Nuclear Peroxisome Proliferator-Activated Receptors (PPARs) as Therapeutic Targets of Resveratrol for Autism Spectrum Disorder

 , , , and
, , , and
Abstract

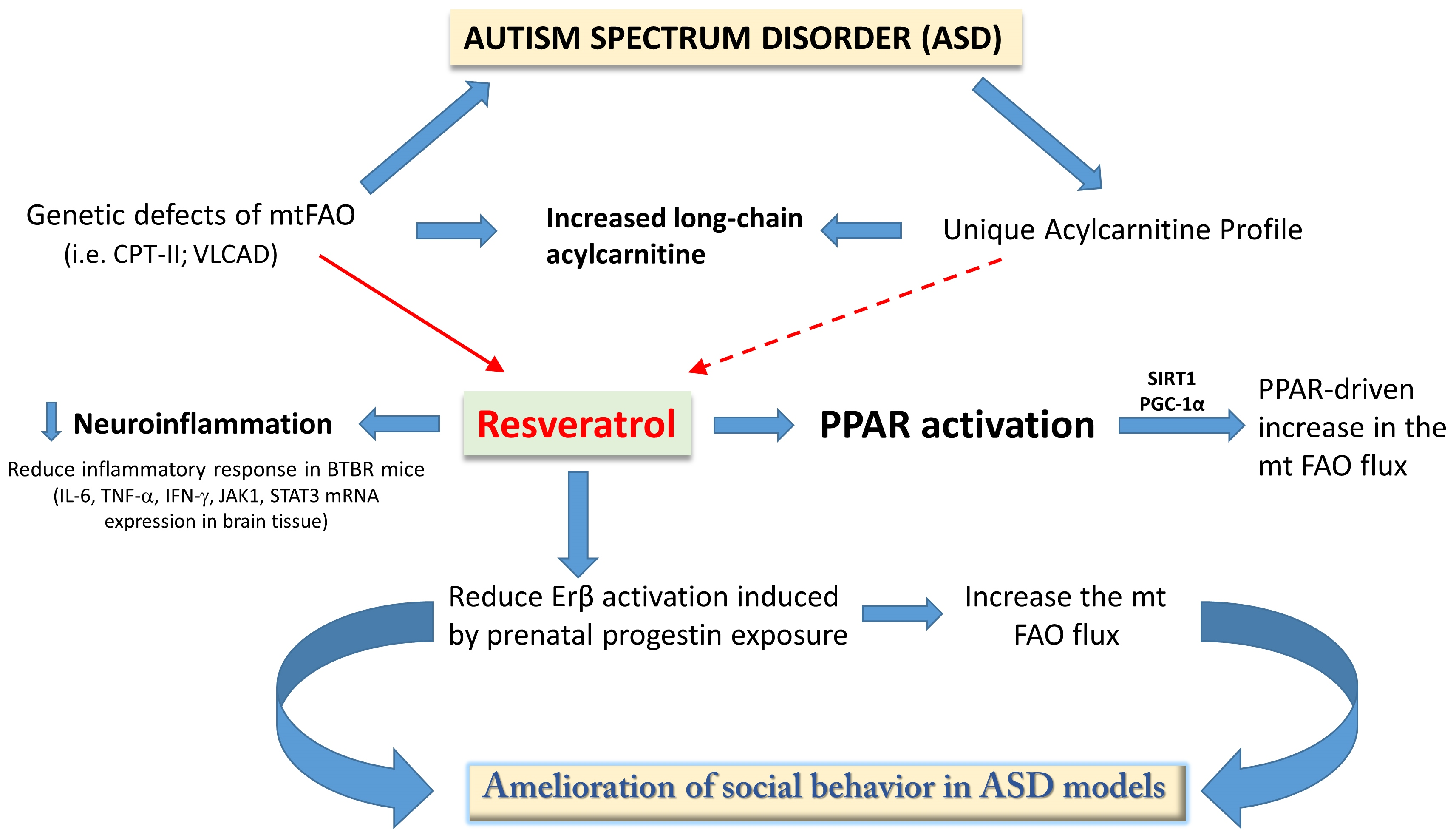{kind=link}
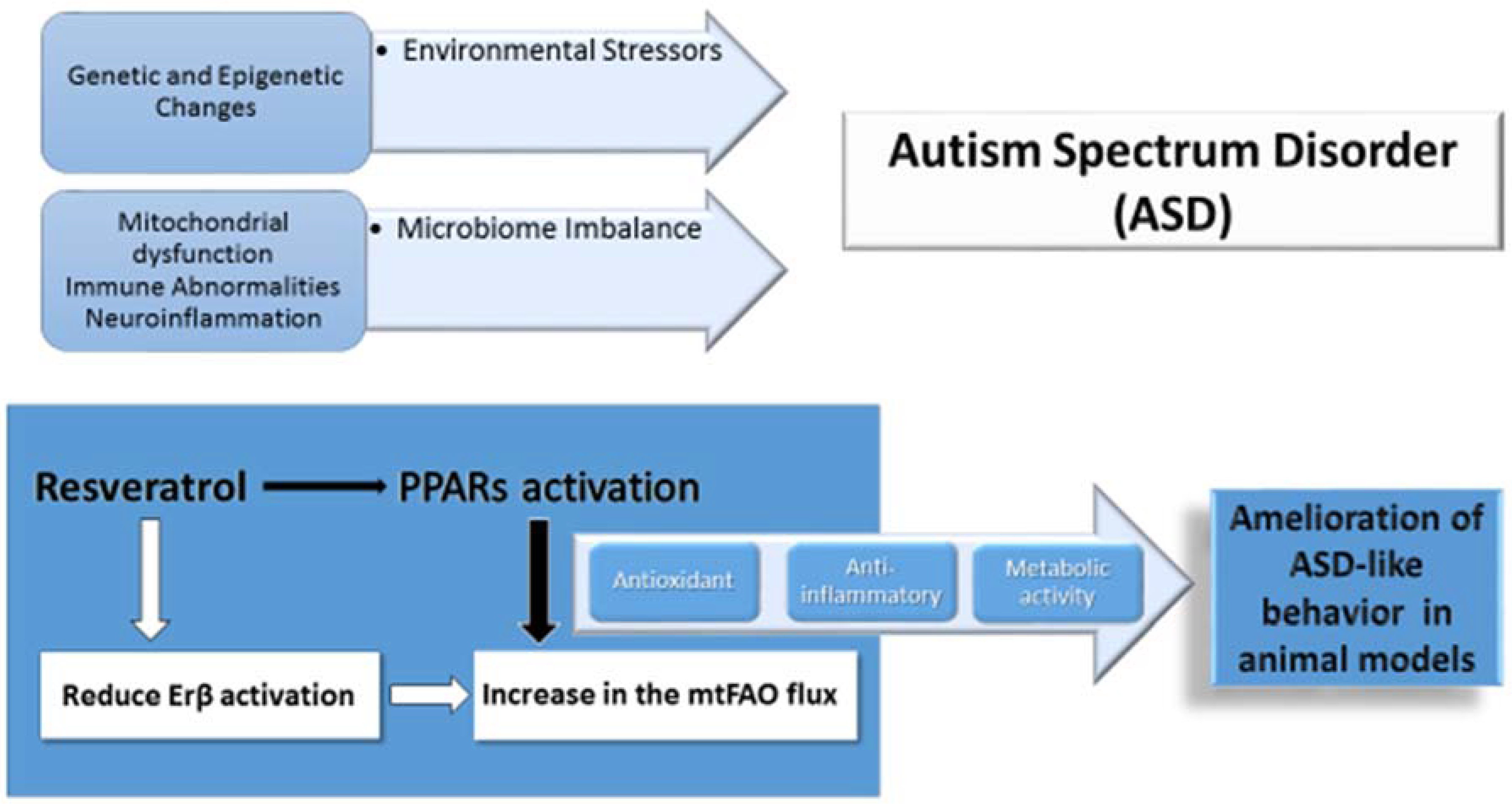{kind=link}
1. Introduction
2. Autism Spectrum Disorder in Genetic Diseases of Mitochondrial Fatty Acid β-Oxidation
3. Fatty Acids in the Energy Metabolism of the Central Nervous System in Autism Spectrum Disorder
Evidence of Dysfunctional Mitochondrial Fatty Acid β-Oxidation in Patients with Idiopathic Autism
4. Molecular Regulation of Mitochondrial FAO: The PPAR Pathway
4.1. PPARs Activators for Treatment of Mitochondrial FAO Defects
4.2. PPARs and Neuropsychiatric Diseases
4.3. Preliminary Evidence of Natural FAO Activator Resveratrol in ASD
4.4. PPARα and Autism Spectrum Disorder
4.5. PPARγ and Autism Spectrum Disorders
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASD | autism spectrum disorder |
| CPT | carnitine palmitoyl transferase |
| CNS | central nervous system |
| ERβ | Estrogen Receptor β |
| ESI-MS/MS | Electrospray ionization tandem mass spectrometry |
| FAO | fatty acid oxidation |
| IAE | influenza associated encephalopathy |
| IEM | inborn errors of metabolism |
| LCFA | long-chain fatty acid |
| LCHAD | long chain 3-hydroxyacyl-CoA dehydrogenase |
| MCAD | medium chain acyl-CoA dehydrogenase |
| MET | met proto-oncogene |
| miRNA | microRNA |
| mtFAO | mitochondrial long-chain fatty acid oxidation |
| NSC | neural stem cells |
| PPAR | peroxisome proliferator-activated receptors |
| PGC-1α | PPAR γ coactivator 1-α |
| PA | propionic acidemia |
| PEA | palmitoylethanolamide |
| PPA | propionic acid |
| RC | respiratory chain |
| RSV | resveratrol |
| SCAD | short chain Acyl-CoA dehydrogenase |
| SIRT1 | sirtuin 1 |
| TCA | tricarboxylic acid |
| TZD | thiazolidinediones |
| VLCAD | very long chain acyl-CoA dehydrogenase |
References
