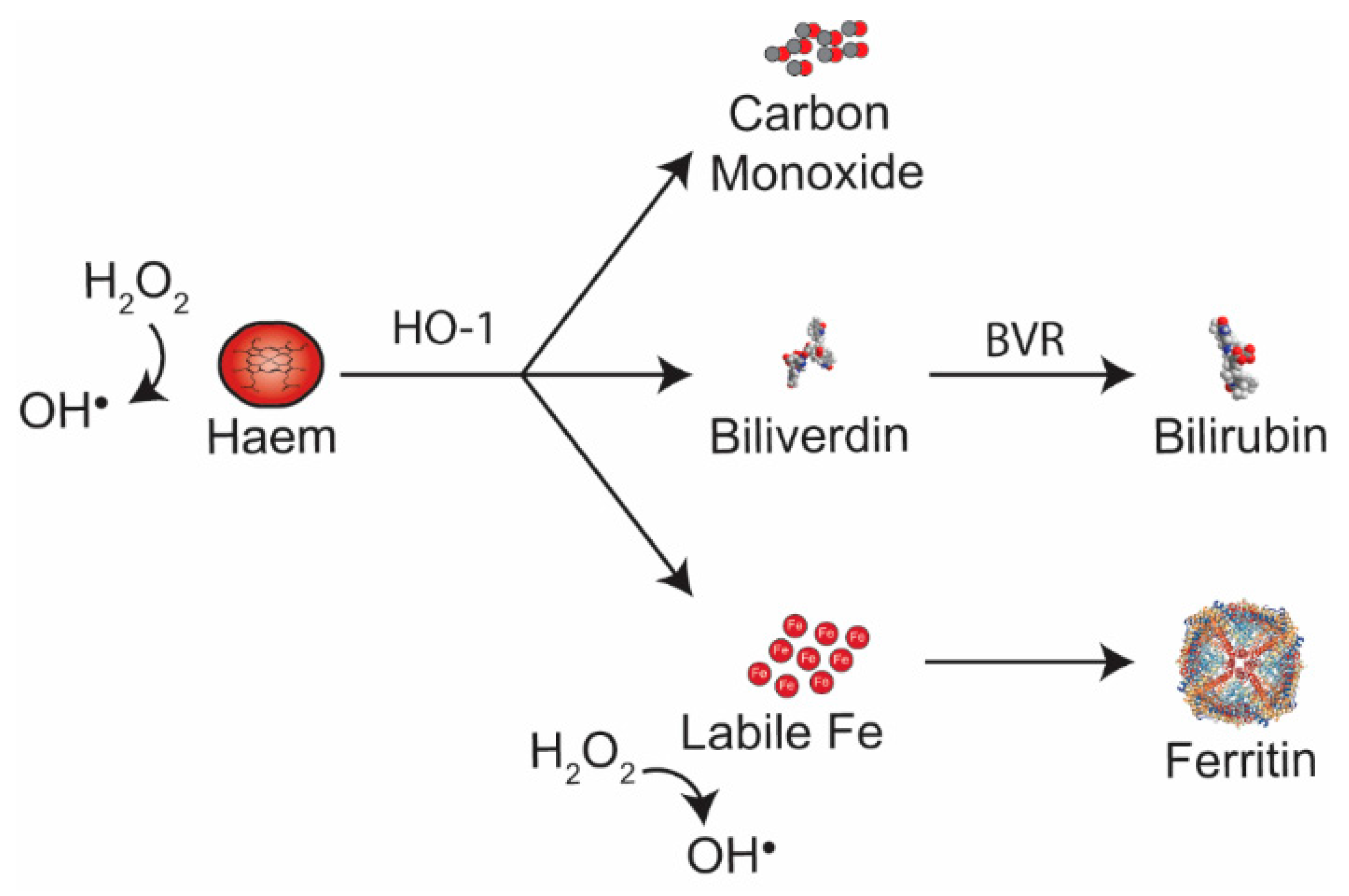{kind=link}
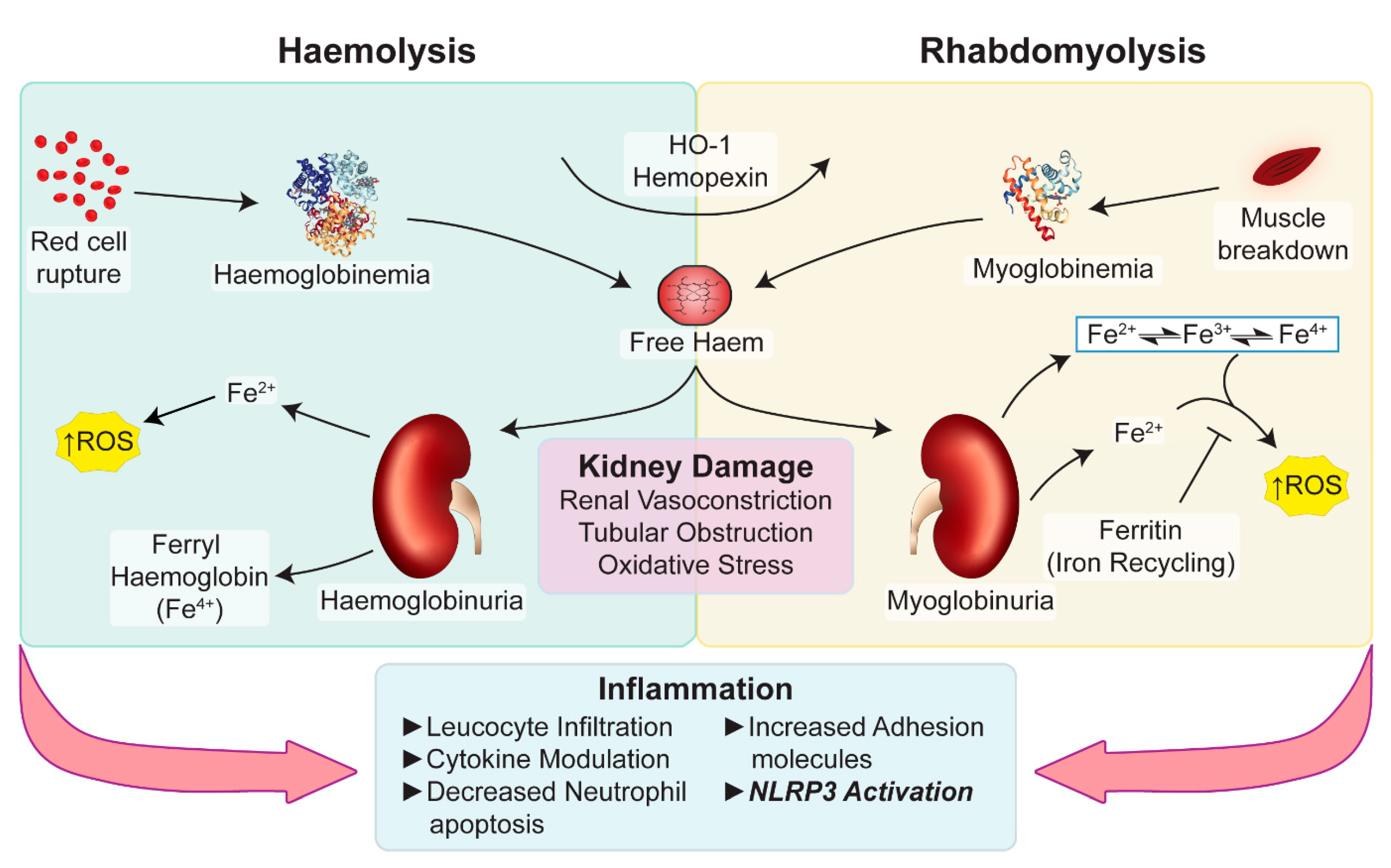{kind=link}
1. Introduction
2. The Nucleotide-Binding domain-Like Receptor Protein 3 (NLRP3) Inflammasome
2.1. Canonical Inflammasome Activation
2.2. Non-Canonical Inflammasome Activation
2.3. Inflammasomes in the Kidney
3. Haem Catabolism and Role in Immune-Mediated Pathology
4. Myoglobin-Mediated Pigment Nephropathy
5. Haemoglobin-Mediated Pigment Nephropathy
6. Inflammasome Inhibition as a Potential Therapeutic Target
6.1. NLRP3 Inflammasome Inhibitors
6.2. Anti-IL-1β and IL-1 Receptor Antagonists
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKI | Acute Kidney Injury |
| ASC | Apoptosis-associated speck-like protein containing a CARD domain |
| ATP | Adenosine Triphosphate |
| CARD | Caspase activation and recruitment domain |
| CD | Cluster of differentiation |
| CKD | Chronic Kidney Disease |
| DAMPs | Damage-associated molecular patterns |
| DC | Dendritic cells |
| DKD | Diabetic kidney disease |
| ESCRT | Endosomal sorting complexes required for transport |
| FMF | Familial Mediterranean Fever |
| GSDMD | Gasdermin-D |
| HAMPs | Homeostasis-altering molecular processes |
| HO | Haem Oxygenase |
| HO-1 | Haem Oxygenase-1 |
| ICAM-l | Intercellular Adhesion Molecule-1 |
| IL | Interleukin |
| IL-1R | IL-1 receptor |
| LPS | Lipopolysaccharide |
| MAVS | Mitochondrial antiviral signal |
| mtDNA | Mitochondrial DNA |
| NADPH | Dihydronicotinamide-adenine dinucleotide phosphate |
| NLRP3 | Nucleotide-binding domain-like receptor protein 3 |
| PAMPs | Pathogen-associated molecular patterns |
| PN | Pigment Nephropathy |
| PRRs | Pattern recognition receptors |
| PTEC | Proximal Tubule Epithelial Cells |
| RBC | Red Blood Cells |
| ROS | Reactive Oxygen Species |
| TLRs | Toll-like receptors |
| UUO | Unilateral ureteral obstruction |
| VCAM-l | Vascular Adhesion Molecule-1 |
| XO | Xanthine Oxidase |
References
- Sikorski, Z.E. Chemical and Functional Properties of Food Components; CRC Press: Boc Raton, FL, USA, 2007. [Google Scholar]
- Paoli, M.; Marles-Wright, J.; Smith, A. Structure–Function Relationships in Heme-Proteins. DNA Cell Biol. 2002, 21, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.; Gouveia, Z.; Soares, M.P.; Gozzelino, R.; Kapitulnik, J.; Hebrew, T.; Ryter, S.W.; Immenschuh, S. Heme cytotoxicity and the pathogenesis of immune-mediated inflammatory diseases. Front. Pharmacol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.J.; Kahraman, A.; Thornton, J.M. Heme proteins-Diversity in structural characteristics, function, and folding. Proteins Struct. Function Bioinform. 2010, 78, 2349–2368. [Google Scholar] [CrossRef] [PubMed]
- Mense, S.M.; Zhang, L. Heme: A versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006, 16, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Immenschuh, S.; Vijayan, V.; Janciauskiene, S.; Gueler, F. Heme as a Target for Therapeutic Interventions. Front Pharmacol. 2017, 8, 146. [Google Scholar] [CrossRef]
