Disruption of Structural Disulfides of Coagulation FXIII-B Subunit; Functional Implications for a Rare Bleeding Disorder

 and
and 
Abstract
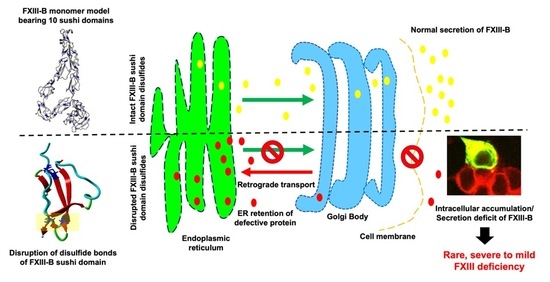
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
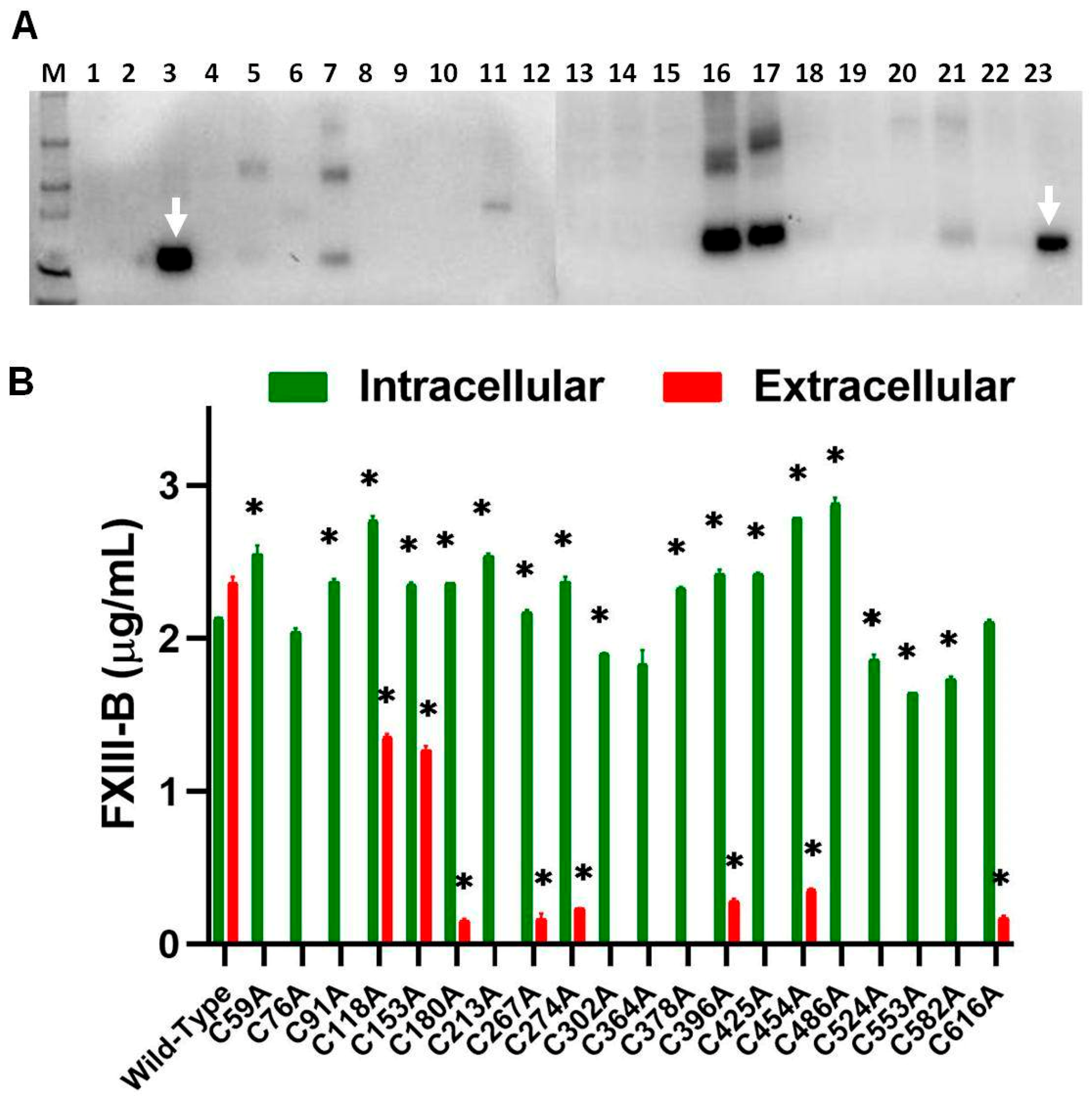
2.1. Disruption of FXIII-B Subunit Structural Disulfides Results in a Secretion Deficit
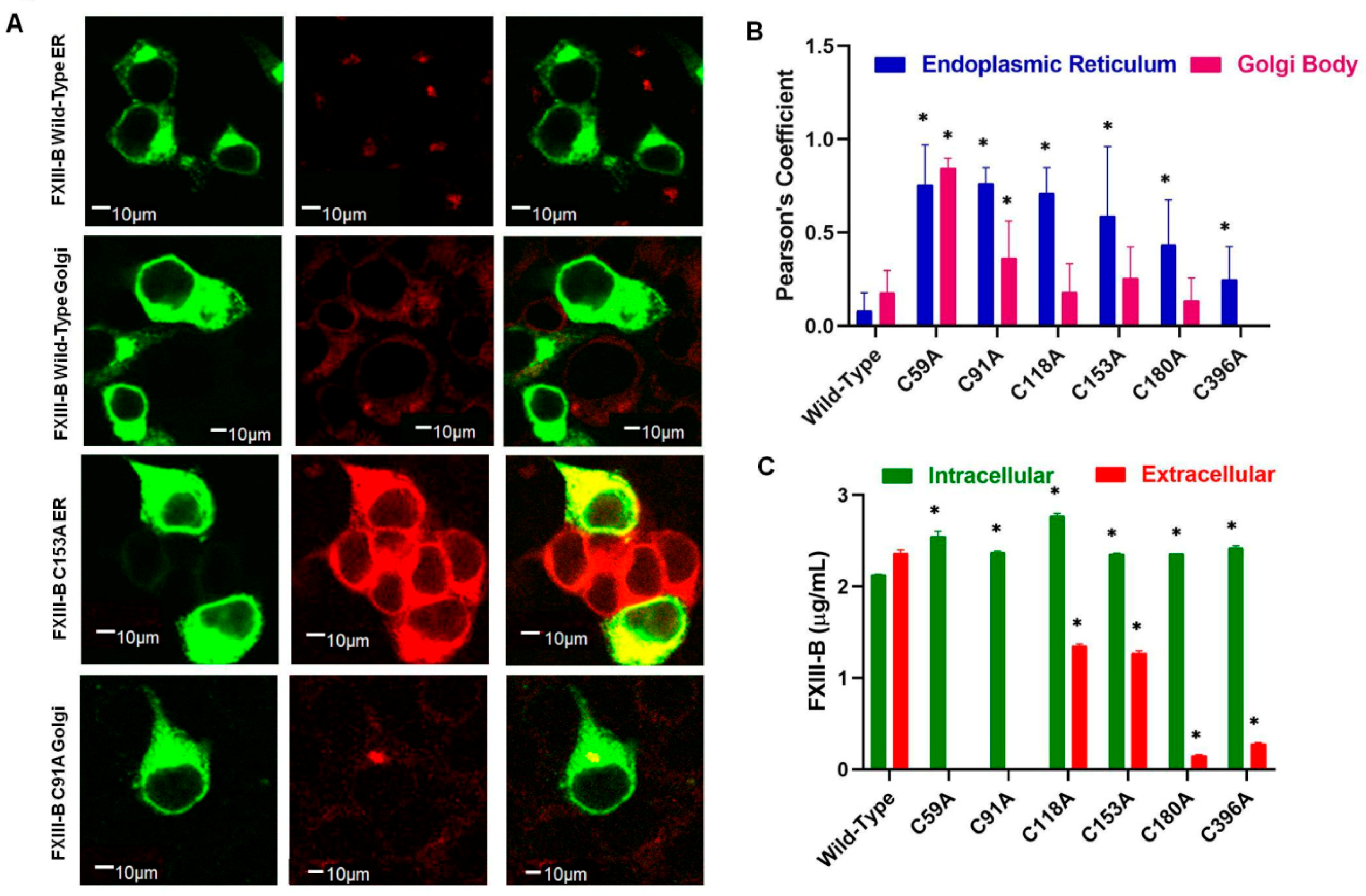
2.2. Disruption of FXIII-B Subunit Structural Disulfides Results Mainly in ER Accumulation
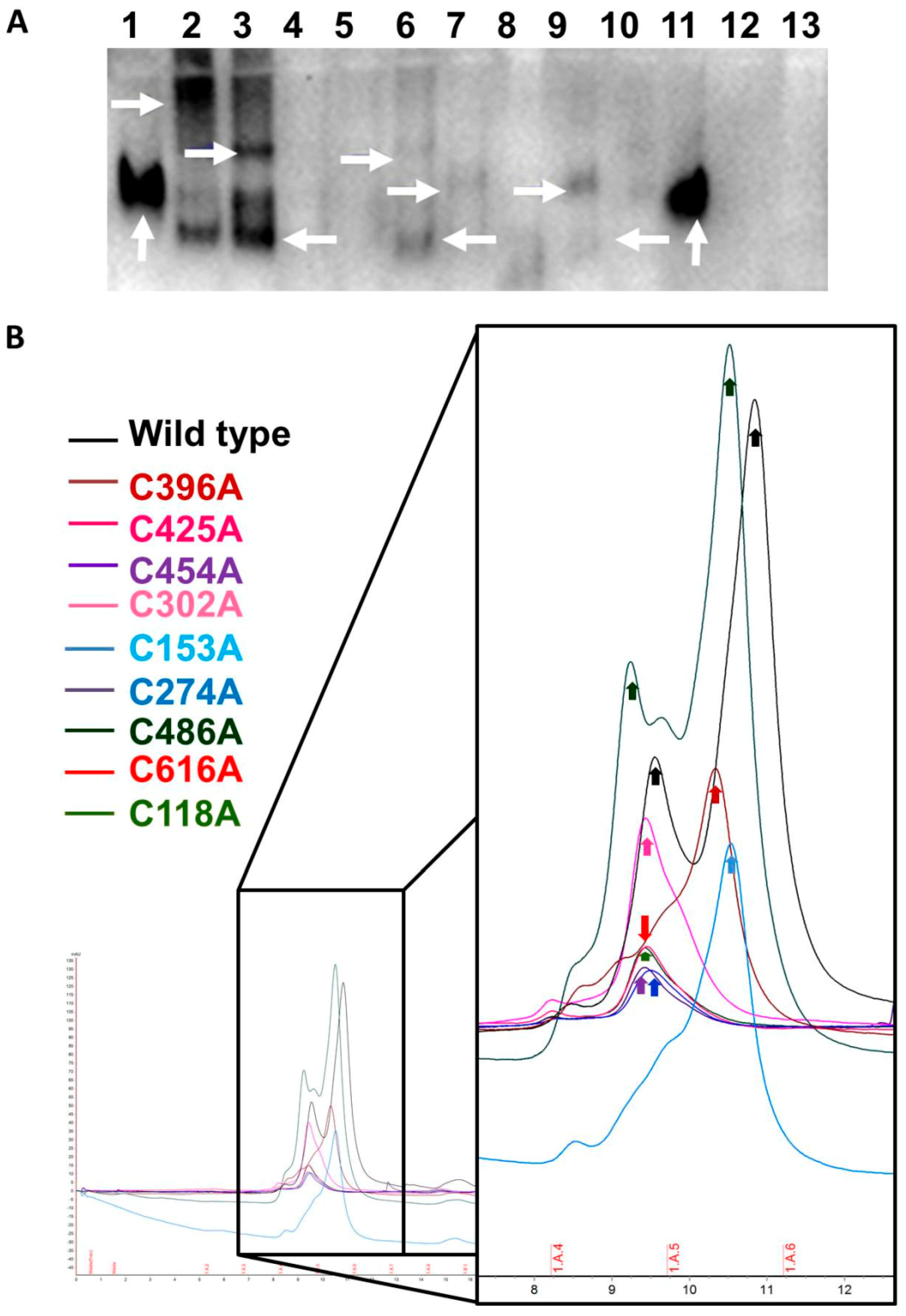
2.3. Secreted FXIII-B Cys-Mutants Show Altered Complexation States and Possible Dimer Disruption
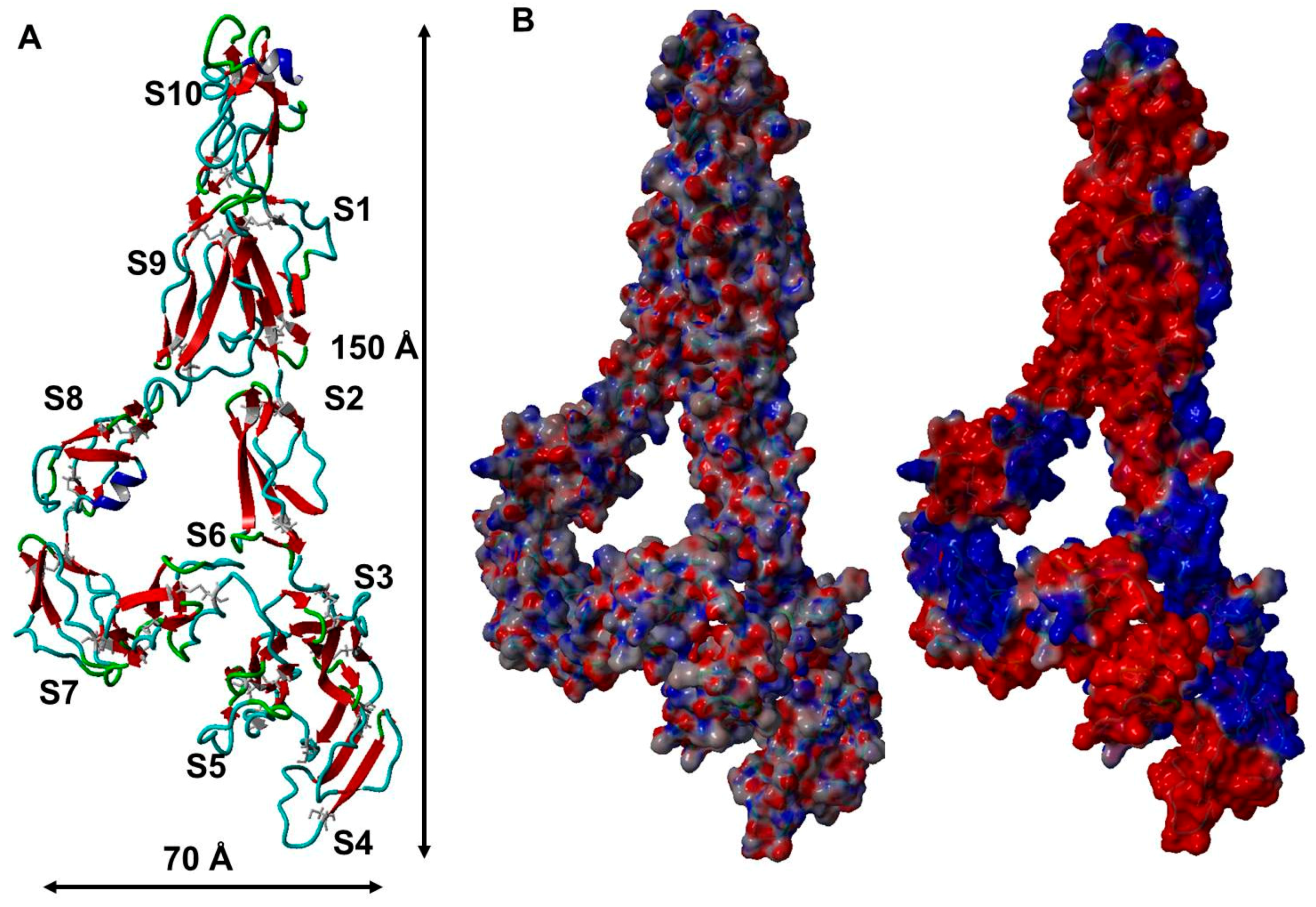
2.4. A Filamentous Monomer FXIII-B Subunit Model with Its N and C Terminals Aligned Close to Each Other
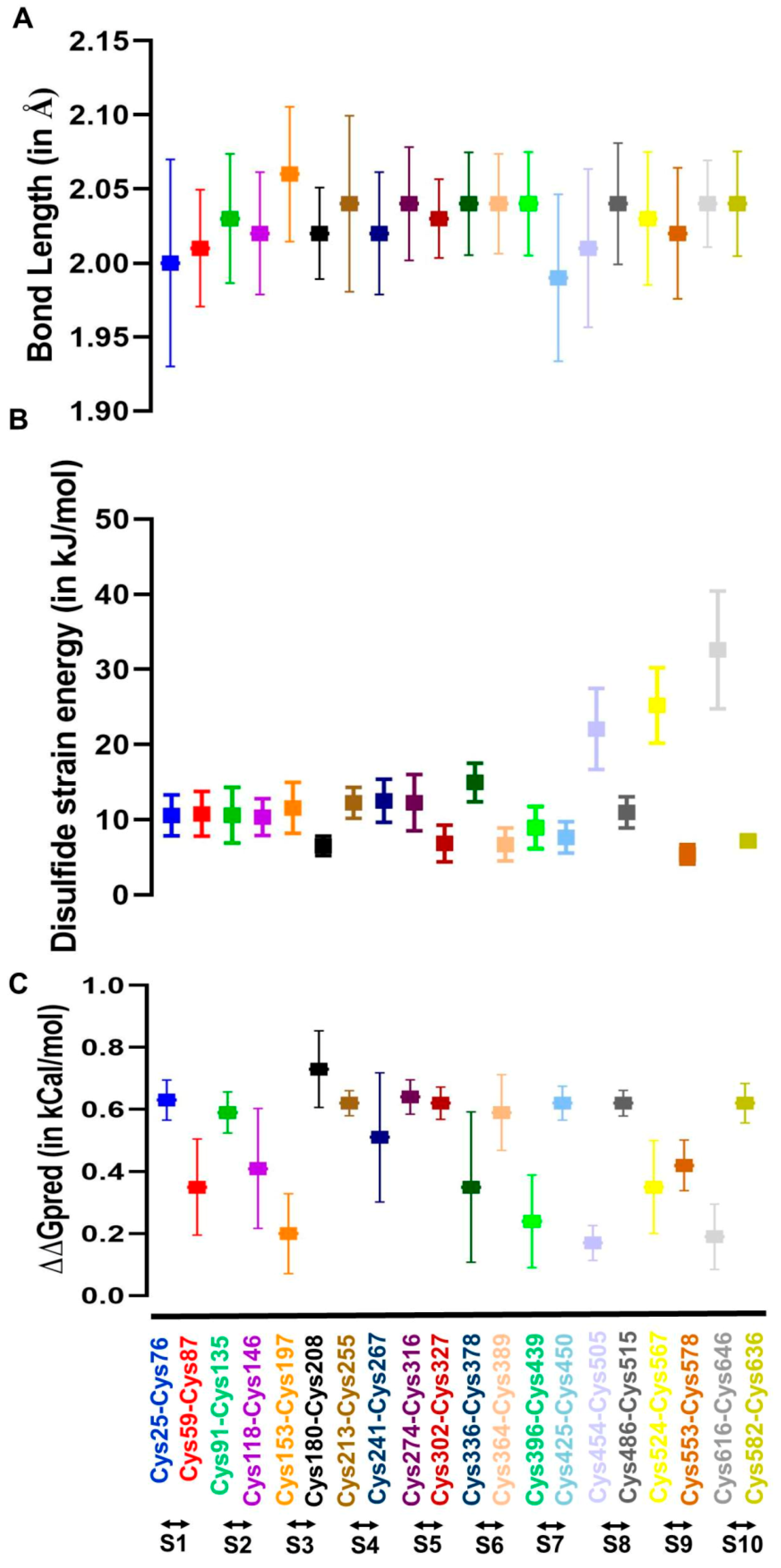
2.5. The FXIII-B Subunit Disulfide Bonds Display Variability in Structural Flexibility, but Ablation of any of These Bonds Leads to a Loss in Stability
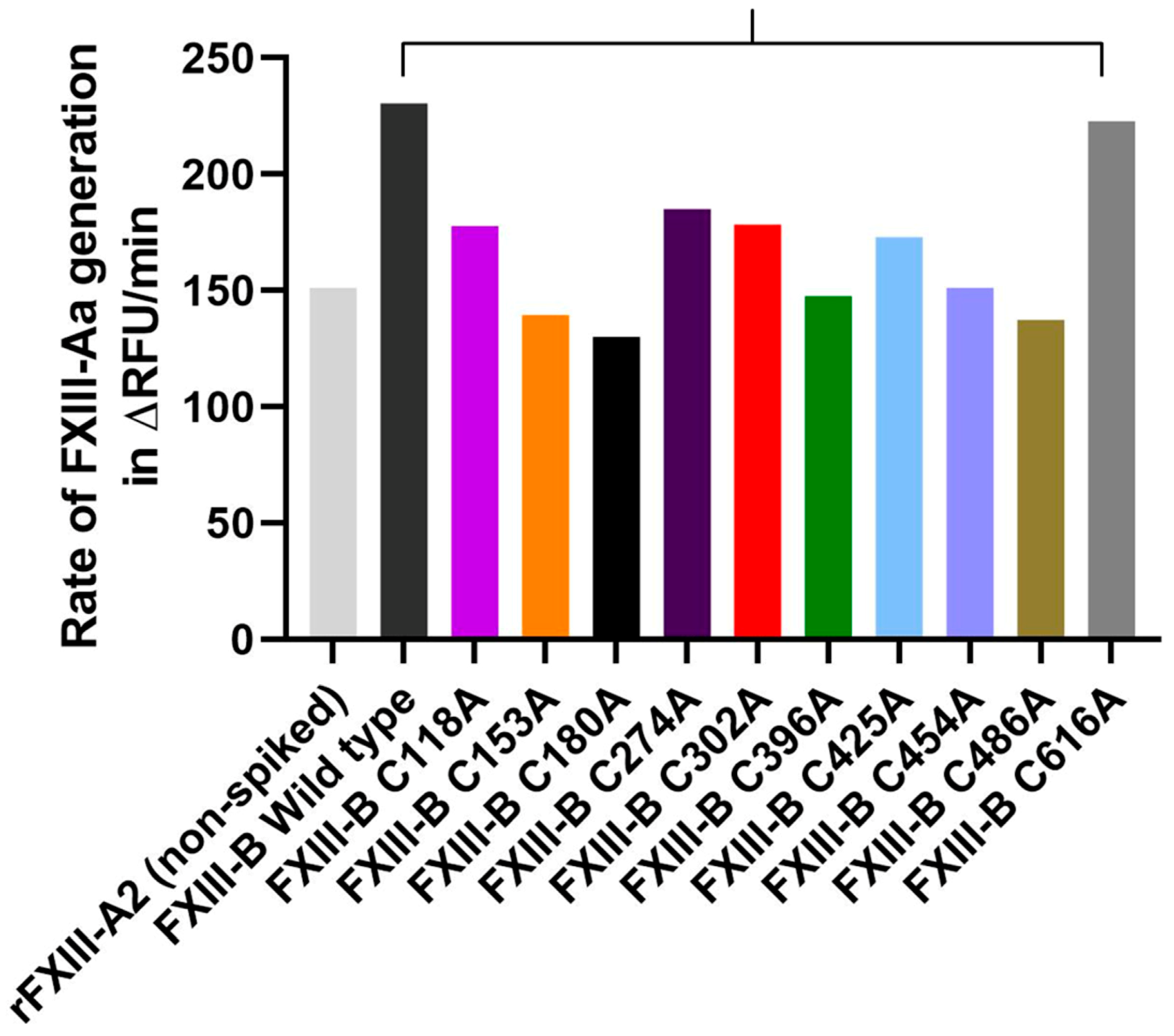
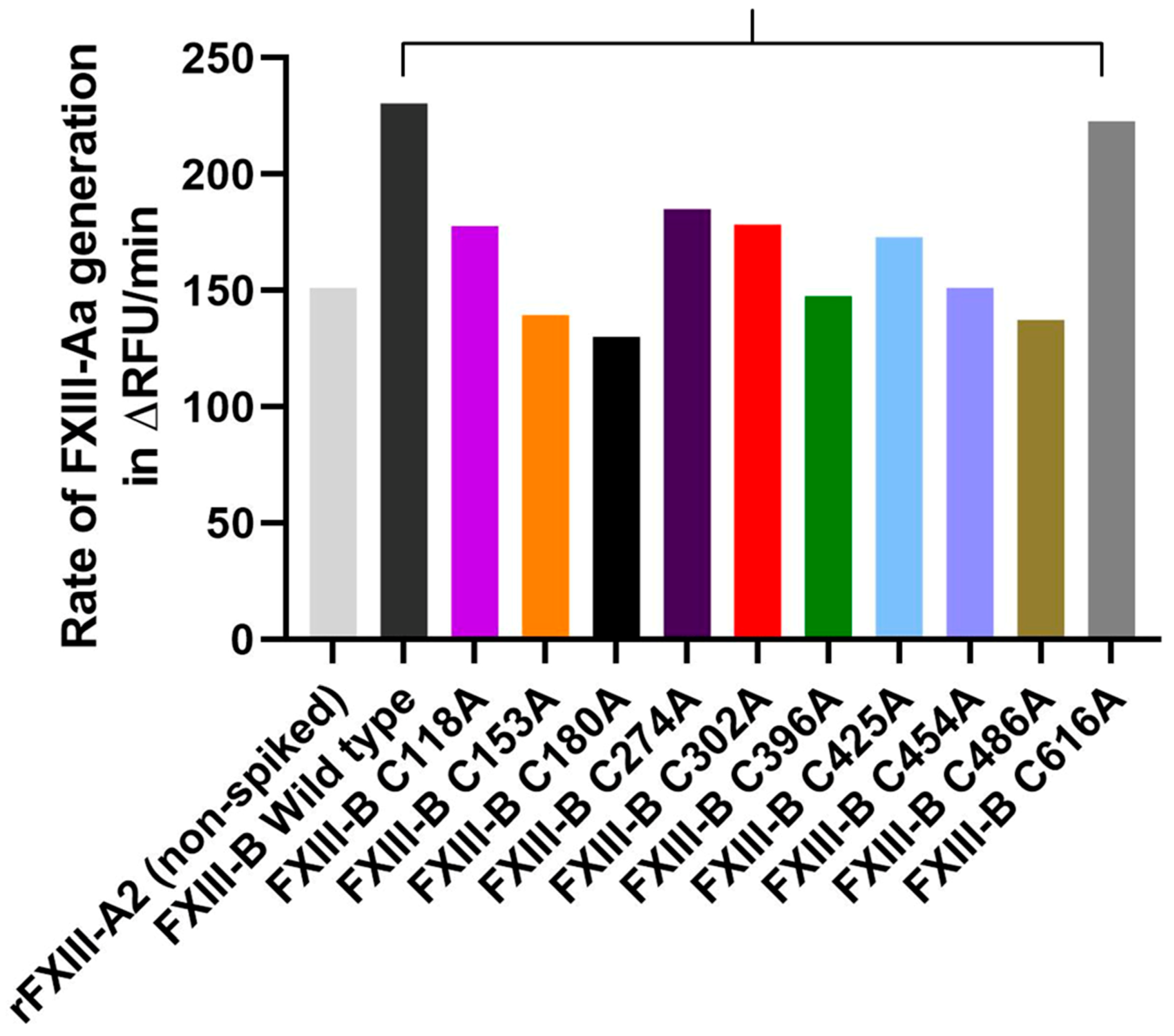
2.6. Disruption of Selected FXIII-B Disulfide Bonds can Affect the Rate of FXIII-Aa Generation
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Cloning and Expression of FXIII-B Cysteine to Alanine Mutants
4.3. Antigenic Quantification of FXIII-B Cysteine Mutants
4.4. Western Blot Analyses
4.5. Confocal Immunofluorescence of Expressed FXIII-B Protein and its Cysteine Mutants