- American Psychiatric Association. Desk Reference to the Diagnostic Criteria from DSM-5™, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013; ISBN 9780890425633. [Google Scholar]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson Rosenberg, C.; White, T.; et al. Prevalence of Autism Spectrum Disorder among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Sturiale, L.; Fiumara, A.; Palmigiano, A.; Bua, R.O.; Rizzo, R.; Zappia, M.; Garozzo, D. CSF N-glycan profile reveals sialylation deficiency in a patient with GM2 gangliosidosis presenting as childhood disintegrative disorder. Autism Res. 2016, 9, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Pellico, A.; Pittalà, A.; Gasperini, S. Neurobehavioral phenotypes of neuronopathic mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 121. [Google Scholar] [CrossRef] [PubMed]
- Kiykim, E.; Zeybek, C.A.; Zubarioglu, T.; Cansever, S.; Yalcinkaya, C.; Soyucen, E.; Aydin, A. Inborn metabolic disorders in Turkish patients with autism spectrum disorders. Autism Res. 2016, 9, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders, a systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Legido, A.; Jethva, R.; Goldenthal, M.J. Mitochondrial dysfunction in autism. Semin. Pediatr. Neurol. 2013, 20, 163–175. [Google Scholar] [CrossRef]
- Giulivi, C.; Zhang, Y.F.; Omanska-Klusek, A.; Ross-Inta, C.; Wong, S.; Hertz-Picciotto, I.; Tassone, F.; Pessah, I.N. Mitochondrial dysfunction in autism. JAMA 2010, 304, 2389–2396. [Google Scholar] [CrossRef]
- Napoli, E.; Wong, S.; Hertz-Picciotto, I.; Giulivi, C. Deficits in bioenergetics and impaired immune response in granulocytes from children with autism. Pediatrics 2014, 133, e1405–e1410. [Google Scholar] [CrossRef]
- Goldenthal, M.J.; Kuruvilla, T.; Damle, S.; Salganicoff, L.; Sheth, S.; Shah, N.; Marks, H.; Khurana, D.; Valencia, I.; Legido, A. Non-invasive evaluation of buccal respiratory chain enzyme dysfunction in mitochondrial disease, Comparison with studies in muscle biopsy. Mol. Genet. Metab. 2012, 105, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.; Dong, Z.; Zhang, Y.; Di Mauro, S.; Peterson, B.S. Mitochondrial dysfunction as a neurobiological subtype of autism spectrum disorder, evidence from brain imaging. JAMA Psychiatry 2014, 71, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Olpin, S.E. Pathophysiology of fatty acid oxidation disorders and resultant phenotypic variability. J. Inherit. Metab. Dis. 2013, 36, 645–658. [Google Scholar] [CrossRef]
