Understanding the Impact of Aberrant Splicing in Coagulation Factor V Deficiency

 and
and 
Abstract
1. Introduction
2. Results
2.1. Clinical Details of the Patients
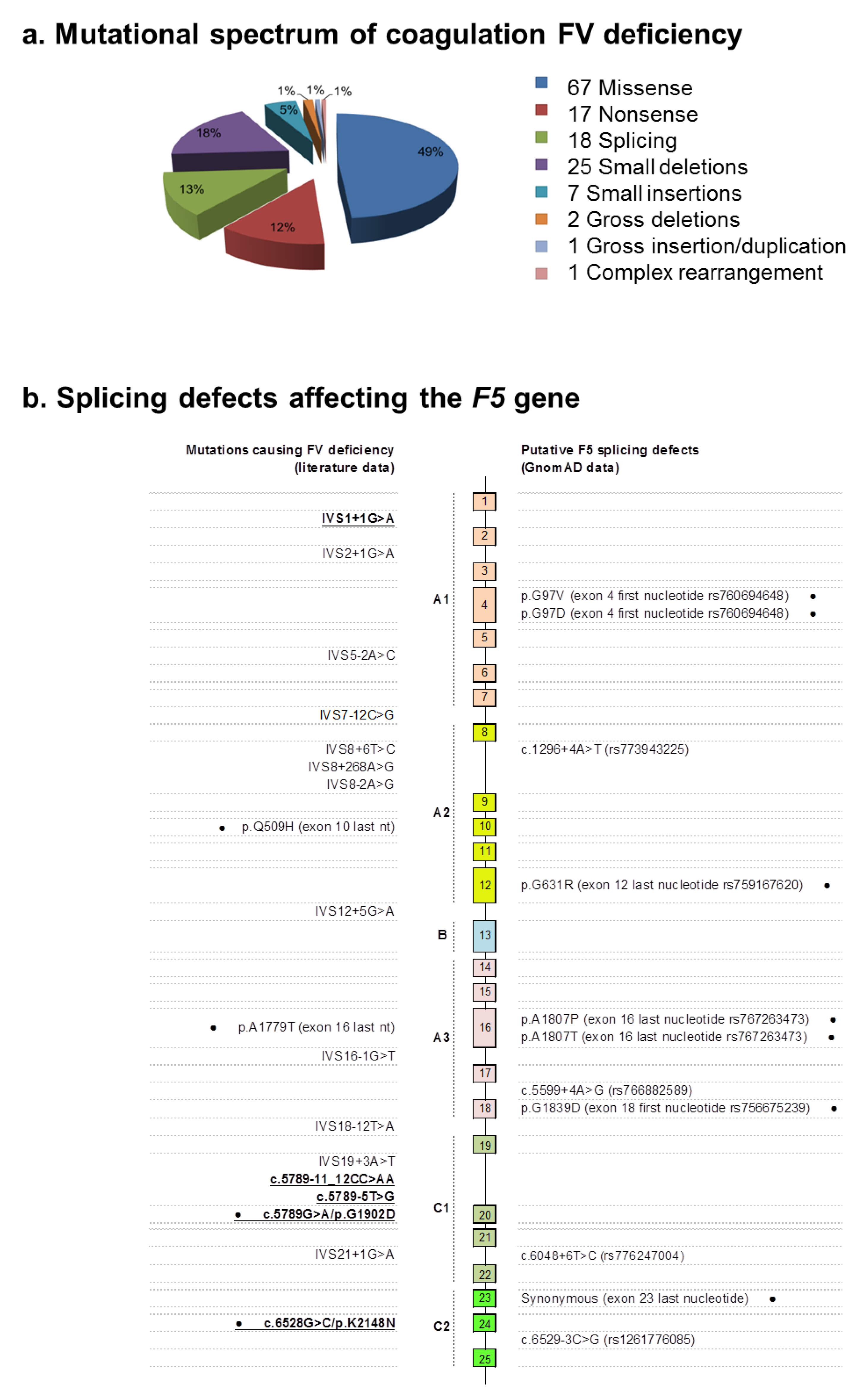
2.2. Identification of Molecular Defects
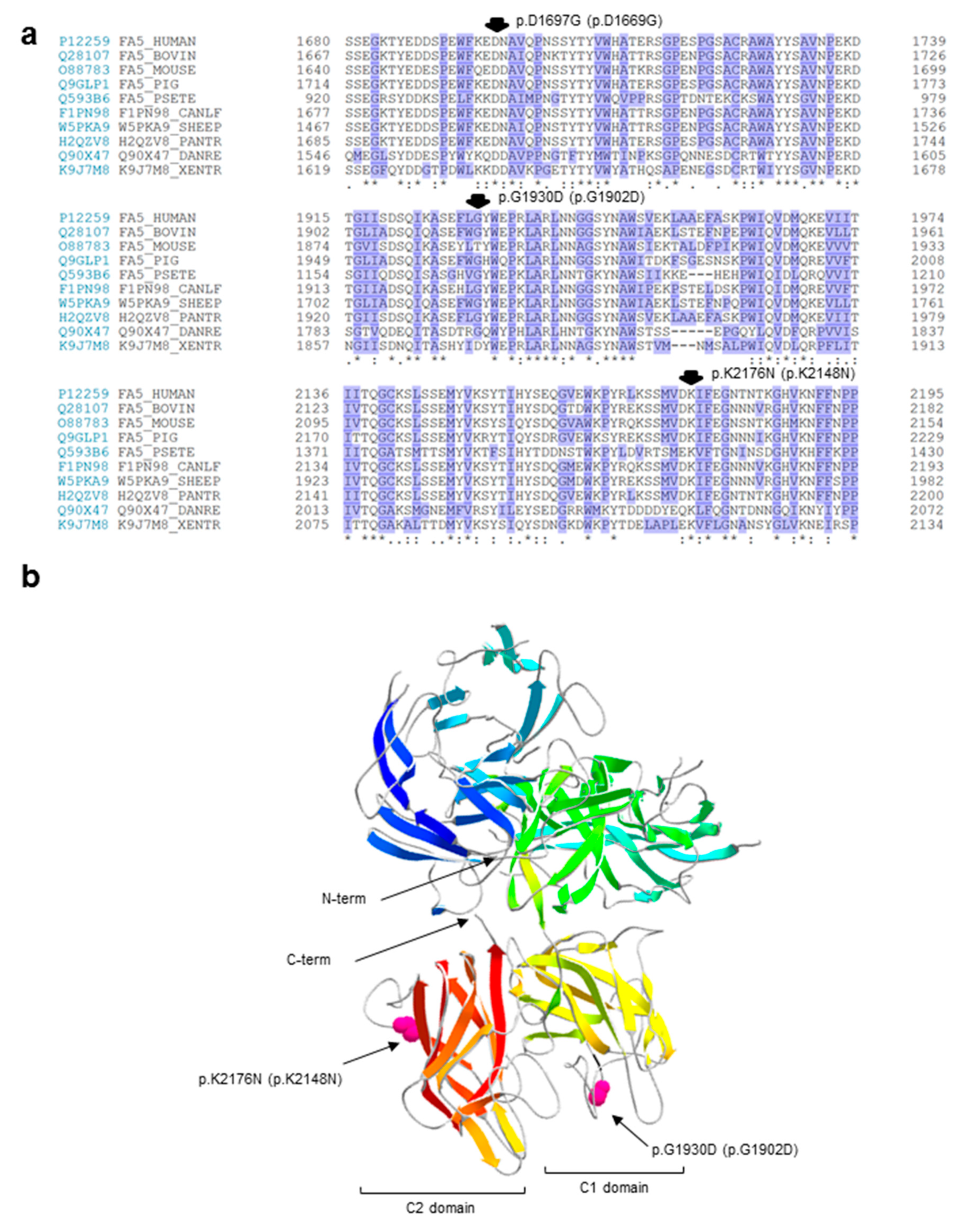
2.3. In Silico Analyses of the Identified Variants
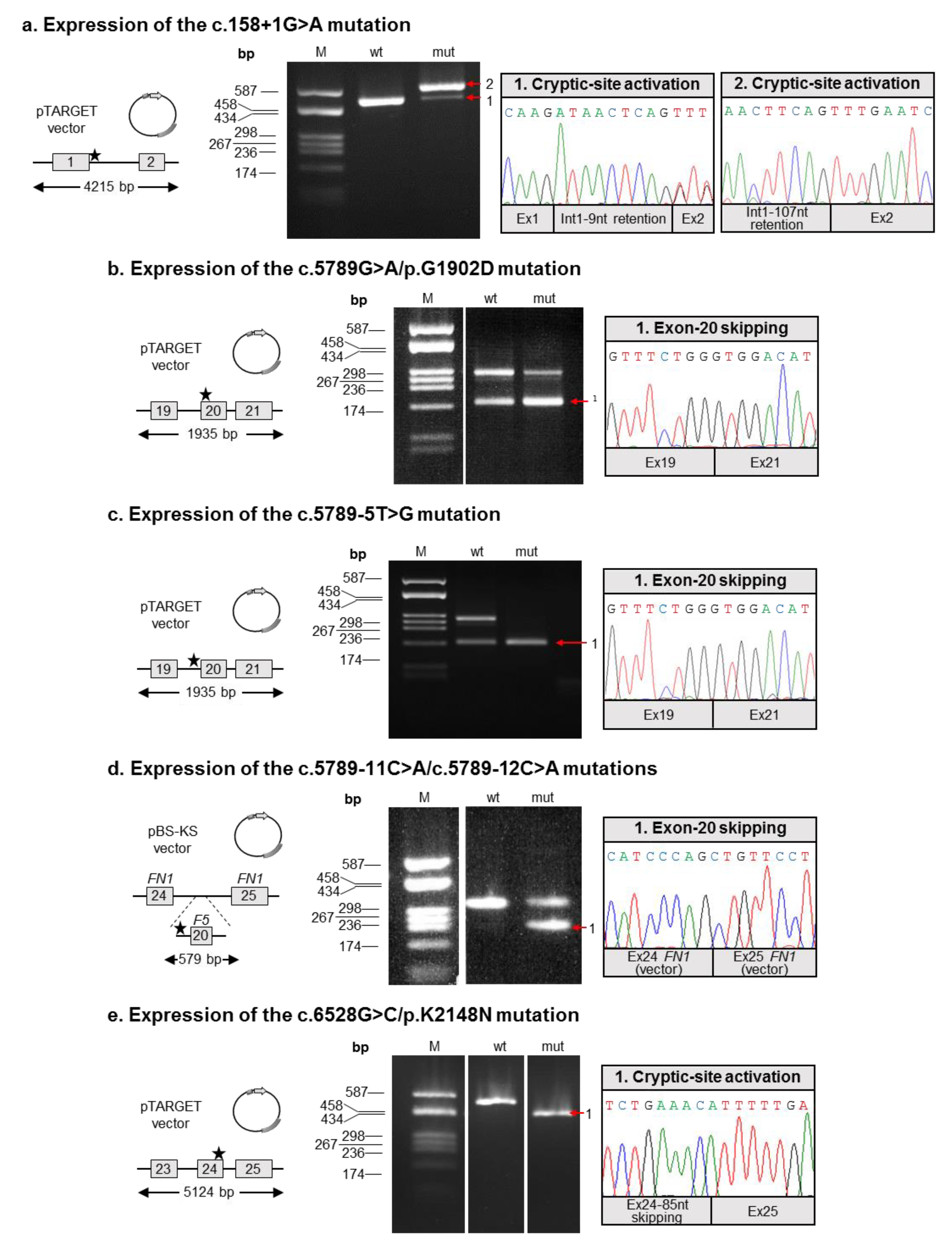
2.4. Molecular Characterization of Splicing Mutations
3. Discussion
- (1)
- We found a complex allele composed of a putative splicing defect (c.158+1G>A) and a missense variant (c.5789G>A/p.G1902D) in two unrelated subjects (P1 and P2). This allele was already described in the literature, but none of the putative mutations were functionally investigated [24];
- (2)
- We identified two missense variants, one involving the first nucleotide (the above-mentioned c.5789G>A/p.G1902D) and the other involving the last nucleotide (c.6528G>C/p.K2148N) of the corresponding exon, hence both potentially interfering with the pre-mRNA splicing;
- (3)
- We disclosed two adjacent nucleotide substitutions in the heterozygous state, in theory contributing to FV deficiency either in cis or in trans.