4.6. Native-PAGE-blot Analysis of Expressed FXIII-B Protein and its Cysteine Mutants
4.7. Purification of Wild Type FXIII-B and its Cysteine Mutants
4.8. In Silico Analysis of FXIII-B Subunit Disulfide Bonds
4.9. Effect of Spiking Wild Type FXIII-B and Its Cysteine Variants into the FXIII-Aa Generation Assay
5. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Biswas, A.; Ivaskevicius, V.; Thomas, A.; Oldenburg, J. Coagulation factor XIII deficiency. Diagnosis, prevalence and management of inherited and acquired forms. Hamostaseologie 2014, 34, 160–166. [Google Scholar] [PubMed]
- Gupta, S.; Biswas, A.; Akhter, M.S.; Krettler, C.; Reinhart, C.; Dodt, J.; Reuter, A.; Philippou, H.; Ivaskevicius, V.; Oldenburg, J. Revisiting the mechanism of coagulation factor XIII activation and regulation from a structure/functional perspective. Sci. Rep. 2016, 6, 30105. [Google Scholar] [Green Version]
- Duckert, F.; Jung, E.; Shmerling, D.H. A hitherto undescribed congenital haemorrhagic diathesis probably due to fibrin stabilizing factor deficiency. Thromb. et Diath. Haemorrh. 1960, 5, 179–186. [Google Scholar] [CrossRef]
- Seitz, R.; Duckert, F.; Lopaciuk, S.; Muszbek, L.; Rodeghiero, F.; Seligsohn, U. ETRO Working Party on Factor XIII questionnaire on congenital factor XIII deficiency in Europe: Status and perspectives. Study Group. Semin. Thromb. and Hemost. 1996, 22, 415–418. [Google Scholar] [CrossRef]