- Bastin, J. Regulation of mitochondrial fatty acid β-oxidation in human, what can we learn from inborn fatty acid β-oxidation deficiencies? Biochimie 2014, 96, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Crowe, L.; Andresen, B.S.; Anderson, V.; Boneh, A. Neurodevelopmental profiles of children with very long chain acyl-CoA dehydrogenase deficiency diagnosed by newborn screening. Mol. Genet. Metab. 2014, 113, 278–282. [Google Scholar] [CrossRef]
- Strandqvist, A.; Haglind, C.B.; Zetterström, R.H.; Nemeth, A.; von Döbeln, U.; Stenlid, M.H.; Nordenström, A. Neuropsychological Development in Patients with Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase (LCHAD) Deficiency. JIMD Rep. 2016, 28, 75–84. [Google Scholar] [PubMed]
- Clark-Taylor, T.; Clark-Taylor, B.E. Is autism a disorder of fatty acid metabolism? Med. Hypotheses 2004, 62, 970–975. [Google Scholar] [CrossRef]
- Filipek, P.A.; Juranek, J.; Nguyen, M.T.; Cummings, C.; Gargus, J.J. Relative carnitine deficiency in autism. J. Autism Dev. Disord. 2004, 34, 615–623. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands, nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Karpe, F.; Ehrenborg, E.E. PPARδ in humans, genetic and pharmacological evidence for a significant metabolic function. Curr. Opin. Lipidol. 2009, 20, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Barroso, E.; Zarei, M.; Botteri, G.; Vázquez-Carrera, M. PPARβ/δ and lipid metabolism in the heart. Biochim. Biophys. Acta 2016, 1861, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Ehrenborg, E.; Krook, A. Regulation of skeletal muscle physiology and metabolism by peroxisome proliferator-activated receptor δ. Pharmacol. Rev. 2009, 61, 373–393. [Google Scholar] [CrossRef]
- Tan, N.S.; Vázquez-Carrera, M.; Montagner, A.; Sng, M.K.; Guillou, H.; Wahli, W. Transcriptional control of physiological and pathological processes by the nuclear receptor PPARβ/δ. Prog. Lipid Res. 2016, 64, 98–122. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond, the diverse biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef]