- (1)
- Both mutations described by Bafunno et al. [24] can be regarded as splicing mutations. Since these two mutations are present in cis on the same allele, it is plausible that their combined effect can be considered a complete loss of function. It is worth noting that should a recombination event involve this allele, it would be responsible for the spreading of two different mutations: one severe (c.158+1G>A), and the other characterized by a milder effect, as we demonstrated that the c.5789G>A defect is associated with a certain degree of wild-type splicing (Figure 2);
- (2)
- Similar to the c.5789G>A/p.G1902D mutation, the c.6528G>C/p.K2148N defect should also be regarded as a splicing rather than a missense mutation;
- (3)
- The two adjacent mutations, c.5789-11C>A and c.5789-12C>A, are present in cis on the same allele, both possibly contributing to the splicing defect. Hence, we propose the c.5789-11_12CC>AA name for this mutation. Indeed, even though the two variants are reported as single alleles in the GnomAD database, inspection of the relevant sequencing reads (accessible from the same website) confirmed the phase of the two variants.
4. Materials and Methods
4.1. Materials
4.2. Coagulation Tests
4.3. DNA Extraction, PCR Amplifications, and Sequencing
4.4. In-Silico Analyses of Splice-Site and Missense Variants
4.5. Mini-Gene Construction
4.6. Cell Cultures, Transfections, and Splicing Assays
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DMEM | Dulbecco’s modified Eagle’s Medium |
| F5 | Coagulation factor V gene |
| FV | Factor V |
| FVa | Activated factor V |
| FV:Ag | Factor V antigen level |
| FVai | Activated protein-C inactivated FVa |
| FV:C | Factor V coagulant activity |
| FXa | Activated factor X |
| GnomAD | Genome Aggregation database |
| HGMD | Human Gene Mutation Database |
| PTC | Premature termination codon |
| RICD | Rare inherited coagulation disorder |
| WT | Wild type |
References
- Mann, K.G.; Kalafatis, M. Factor V: A combination of Dr Jekyll and Mr Hyde. Blood 2003, 101, 20–30. [Google Scholar] [CrossRef]
- Duga, S.; Asselta, R.; Tenchini, M.L. Coagulation factor V. Int. J. Biochem. Cell Biol. 2004, 36, 1393–1399. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Tenchini, M.L.; Duga, S. Inherited defects of coagulation factor V: The hemorrhagic side. J. Thromb. Haemost. 2006, 4, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Dahlbäck, B. Factor V and protein S as synergistic cofactors to activated protein C in degradation of factor VIIIa. J. Biol. Chem. 1994, 269, 18735–18738. [Google Scholar] [PubMed]
- Owren, P. Parahaemophilia: Haemorrhagic diathesis due to absence of a previously unknown clotting factor. Lancet 1947, 249, 446–448. [Google Scholar] [CrossRef]
- Kingsley, C.S. Familial factor V deficiency: The pattern of heredity. Quart. J. Med. 1954, 23, 323–329. [Google Scholar]
- Nesheim, M.E.; Myrmel, K.H.; Hibbard, L.; Mann, K.G. Isolation and characterization of single chain bovine factor V. J. Biol. Chem. 1979, 254, 508–517. [Google Scholar]
- Mannucci, P.M.; Duga, S.; Peyvandi, F. Recessively inherited coagulation disorders. Blood 2004, 104, 1243–1252. [Google Scholar] [CrossRef]
- Lak, M.; Sharifian, R.; Peyvandi, F.; Mannucci, P.M. Symptoms of inherited factor V deficiency in 35 Iranian patients. Br. J. Haematol. 1998, 103, 1067–1069. [Google Scholar] [CrossRef]