- Nangaku, M. Hypoxia and Tubulointerstitial Injury: A Final Common Pathway to End-Stage Renal Failure. Nephron Exp. Nephrol. 2004, 98, e8–e12. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef]
- Perazella, M.A.; Rosner, M.H. Clinical features and diagnosis of heme pigment-induced acute kidney injury. Available online: https://www.uptodate.com/contents/clinical-features-and-diagnosis-of-heme-pigment-induced-acute-kidney-injury (accessed on 7 February 2019).
- Gois, P.H.F.; Canale, D.; Volpini, R.A.; Ferreira, D.; Veras, M.M.; Andrade-Oliveira, V.; Câmara, N.O.S.; Shimizu, M.H.M.; Seguro, A.C. Allopurinol attenuates rhabdomyolysis-associated acute kidney injury: Renal and muscular protection. Free Radic. Biol. Med. 2016, 101, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Rosen, S.; Fuchs, S.; Epstein, F.H.; Brezis, M. Myoglobinuric acute renal failure in the rat: a role for medullary hypoperfusion, hypoxia, and tubular obstruction. J. Am. Soc. Nephrol. 1996, 7, 1066–1074. [Google Scholar]
- Moore, K.P.; Holt, S.G.; Patel, R.P.; Svistunenko, D.A.; Zackert, W.; Goodier, D.; Reeder, B.J.; Clozel, M.; Anand, R.; Cooper, C.E.; et al. A causative role for redox cycling of myoglobin and its inhibition by alkalinization in the pathogenesis and treatment of rhabdomyolysis-induced renal failure. J. Biol. Chem. 1998, 273, 31731–31737. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A.; Burkhart, K. Myoglobin toxicity in proximal human kidney cells: roles of Fe, Ca2+, H2O2, and terminal mitochondrial electron transport. Kidney Int. 1997, 51, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.F.; Bozza, M.T. Heme on innate immunity and inflammation. Front. Pharmacol. 2014, 5, 115. [Google Scholar] [CrossRef]
- Erdei, J.; Tóth, A.; Balogh, E.; Nyakundi, B.B.; Bányai, E.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxid. Med. Cell. Longev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Usui, F.; Kawashima, A.; Kimura, H.; Karasawa, T.; Inoue, Y.; Kobayashi, M.; Mizushina, Y.; Kasahara, T.; Taniguchi, S.I.; et al. Role of NLRP3 inflammasomes for rhabdomyolysis-induced acute kidney injury. Sci. Rep. 2015. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Brähler, S.; Zinselmeyer, B.H.; Raju, S.; Nitschke, M.; Suleiman, H.; Saunders, B.T.; Johnson, M.W.; Böhner, A.M.C.; Viehmann, S.F.; Theisen, D.J.; et al. Opposing Roles of Dendritic Cell Subsets in Experimental GN. J. Am. Soc. Nephrol. 2017. [Google Scholar] [CrossRef]
- Ludwig-Portugall, I.; Bartok, E.; Dhana, E.; Evers, B.D.G.; Primiano, M.J.; Hall, J.P.; Franklin, B.S.; Knolle, P.A.; Hornung, V.; Hartmann, G.; et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2016, 90, 525–539. [Google Scholar] [CrossRef]