- D’Agostino, G.; Cristiano, C.; Lyons, D.J.; Citraro, R.; Russo, E.; Avagliano, C.; Russo, R.; Raso, G.M.; Meli, R.; De Sarro, G.; et al. Peroxisome proliferator-activated receptor α plays a crucial role in behavioral repetition and cognitive flexibility in mice. Mol. Metab. 2015, 4, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Boris, M.; Kaiser, C.C.; Goldblatt, A.; Elice, M.W.; Edelson, S.M.; Adams, J.B.; Feinstein, D.L. Effect of pioglitazone treatment on behavioral symptoms in autistic children. J. Neuroinflamm. 2007, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Ghaleiha, A.; Rasa, S.M.; Nikoo, M.; Farokhnia, M.; Mohammadi, M.R.; Akhondzadeh, S. A pilot double-blind placebo-controlled trial of pioglitazone as adjunctive treatment to risperidone, effects on aberrant behavior in children with autism. Psychiatry Res. 2015, 229, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Blunder, M.; Liu, X.; Malainer, C.; Blazevic, T.; Schwaiger, S.; Rollinger, J.M.; Heiss, E.H.; et al. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ), a review. Biochem. Pharmacol. 2014, 92, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Nakata, R.; Takahashi, S.; Inoue, H. Recent advances in the study on resveratrol. Biol. Pharm. Bull. 2012, 35, 273–279. [Google Scholar] [CrossRef]
- Wąsik, A.; Antkiewicz-Michaluk, L. The mechanism of neuroprotective action of natural compounds. Pharmacol. Rep. 2017, 69, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, G.; Pennisi, M.; Bertino, G.; Motta, M.; Borzì, A.M.; Vicari, E.; Bella, R.; Drago, F.; Malaguarnera, M. Resveratrol in patients with minimal hepatic encephalopathy. Nutrients 2018, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol, the in vivo evidence. Nat. Rev. Drug. Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Tomé-Carneiro, J.; Larrosa, M.; González-Sarrías, A.; Tomás-Barberán, F.A.; García-Conesa, M.T.; Espín, J.C. Resveratrol and clinical trials, the crossroad from in vitro studies to human evidence. Curr. Pharm. Des. 2013, 19, 6064–6093. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Pezzuto, J.M. The pharmacology of resveratrol in animals and humans. Biochim. Biophys. Acta 2015, 1852, 1071–1113. [Google Scholar] [CrossRef]