- Asselta, R.; Peyvandi, F. Factor V deficiency. Semin. Thromb. Hemost. 2009, 35, 382–389. [Google Scholar] [CrossRef]
- Peyvandi, F.; Palla, R.; Menegatti, M.; Siboni, S.M.; Halimeh, S.; Faeser, B.; Pergantou, H.; Platokouki, H.; Giangrande, P.; Peerlinck, K.; et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: Results from the European Network of Rare Bleeding Disorders. J. Thromb. Haemost. 2012, 10, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Guasch, J.F.; Lensen, R.P.; Bertina, R.M. Molecular characterization of a type I quantitative factor V deficiency in a thrombosis patient that is “pseudo homozygous” for activated protein C resistance. Thromb. Haemost. 1997, 77, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Duga, S.; Akhavan, S.; Mannucci, P.M. Rare coagulation deficiencies. Haemophilia 2002, 8, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Schrijver, I.; Koerper, M.A.; Jones, C.D.; Zehnder, J.L. Homozygous factor V splice site mutation associated with severe factor V deficiency. Blood 2002, 99, 3063–3065. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Montefusco, M.C.; Duga, S.; Malcovati, M.; Peyvandi, F.; Mannucci, P.M.; Tenchini, M.L. Severe factor V deficiency: Exon skipping in the factor V gene causing a partial deletion of the C1 domain. J. Thromb. Haemost. 2003, 1, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.J.; Hou, J.; Wang, D.X.; Yu, R.Q. A novel molecular mechanism of congenital FV deficiency: Mutation in the intron acceptor splice site of human blood coagulation FV gene. Zhonghua Yi Xue Za Zhi 2003, 83, 24–26. [Google Scholar] [PubMed]
- Zheng, W.D.; Liu, Y.H.; Liu, H.F.; Chen, Z.H.; Wang, Y. Identification of two novel mutations of human blood coagulation factor V gene in a Chinese family with congenital factor V deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2006, 23, 515–518. [Google Scholar]
- Dall’Osso, C.; Guella, I.; Duga, S.; Locatelli, N.; Paraboschi, E.M.; Spreafico, M.; Afrasiabi, A.; Pechlaner, C.; Peyvandi, F.; Tenchini, M.L.; et al. Molecular characterization of three novel splicing mutations causing factor V deficiency and analysis of the F5 gene splicing pattern. Haematologica 2008, 93, 1505–1513. [Google Scholar] [CrossRef]
- Lunghi, B.; Pinotti, M.; Maestri, I.; Batorova, A.; Bernardi, F. Evaluation of factor V mRNA to define the residual factor V expression levels in severe factor V deficiency. Haematologica 2008, 93, 477–478. [Google Scholar] [CrossRef]
- Delev, D.; Pavlova, A.; Heinz, S.; Seifried, E.; Oldenburg, J. Factor 5 mutation profile in German patients with homozygous and heterozygous factor V deficiency. Haemophilia 2009, 15, 1143–1153. [Google Scholar] [CrossRef]
- Cutler, J.A.; Patel, R.; Rangarajan, S.; Tait, R.C.; Mitchell, M.J. Molecular characterization of 11 novel mutations in patients with heterozygous and homozygous FV deficiency. Haemophilia 2010, 16, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Guella, I.; Paraboschi, E.M.; van Schalkwyk, W.A.; Asselta, R.; Duga, S. Identification of the first Alu-mediated large deletion involving the F5 gene in a compound heterozygous patient with severe factor V deficiency. Thromb. Haemost. 2011, 106, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, E.; Duckers, C.; Radu, C.; Spiezia, L.; Rossetto, V.; Tagariello, G.; Rosing, J.; Simioni, P. Homozygous F5 deep-intronic splicing mutation resulting in severe factor V deficiency and undetectable thrombin generation in platelet-rich plasma. J. Thromb. Haemost. 