- Anders, H.J.; Muruve, D.A. The Inflammasomes in Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 1007–1018. [Google Scholar] [CrossRef]
- Wilson, G.J.; Gois, P.H.F.; Zhang, A.; Wang, X.; Law, B.M.P.; Kassianos, A.J.; Healy, H.G. The Role of Oxidative Stress and Inflammation in Acute Oxalate Nephropathy Associated with Ethylene Glycol Intoxication. Kidney Int. Rep. 2018, 3, 1217–1221. [Google Scholar] [CrossRef]
- Knauf, F.; Asplin, J.R.; Granja, I.; Schmidt, I.M.; Moeckel, G.W.; David, R.J.; Flavell, R.A.; Aronson, P.S. NALP3-mediated inflammation is a principal cause of progressive renal failure in oxalate nephropathy. Kidney Int. 2013, 84, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Katsiki, N.; Butler, A.E.; Sahebkar, A. Effects of antidiabetic drugs on NLRP3 inflammasome activity, with a focus on diabetic kidneys. Drug Discov. Today 2018. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Kulkarni, O.P.; Rupanagudi, K.V.; Migliorini, A.; Darisipudi, M.N.; Vilaysane, A.; Muruve, D.; Shi, Y.; Munro, F.; Liapis, H.; et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J. Clin. Investig. 2013, 123, 236–246. [Google Scholar] [CrossRef]
- Rajamäki, K.; Lappalainen, J.; Öörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol Crystals Activate the NLRP3 Inflammasome in Human Macrophages: A Novel Link between Cholesterol Metabolism and Inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357. [Google Scholar] [CrossRef] [PubMed]
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef]
- Amores-Iniesta, J.; Barberà-Cremades, M.; Martínez, C.M.; Pons, J.A.; Revilla-Nuin, B.; Martínez-Alarcón, L.; Di Virgilio, F.; Parrilla, P.; Baroja-Mazo, A.; Pelegrín, P. Extracellular ATP Activates the NLRP3 Inflammasome and Is an Early Danger Signal of Skin Allograft Rejection. Cell Rep. 2017, 21, 3414–3426. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, J.; Zhang, W.; Zhang, J.; Yang, J.; Li, K.; He, Y. ATP-P2X4 signaling mediates NLRP3 inflammasome activation: A novel pathway of diabetic nephropathy. Int. J. Biochem. Cell Biol. 2013, 45, 932–943. [Google Scholar] [CrossRef]
- Sadatomi, D.; Nakashioya, K.; Mamiya, S.; Honda, S.; Tanimura, S.; Yamamura, Y.; Kameyama, Y.; Takeda, K. Mitochondrial function is required for extracellular ATP-induced NLRP3 inflammasome activation. J. Biochem. 2017, 161, 503–512. [Google Scholar] [CrossRef]
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.j.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-M.; Hu, W.; Troutman, T.D.; Jennings, M.; Brewer, T.; Li, X.; Nanda, S.; Cohen, P.; Thomas, J.A.; Pasare, C. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. USA 2014, 111, 775–780. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Hari, A.; Zhang, Y.; Tu, Z.; Detampel, P.; Stenner, M.; Ganguly, A.; Shi, Y. Activation of NLRP3 inflammasome by crystalline structures via cell surface contact. Sci. Rep. 2014, 4, 7281. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847. [Google Scholar] [CrossRef] [PubMed]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nuñez, G.; He, Y.; Yin, X.-M.; O’Riordan, M.X.D. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018. [Google Scholar] [CrossRef]