- Biswas, A.; Ivaskevicius, V.; Seitz, R.; Thomas, A.; Oldenburg, J. An update of the mutation profile of Factor 13 A and B genes. Blood Rev. 2011, 25, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, T.; Ichinose, A. Molecular and cellular basis of deficiency of the b subunit for factor XIII secondary to a Cys430-Phe mutation in the seventh Sushi domain. J. Clin. Investig. 1995, 95, 1002–1008. [Google Scholar] [CrossRef]
- Hashiguchi, T.; Saito, M.; Morishita, E.; Matsuda, T.; Ichinose, A. Two genetic defects in a patient with complete deficiency of the b-subunit for coagulation factor XIII. Blood 1993, 82, 145–150. [Google Scholar] [PubMed]
- Izumi, T.; Hashiguchi, T.; Castaman, G.; Tosetto, A.; Rodeghiero, F.; Girolami, A.; Ichinose, A. Type I factor XIII deficiency is caused by a genetic defect of its b subunit: Insertion of triplet AAC in exon III leads to premature termination in the second Sushi domain. Blood 1996, 87, 2769–2774. [Google Scholar]
- Souri, M.; Izumi, T.; Higashi, Y.; Girolami, A.; Ichinose, A. A founder effect is proposed for factor XIII B subunit deficiency caused by the insertion of triplet AAC in exon III encoding the second Sushi domain. Thromb. Haemost. 1998, 80, 211–213. [Google Scholar] [CrossRef]
- Wada, H.; Souri, M.; Matsumoto, R.; Sugihara, T.; Ichinose, A. Alloantibodies against the B subunit of plasma factor XIII developed in its congenital deficiency. Thromb. Haemost. 2013, 109, 661–668. [Google Scholar] [PubMed]