2011, 9, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Bafunno, V.; Favuzzi, G.; Fierro, T.; Chetta, M.; Mastrodicasa, E.; Chinni, E.; Grandone, E.; Margaglione, M.; Gresele, P. Coinheritance of three novel FV gene mutations in a patient with a severe FV deficiency. Haemophilia 2012, 18, e51–e53. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.E.; Hockin, M.F.; Mann, K.G.; Everse, S.J. The crystal structure of activated protein C-inactivated bovine factor Va: Implications for cofactor function. Proc. Natl. Acad. Sci. USA 2004, 101, 8918–8923. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Maquat, L.E. Leveraging Rules of Nonsense-Mediated mRNA Decay for Genome Engineering and Personalized Medicine. Cell 2016, 165, 1319–1322. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Paraboschi, E.M.; Rimoldi, V.; Menegatti, M.; Peyvandi, F.; Salomon, O.; Duga, S. Exploring the global landscape of genetic variation in coagulation factor XI deficiency. Blood 2017, 130, e1–e6. [Google Scholar] [CrossRef]
- Paraboschi, E.M.; Duga, S.; Asselta, R. Fibrinogen as a Pleiotropic Protein Causing Human Diseases: The Mutational Burden of Aα, Bβ, and γ Chains. Int. J. Mol. Sci. 2017, 18, 2711. [Google Scholar] [CrossRef]
- Ter Avest, P.C.; Fischer, K.; Mancuso, M.E.; Santagostino, E.; Yuste, V.J.; van den Berg, H.M.; van der Bom, J.G.; CANAL Study Group. Risk stratification for inhibitor development at first treatment for severe hemophilia A: A tool for clinical practice. J. Thromb. Haemost. 2008, 6, 2048–2054. [Google Scholar] [CrossRef]
- Pinotti, M.; Bernardi, F.; Dal Mas, A.; Pagani, F. RNA-based therapeutic approaches for coagulation factor deficiencies. J. Thromb. Haemost. 2011, 9, 2143–2152. [Google Scholar] [CrossRef]
- Latorre, E.; Harries, L.W. Splicing regulatory factors, ageing and age-related disease. Ageing Res. Rev. 2017, 36, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Baralle, M.; Baralle, D.; De Conti, L.; Mattocks, C.; Whittaker, J.; Knezevich, A.; Ffrench-Constant, C.; Baralle, F.E. Identification of a mutation that perturbs NF1 a gene splicing using genomic DNA samples and a minigene assay. J. Med. Genet. 2003, 40, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Montefusco, M.C.; Duga, S.; Asselta, R.; Malcovati, M.; Peyvandi, F.; Santagostino, E.; Mannucci, P.M.; Tenchini, M.L. Clinical and molecular characterization of 6 patients affected by severe deficiency of coagulation factor V: Broadening of the mutational spectrum of factor V gene and in vitro analysis of the newly identified missense mutations. Blood 2003, 102, 3210–3216. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouzé, P.; Brunak, S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Culp, M.; Johnson, K.; Michailidis, G. ada: An R package for stochastic boosting. J. Stat. Softw. 2006, 17, 1–27. [Google Scholar] [CrossRef][Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sciences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modelling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Origin | Present Age (Years) | Sex | FV:C (%) | FV:Ag (%) | Main Hemorrhagic Manifestations/Challenges |
|---|---|---|---|---|---|---|
| P1 | Italy | 29 | F | 3 | 2 | Ecchymoses; Surgery for cysts removal on knee, resulting in post-surgery bleeding and consequent anemia (no treatment used) |