- Monteleone, M.; Stanley, A.C.; Chen, K.W.; Brown, D.L.; Bezbradica, J.S.; von Pein, J.B.; Holley, C.L.; Boucher, D.; Shakespear, M.R.; Kapetanovic, R.; et al. Interleukin-1β Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep. 2018, 24, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Sborgi, L.; Rühl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Müller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Rühl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef]
- Pellegrini, C.; Antonioli, L.; Lopez-Castejon, G.; Blandizzi, C.; Fornai, M. Canonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front. Immunol. 2017, 8, 36. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Knodler, L.A.; Crowley, S.M.; Sham, H.P.; Yang, H.; Wrande, M.; Ma, C.; Ernst, R.K.; Steele-Mortimer, O.; Celli, J.; Vallance, B.A. Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses against Enteric Bacterial Pathogens. Cell Host Microbe 2014, 16, 249–256. [Google Scholar] [CrossRef]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 Inflammasome Promotes Renal Inflammation and Contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef]
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.D.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Gersch, M.S.; Johnson, R.J. Uric acid and the immune response. Nephrol. Dial. Transplant. 2006, 21, 3046–3047. [Google Scholar] [CrossRef] [PubMed]
- Ives, A.; Nomura, J.; Martinon, F.; Roger, T.; LeRoy, D.; Miner, J.N.; Simon, G.; Busso, N.; So, A. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat. Commun. 2015, 6, 6555. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, Y.G.; Kim, D.J.; Park, S.H.; Jeong, K.H.; Lee, Y.H.; Lim, S.J.; Lee, S.H.; Moon, J.Y. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef] [PubMed]
- Kassianos, A.J.; Wang, X.; Sampangi, S.; Muczynski, K.; Healy, H.; Wilkinson, R. Increased tubulointerstitial recruitment of human CD141hi CLEC9A+ and CD1c+ myeloid dendritic cell subsets in renal fibrosis and chronic kidney disease. AJP Renal Physiol. 2013, 305, F1391–F1401. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.M.; Ling, Y.H.; Huuskes, B.M.; Ferens, D.M.; Saini, N.; Chan, C.T.; Diep, H.; Kett, M.M.; Samuel, C.S.; Kemp-Harper, B.K.; et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc. Res. 2019, 115, 776–787. [Google Scholar] [CrossRef]
- Paller, M.S. Hemoglobin- and myoglobin-induced acute renal failure in rats: Role of iron in nephrotoxicity. Am. J. Physiol. 1988, 255, F539–F544. [Google Scholar] [CrossRef]
- Zager, R.A.; Gamelin, L.M. Pathogenetic mechanisms in experimental hemoglobinuric acute renal failure. Am. J. Physiol. 1989, 256, F446–F455. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef]