- Saito, M.; Asakura, H.; Yoshida, T.; Ito, K.; Okafuji, K.; Matsuda, T. A familial factor XIII subunit B deficiency. Br. J. Haematol. 1990, 74, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Tahlan, A.; Ahluwalia, J. Factor XIII: Congenital deficiency factor XIII, acquired deficiency, factor XIII A-subunit, and factor XIII B-subunit. Arch. Pathol. Lab. Med. 2014, 138, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Ivaskevicius, V.; Biswas, A.; Loreth, R.; Schroeder, V.; Ohlenforst, S.; Rott, H.; Krause, M.; Kohler, H.-P.; Scharrer, I.; Oldenburg, J. Mutations affecting disulphide bonds contribute to a fairly common prevalence of F13B gene defects: Results of a genetic study in 14 families with factor XIII B deficiency. Haemophilia 2010, 16, 675–682. [Google Scholar]
- Thomas, A.; Biswas, A.; Ivaskevicius, V.; Oldenburg, J. Structural and functional influences of coagulation factor XIII subunit B heterozygous missense mutants. Mol. Genet. Genom. Med. 2015, 3, 258–271. [Google Scholar] [CrossRef] [Green Version]
- Ivaskevicius, V.; Windyga, J.; Baran, B.; Schroeder, V.; Junen, J.; Bykowska, K.; Seifried, E.; Kohler, H.P.; Oldenburg, J. Phenotype-genotype correlation in eight Polish patients with inherited Factor XIII deficiency: Identification of three novel mutations. Haemophilia 2007, 13, 649–657. [Google Scholar] [CrossRef]