| P2 | Italy | 52 | F | 55 | 53 | Two pregnancies, both with premature delivery, and in one case post-delivery bleeding (two days after the event; no treatment); At age four, appendectomy (no complications); At age 32, saphenectomy (no complications); Menorrhagia (no treatment); Implantology surgery (antifibrinolytic treatment); |
| P3 | Italy | 73 | F | 5 | n.a. | Menorrhagia (no treatment); Tooth extractions performed under prophylactic treatment, but still resulting in bleeding; Two pregnancies (no complications); At age 49, polyp removal from vocal cord (antifibrinolytic treatment); At age 56, polyp removal from uterus (under prophylactic treatment with FFP); At age 59, dilation and curettage procedure (under prophylactic treatment with FFP) |
| P4 | Iran | n.a. | F | 4 | n.a. | n.a. |
| Patient | Exon/Intron | Type of Mutation | cDNA Level * | Native Protein | Mature Protein | Status | Gnom AD ** | Reference |
|---|---|---|---|---|---|---|---|---|
| P1 | In1 Ex20 Ex24 | Splicing Missense or splicing? Missense or splicing? | c.6528G>C | - p.G1930D p.K2176N | - p.G1902D p.K2148N | Hetero Hetero Hetero | - 4 - | [24] [24] Novel |
| P2 | In1 Ex20 | Splicing Missense or splicing? | - p.G1930D | - p.G1902D | Hetero Hetero | - 4 | [24] [24] | |
| P3 | Ex15 In19 In19 | Missense Splicing Splicing | c.5090A>G | p.D1697G - - | p.D1669G - - | Hetero Hetero Hetero | - 1 1 | Novel Novel Novel |
| P4 | In19 | Splicing | c.5789-5T>G | - | - | Homo *** | 2 | Novel |
| Variant | Splice-Site Predictions | Missense-Variant Predictions | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HSF | NetGene2 | SSPNN | ADA | SIFT | HumVar | HumDiv | Mutation Taster | LRT | |
| c.158+1G>A | 60.20 (87.03) | disrupted (0.82) | disrupted (1.00) | 0.99 | n.p. | n.p. | n.p. | n.p. | n.p. |
| c.5789G>A/p.G1902D | 75.80 (79.96) | 0.27 (0.53) | 0.61 (0.92) | 0.98 | ND | ND | ND | D | ND |
| c.6528G>C/p.K2148N | 65.59 (76.61) | disrupted (0.00) | disrupted (0.68) | 0.99 | D | D | D | D | D |
| p.D1669G | n.p. | n.p. | n.p. | n.p. | ND | D | D | D | D |
| c.5789-11C>A | 61.63 (68.5) | 0.30 (0.53) | 0.86 (0.92) | 4.97E-04 | n.p. | n.p. | n.p. | n.p. | n.p. |
| c.5789-12C>A | 64.25 (68.5) | 0.33 (0.53) | 0.77 (0.92) | 0.0078 | n.p. | n.p. | n.p. | n.p. | n.p. |
| c.5789-5T>G | 76.33 (79.96) | disrupted (0.53) | disrupted (0.92) | 0.45 | n.p. | n.p. | n.p. | n.p. | n.p. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paraboschi, E.M.; Menegatti, M.; Peyvandi, F.; Duga, S.; Asselta, R. Understanding the Impact of Aberrant Splicing in Coagulation Factor V Deficiency. Int. J. Mol. Sci. 2019, 20, 910. https://doi.org/10.3390/ijms20040910
Paraboschi EM, Menegatti M, Peyvandi F, Duga S, Asselta R. Understanding the Impact of Aberrant Splicing in Coagulation Factor V Deficiency. International Journal of Molecular Sciences. 2019; 20(4):910. https://doi.org/10.3390/ijms20040910
Chicago/Turabian StyleParaboschi, Elvezia Maria, Marzia Menegatti, Flora Peyvandi, Stefano Duga, and Rosanna Asselta. 2019. "Understanding the Impact of Aberrant Splicing in Coagulation Factor V Deficiency" International Journal of Molecular Sciences 20, no. 4: 910. https://doi.org/10.3390/ijms20040910
APA StyleParaboschi, E. M., Menegatti, M., Peyvandi, F., Duga, S., & Asselta, R. (2019). Understanding the Impact of Aberrant Splicing in Coagulation Factor V Deficiency. International Journal of Molecular Sciences, 20(4), 910. https://doi.org/10.3390/ijms20040910