- Qian, Q.; Nath, K.A.; Wu, Y.; Daoud, T.M.; Sethi, S. Hemolysis and acute kidney failure. Am. J. Kidney Dis. 2010. [Google Scholar] [CrossRef] [PubMed]
- Tracz, M.J.; Alam, J.; Nath, K.A. Physiology and Pathophysiology of Heme: Implications for Kidney Disease. J. Am. Soc. Nephrol. 2007. [Google Scholar] [CrossRef]
- Wagener, F.A.D.T.G.; Eggert, A.; Boerman, O.C.; Oyen, W.J.G.; Verhofstad, A.; Abraham, N.G.; Adema, G.; Van Kooyk, Y.; De Witte, T.; Figdor, C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001. [Google Scholar] [CrossRef]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [PubMed]
- Porto, B.N.; Alves, L.S.; Fernández, P.L.; Dutra, T.P.; Figueiredo, R.T.; Graça-Souza, A.V.; Bozza, M.T. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J. Biol. Chem. 2007. [Google Scholar] [CrossRef]
- Graça-Souza, A.V.; Arruda, M.A.B.; De Freitas, M.S.; Barja-Fidalgo, C.; Oliveira, P.L. Neutrophil activation by heme: Implications for inflammatory processes. Blood 2002. [Google Scholar] [CrossRef]
- Arruda, M.A.; Rossi, A.G.; de Freitas, M.S.; Barja-Fidalgo, C.; Graça-Souza, A.V. Heme inhibits human neutrophil apoptosis: Involvement of phosphoinositide 3-kinase, MAPK, and NF-κB. J. Immunol. 2004, 173, 2023–2030. [Google Scholar] [CrossRef]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graça-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of toll-like receptor 4. J. Biolog. Chem. 2007. [Google Scholar] [CrossRef]
- Wagener, F.A.; Feldman, E.; de Witte, T.; Abraham, N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1997, 216, 456–463. [Google Scholar] [CrossRef]
- Lv, J.; Su, W.; Yu, Q.; Zhang, M.; Di, C.; Lin, X.; Wu, M.; Xia, Z. Heme oxygenase-1 protects airway epithelium against apoptosis by targeting the proinflammatory NLRP3–RXR axis in asthma. J. Biol. Chem. 2018, 293, 18454–18465. [Google Scholar] [CrossRef]
- Lorenz, G.; Darisipudi, M.N.; Anders, H.J. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol. Dial. Transplant. 2014, 29, 41–48. [Google Scholar] [CrossRef]
- Bosch, X.; Poch, E.; Grau, J.M. Rhabdomyolysis and Acute Kidney Injury. N. Engl. J. Med. 2009, 361, 62–72. [Google Scholar] [CrossRef]
- Korrapati, M.C.; Shaner, B.E.; Schnellmann, R.G. Recovery from Glycerol-Induced Acute Kidney Injury Is Accelerated by Suramin. J. Pharmacol. Exp. Ther. 2012. [Google Scholar] [CrossRef]
- Desforges, J.F.; Better, O.S.; Stein, J.H. Early Management of Shock and Prophylaxis of Acute Renal Failure in Traumatic Rhabdomyolysis. N. Engl. J. Med. 1990, 322, 825–829. [Google Scholar] [CrossRef]
- Zager, R.A. Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney Int 1996, 49, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Homsi, E.; Janino, P.; De Faria, J.B.L. Role of caspases on cell death, inflammation, and cell cycle in glycerol-induced acute renal failure. Kidney Int. 2006, 69, 1385–1392. [Google Scholar] [CrossRef]