- Biswas, A.; Thomas, A.; Bevans, C.G.; Ivaskevicius, V.; Oldenburg, J. In vitro secretion deficits are common among human coagulation factor XIII subunit B missense mutants: Correlations with patient phenotypes and molecular models. Hum. Mutat. 2013, 34, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Souri, M.; Kaetsu, H.; Ichinose, A. Sushi domains in the B subunit of factor XIII responsible for oligomer assembly. Biochemistry 2008, 47, 8656–8664. [Google Scholar] [CrossRef]
- Schroeder, V.; Kohler, H.P. Factor XIII: Structure and Function. Semin. Thromb. Hemost. 2016, 42, 422–428. [Google Scholar]
- Nagy, J.A.; Henriksson, P.; McDonagh, J. Biosynthesis of factor XIII B subunit by human hepatoma cell lines. Blood 1986, 68, 1272–1279. [Google Scholar]
- Komaromi, I.; Bagoly, Z.; Muszbek, L. Factor XIII: Novel structural and functional aspects. J. Thromb. Haemost. JTH 2011, 9, 9–20. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science (New York, N.Y.) 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Norman, D.G.; Barlow, P.N.; Baron, M.; Day, A.J.; Sim, R.B.; Campbell, I.D. Three-dimensional structure of a complement control protein module in solution. J. Mol. Biol. 1991, 219, 717–725. [Google Scholar] [CrossRef]
- Souri, M.; Osaki, T.; Ichinose, A. The Non-catalytic B Subunit of Coagulation Factor XIII Accelerates Fibrin Cross-linking. J. Biol. Chem. 2015, 290, 12027–12039. [Google Scholar] [CrossRef]
- Byrnes, J.R.; Wilson, C.; Boutelle, A.M.; Brandner, C.B.; Flick, M.J.; Philippou, H.; Wolberg, A.S. The interaction between fibrinogen and zymogen FXIII-A2B2 is mediated by fibrinogen residues gamma390-396 and the FXIII-B subunits. Blood 2016, 128, 1969–1978. [Google Scholar] [CrossRef]