- Zager, R.A.; Foerder, C.A. Effects of inorganic iron and myoglobin on in vitro proximal tubular lipid peroxidation and cytotoxicity. J. Clin. Investig. 1992, 89, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Belliere, J.; Casemayou, A.; Ducasse, L.; Zakaroff-Girard, A.; Martins, F.; Iacovoni, J.S.; Guilbeau-Frugier, C.; Buffin-Meyer, B.; Pipy, B.; Chauveau, D.; et al. Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J. Am. Soc. Nephrol. 2015, 26, 1363–1377. [Google Scholar] [CrossRef]
- Lichtnekert, J.; Kulkarni, O.P.; Mulay, S.R.; Rupanagudi, K.V.; Ryu, M.; Allam, R.; Vielhauer, V.; Muruve, D.; Lindenmeyer, M.T.; Cohen, C.D.; et al. Anti-GBM Glomerulonephritis Involves IL-1 but Is Independent of NLRP3/ASC Inflammasome-Mediated Activation of Caspase-1. PLoS ONE 2011, 6, e26778. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Takahashi, M. The crystal-induced activation of NLRP3 inflammasomes in atherosclerosis. Inflamm. Regen. 2017, 37, 18. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Murali, N.S. Myoglobinuric and Hemoglobinuric Acute Kidney Injury, 5th ed.; Saunders Elsevier: Philadelphia, PA, USA, 2009; pp. 298–304. [Google Scholar]
- Anuradha, S.; Arora, S.; Mehrotra, S.; Arora, A.; Kar, P. Acute renal failure following para-phenylenediamine (PPD) poisoning: A case report and review. Renal Fail. 2004, 26, 329–332. [Google Scholar] [CrossRef]
- Fernandez, P.L.; Dutra, F.F.; Alves, L.; Figueiredo, R.T.; Mourão-Sa, D.; Fortes, G.B.; Bergstrand, S.; Lönn, D.; Cevallos, R.R.; Pereira, R.M.S.; et al. Heme Amplifies the Innate Immune Response to Microbial Molecules through Spleen Tyrosine Kinase (Syk)-dependent Reactive Oxygen Species Generation. J. Biol. Chem. 2010, 285, 32844–32851. [Google Scholar] [CrossRef]
- Gois, P.H.F.; Martines, M.S.; Ferreira, D.; Volpini, R.; Canale, D.; Malaque, C.; Crajoinas, R.; Girardi, A.C.C.; Massola Shimizu, M.H.; Seguro, A.C. Allopurinol attenuates acute kidney injury following Bothrops jararaca envenomation. PLoS Negl. Trop. Dis. 2017, 11, e0006024. [Google Scholar] [CrossRef]
- Mate-Kole, M.O.; Yeboah, E.D.; Affram, R.K.; Adu, D. Blackwater fever and acute renal failure in expatriates in Africa. Renal Fail. 1996, 18, 525–531. [Google Scholar] [CrossRef]
- Schrier, S.L. Diagnosis of Hemolytic Anemia in the Adult. Available online: https://www.uptodate.com/contents/diagnosis-of-hemolytic-anemia-in-the-adult (accessed on 12 March 2019).
- Viraraghavan, R.; Chakravarty, A.G.; Soreth, J. Cefotetan-induced haemolytic anaemia: A review of 85 cases. Adverse Drug React. Toxicol. Rev. 2002, 21, 101–107. [Google Scholar] [CrossRef]
- Chapman, A.B.; Rahbari-Oskoui, F.F.; Bennett, W.M. Acquired cystic disease of the kidney in adults. Available online: https://www.uptodate.com/contents/acquired-cystic-disease-of-the-kidney-in-adults (accessed on 11 March 2019).
- Abreu, P.A.E.; Seguro, A.C.; Canale, D.; Silva, A.M.G.d.; Matos, L.d.R.B.; Gotti, T.B.; Monaris, D.; Jesus, D.A.d.; Vasconcellos, S.A.; de Brito, T.; et al. Lp25 membrane protein from pathogenic Leptospira spp. is associated with rhabdomyolysis and oliguric acute kidney injury in a guinea pig model of leptospirosis. PLoS Negl. Trop. Dis. 2017, 11, e0005615. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, P.L.; Jacinto, C.N.; Silva Junior, G.B.; Lima, J.B.; Veras, M.d.S.B.; Daher, E.F.; Daher, E.F. Acute kidney injury caused by Crotalus and Bothrops snake venom: A review of epidemiology, clinical manifestations and treatment. Rev. Inst. Med. Trop. Sao Paulo 2013, 55, 295–301. [Google Scholar] [CrossRef]
- De Bragança, A.C.; Moreau, R.L.M.; De Brito, T.; Shimizu, M.H.M.; Canale, D.; De Jesus, D.A.; Silva, A.M.G.; Gois, P.H.; Seguro, A.C.; Magaldi, A.J. Ecstasy induces reactive oxygen species, kidney water absorption and rhabdomyolysis in normal rats. Effect of N-acetylcysteine and Allopurinol in oxidative stress and muscle fiber damage. PLoS ONE 2017, 12, e0179199. [Google Scholar] [CrossRef]
- Trowbridge, A.A.; Green, J.B.; Bonnett, J.D.; Shohet, S.B.; Ponnappa, B.D.; McCombs, W.B. Hemolytic anemia associated with leptospirosis. Morphologic and lipid studies. Am. J. Clin. Pathol. 1981, 76, 493–498. [Google Scholar] [CrossRef]
- Da Silva Junior, G.B.; Pinto, J.R.; Barros, E.J.G.; Farias, G.M.N.; Daher, E.D.F. Kidney involvement in malaria: An update. Rev. Inst. Med. Trop. Sao Paulo 2017. [Google Scholar] [CrossRef]
- Mendonça, R.; Silveira, A.A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm. Res. 2016. [Google Scholar] [CrossRef]
- Wagener, F.A.; Abraham, N.G.; Van Kooyk, Y.; De Witte, T.; Figdor, C.G. Heme-induced cell adhesion in the pathogenesis of sickle-cell disease and inflammation. Trends Pharmacol. Sci. 2001. [Google Scholar] [CrossRef]
- Nyakundi, B.B.; Tóth, A.; Balogh, E.; Nagy, B.; Erdei, J.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Oxidized hemoglobin forms contribute to NLRP3 inflammasome-driven IL-1β production upon intravascular hemolysis. Biochim. Biophys. Acta Mol. Basis Dis. 2019. [Google Scholar] [CrossRef]
- López-Castejón, G.; Pelegrín, P. Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin. Investig. Drugs 2012, 21, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.G.; Brough, D.; Freeman, S. Inhibiting the Inflammasome: A Chemical Perspective. J. Med. Chem. 2016, 59, 1691–1710. [Google Scholar] [CrossRef] [PubMed]
- Diwan, V.; Gobe, G.; Brown, L. Glibenclamide improves kidney and heart structure and function in the adenine-diet model of chronic kidney disease. Pharmacol. Res. 2014, 79, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Berns, J.S.; Glickman, J.D. Management of Hyperglycemia in Patients with Type 2 Diabetes and Pre-Dialysis Chronic Kidney Disease or End-Stage Renal Disease. Available online: https://www.uptodate.com/contents/management-of-hyperglycemia-in-patients-with-type-2-diabetes-and-pre-dialysis-chronic-kidney-disease-or-end-stage-renal-disease (accessed on 15 March 2019).
- Meloni, G.; Meloni, T. Glyburide-induced acute haemolysis in a G6PD-deficient patient with NIDDM. Br. J. Haematol. 1996, 92, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Vinzio, S.; Andrès, E.; Perrin, A.-E.; Schlienger, J.-L.; Goichot, B. Glibenclamide-induced acute haemolytic anaemia revealing a G6PD-deficiency. Diabetes Res. Clin. Pract. 2004, 64, 181–183. [Google Scholar] [CrossRef]
- Perregaux, D.G.; McNiff, P.; Laliberte, R.; Hawryluk, N.; Peurano, H.; Stam, E.; Eggler, J.; Griffiths, R.; Dombroski, M.A.; Gabel, C.A. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J. Pharmacol. Exp. Ther. 2001, 299, 187–197. [Google Scholar] [PubMed]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Perera, A.P.; Fernando, R.; Shinde, T.; Gundamaraju, R.; Southam, B.; Sohal, S.S.; Robertson, A.A.B.; Schroder, K.; Kunde, D.; Eri, R. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci. Rep. 2018, 8, 8618. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef]