- HOGG, P.J. Contribution of allosteric disulfide bonds to regulation of hemostasis. J. Thromb. Haemost. 2009, 7, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Carrell, N.A.; Erickson, H.P.; McDonagh, J. Electron microscopy and hydrodynamic properties of factor XIII subunits. J. of Biol. Chem. 1989, 264, 551–556. [Google Scholar]
- Katona, E.; Penzes, K.; Csapo, A.; Fazakas, F.; Udvardy, M.L.; Bagoly, Z.; Orosz, Z.Z.; Muszbek, L. Interaction of factor XIII subunits. Blood 2014, 123, 1757–1763. [Google Scholar] [CrossRef] [Green Version]
- Geva, Y.; Schuldiner, M. The back and forth of cargo exit from the endoplasmic reticulum. Curr. Biol. 2014, 24, 130–136. [Google Scholar] [CrossRef]
- Aslam, M.; Perkins, S.J. Folded-back solution structure of monomeric factor H of human complement by synchrotron X-ray and neutron scattering, analytical ultracentrifugation and constrained molecular modelling. J. Mol. Biol. 2001, 309, 1117–1138. [Google Scholar] [CrossRef]
- Okemefuna, A.I.; Nan, R.; Gor, J.; Perkins, S.J. Electrostatic interactions contribute to the folded-back conformation of wild type human factor H. J. Mol. Biol. 2009, 391, 98–118. [Google Scholar] [CrossRef]
- Sevier, C.S.; Kaiser, C.A. Formation and transfer of disulphide bonds in living cells. Nature reviews. Mol. Cell Biol. 2002, 3, 836–847. [Google Scholar]