- Zhai, Y.; Meng, X.; Ye, T.; Xie, W.; Sun, G.; Sun, X. Inhibiting the NLRP3 Inflammasome Activation with MCC950 Ameliorates Diabetic Encephalopathy in db/db Mice. Molecules 2018, 23, 1–14. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Vitale, A.; Insalaco, A.; Sfriso, P.; Lopalco, G.; Emmi, G.; Cattalini, M.; Manna, R.; Cimaz, R.; Priori, R.; Talarico, R.; et al. A Snapshot on the On-Label and Off-Label Use of the Interleukin-1 Inhibitors in Italy among Rheumatologists and Pediatric Rheumatologists: A Nationwide Multi-Center Retrospective Observational Study. Front. Pharmacol. 2016, 7, 380. [Google Scholar] [CrossRef]
- Alten, R.; Gram, H.; Joosten, L.A.; Berg, W.B.v.d.; Sieper, J.; Wassenberg, S.; Burmester, G.; van Riel, P.; Diaz-Lorente, M.; Bruin, G.J.M.; et al. The human anti-IL-1β monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof-of-concept study in patients with rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R67. [Google Scholar] [CrossRef]
- Rondeau, J.-M.; Ramage, P.; Zurini, M.; Gram, H. The molecular mode of action and species specificity of canakinumab, a human monoclonal antibody neutralizing IL-1β. mAbs 2015, 7, 1151–1160. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Koenig, W.; Libby, P.; Everett, B.M.; Lefkowitz, M.; Thuren, T.; Cornel, J.H. Inhibition of Interleukin-1beta by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. J. Am. Coll. Cardiol. 2018, 71, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.H.; Glynn, R.J.; MacFadyen, J.G.; Libby, P.; Thuren, T.; Everett, B.M.; Ridker, P.M. Relationship of Interleukin-1β Blockade With Incident Gout and Serum Uric Acid Levels: Exploratory Analysis of a Randomized Controlled TrialInterleukin-1β Blockade, Incident Gout, and Serum Uric Acid Levels. Ann. Intern. Med. 2018, 169, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, G.; Parker, T.; Turner, D.; Parker, M.; Scales, J.; Harnett, P.; Harrison, M.; Ahmed, K.; Bhagat, S.; Marianayagam, T.; et al. Feasibility randomised multicentre, double-blind, double-dummy controlled trial of anakinra, an interleukin-1 receptor antagonist versus intramuscular methylprednisolone for acute gout attacks in patients with chronic kidney disease (ASGARD): Protocol study. BMJ Open 2017, 7, e017121. [Google Scholar] [PubMed]
- Ben-Zvi, I.; Kukuy, O.; Giat, E.; Pras, E.; Feld, O.; Kivity, S.; Perski, O.; Bornstein, G.; Grossman, C.; Harari, G.; et al. Anakinra for Colchicine-Resistant Familial Mediterranean Fever: A Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2017, 69, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Ugurlu, S.; Ergezen, B.; Ozdogan, H. Anakinra treatment in patients with Familial Mediterranean Fever: A single-center experience. Pediatr. Rheumatol. 2015, 13, P123. [Google Scholar] [CrossRef]
- Moghaddas, F.; Llamas, R.; De Nardo, D.; Martinez-Banaclocha, H.; Martinez-Garcia, J.J.; Mesa-del-Castillo, P.; Baker, P.J.; Gargallo, V.; Mensa-Vilaro, A.; Canna, S.; et al. A novel Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis mutation further defines 14-3-3 binding of pyrin and distinction to Familial Mediterranean Fever. Ann. Rheum. Dis. 2017, 76, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Lagou, V.; Jéru, I.; Baker, P.J.; Van Eyck, L.; Parry, D.A.; Lawless, D.; De Nardo, D.; Garcia-Perez, J.E.; Dagley, L.F.; et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci. Transl. Med. 2016, 8, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Curiel, R.V.; Guzman, N.J. Challenges Associated with the Management of Gouty Arthritis in Patients with Chronic Kidney Disease: A Systematic Review. Semin. Arthritis Rheum. 2012, 42, 166–178. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giuliani, K.T.K.; Kassianos, A.J.; Healy, H.; Gois, P.H.F. Pigment Nephropathy: Novel Insights into Inflammasome-Mediated Pathogenesis. Int. J. Mol. Sci. 2019, 20, 1997. https://doi.org/10.3390/ijms20081997
Giuliani KTK, Kassianos AJ, Healy H, Gois PHF. Pigment Nephropathy: Novel Insights into Inflammasome-Mediated Pathogenesis. International Journal of Molecular Sciences. 2019; 20(8):1997. https://doi.org/10.3390/ijms20081997
Chicago/Turabian StyleGiuliani, Kurt T. K., Andrew J. Kassianos, Helen Healy, and Pedro H. F. Gois. 2019. "Pigment Nephropathy: Novel Insights into Inflammasome-Mediated Pathogenesis" International Journal of Molecular Sciences 20, no. 8: 1997. https://doi.org/10.3390/ijms20081997
APA StyleGiuliani, K. T. K., Kassianos, A. J., Healy, H., & Gois, P. H. F. (2019). Pigment Nephropathy: Novel Insights into Inflammasome-Mediated Pathogenesis. International Journal of Molecular Sciences, 20(8), 1997. https://doi.org/10.3390/ijms20081997