- Dodt, J.; Volkers, P.; Seitz, R. Factor XIIIa generation assay: A tool for studying factor XIII function in plasma. Anal. Biochem. 2013, 439, 145–151. [Google Scholar] [CrossRef]
- Chiu, J.; Hogg, P.J. Allosteric disulfides: Sophisticated molecular structures enabling flexible protein regulation. J. Biol. Chem. 2019, 294, 2949–2960. [Google Scholar] [CrossRef]
- Xu, D.; Jaroszewski, L.; Li, Z.; Godzik, A. AIDA: Ab initio domain assembly server. Nucleic Acids Res. 2014, 42, W308–W313. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View - molecular graphics for all devices - from smartphones to workstations. Bioinformatics (Oxford, England) 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins 2004, 57, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations: Amber Force Field. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Hooft, R.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in protein structures. Nature 1996, 381, 272. [Google Scholar] [CrossRef]
- Katz, B.A.; Kossiakoff, A. The crystallographically determined structures of atypical strained disulfides engineered into subtilisin. J. Biol. Chem. 1986, 261, 15480–15485. [Google Scholar]
- Laimer, J.; Hofer, H.; Fritz, M.; Wegenkittl, S.; Lackner, P. MAESTRO--multi agent stability prediction upon point mutations. BMC Bioinform. 2015, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Kahm, M.; Hasenbrink, G.; Lichtenberg-Fraté, H.; Ludwig, J.; Kschischo, M. grofit: Fitting Biological Growth Curves with R. J. Stat. Softw. 2010, 33, 7–10. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Akhter, M.S.; Dodt, J.; Sharma, A.; Kaniyappan, S.; Yadegari, H.; Ivaskevicius, V.; Oldenburg, J.; Biswas, A. Disruption of Structural Disulfides of Coagulation FXIII-B Subunit; Functional Implications for a Rare Bleeding Disorder. Int. J. Mol. Sci. 2019, 20, 1956. https://doi.org/10.3390/ijms20081956
Singh S, Akhter MS, Dodt J, Sharma A, Kaniyappan S, Yadegari H, Ivaskevicius V, Oldenburg J, Biswas A. Disruption of Structural Disulfides of Coagulation FXIII-B Subunit; Functional Implications for a Rare Bleeding Disorder. International Journal of Molecular Sciences. 2019; 20(8):1956. https://doi.org/10.3390/ijms20081956
Chicago/Turabian StyleSingh, Sneha, Mohammad Suhail Akhter, Johannes Dodt, Amit Sharma, Senthilvelrajan Kaniyappan, Hamideh Yadegari, Vytautas Ivaskevicius, Johannes Oldenburg, and Arijit Biswas. 2019. "Disruption of Structural Disulfides of Coagulation FXIII-B Subunit; Functional Implications for a Rare Bleeding Disorder" International Journal of Molecular Sciences 20, no. 8: 1956. https://doi.org/10.3390/ijms20081956
APA StyleSingh, S., Akhter, M. S., Dodt, J., Sharma, A., Kaniyappan, S., Yadegari, H., Ivaskevicius, V., Oldenburg, J., & Biswas, A. (2019). Disruption of Structural Disulfides of Coagulation FXIII-B Subunit; Functional Implications for a Rare Bleeding Disorder. International Journal of Molecular Sciences, 20(8), 1956. https://doi.org/10.3390/ijms20081956