- Bambini-Junior, V.; Zanatta, G.; Della Flora Nunes, G.; Mueller de Melo, G.; Michels, M.; Fontes-Dutra, M.; Nogueira Freire, V.; Riesgo, R.; Gottfried, C. Resveratrol prevents social deficits in animal model of autism induced by valproic acid. Neurosci. Lett. 2014, 583, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Bakheet, S.A.; Alzahrani, M.Z.; Nadeem, A.; Ansari, M.A.; Zoheir, K.M.A.; Attia, S.M.; Al-Ayadhi, L.Y.; Ahmad, S.F. Resveratrol treatment attenuates chemokine receptor expression in the BTBR T+tf/J mouse model of autism. Mol. Cell. Neurosci. 2016, 77, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Dutra, M.; Santos-Terra, J.; Deckmann, I.; Brum Schwingel, G.; Della Flora Nunes, G.; Hirsch, M.M.; Bauer-Negrini, G.; Riesgo, R.S.; Bambini-Júnior, V.; Hedin-Pereira, C.; et al. Resveratrol prevents cellular and behavioral sensory alterations in the animal model of autism induced by valproic acid. Front. Synaptic Neurosci. 2018, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, M.M.; Deckmann, I.; Fontes-Dutra, M.; Bauer-Negrini, G.; Della Flora Nunes, G.; Nunes, W.; Rabelo, B.; Riesgo, R.; Margis, R.; Bambini-Junior, V.; et al. Behavioral alterations in autism model induced by valproic acid and translational analysis of circulating microRNA. Food Chem. Toxicol. 2018, 115, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Ge, X.; Li, L.; Yao, A.; Wang, X.; Li, M.; Gong, X.; Chu, Z.; Lu, Z.; Huang, X.; et al. Resveratrol ameliorates prenatal progestin exposure-induced autism-like behavior through ERβ activation. Mol. Autism 2018, 9, 43. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Pougovkina, O.; te Brinke, H.; Ofman, R.; van Cruchten, A.G.; Kulik, W.; Wanders, R.J.; Houten, S.M.; de Boer, V.C. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum. Mol. Genet. 2014, 23, 3513–3522. [Google Scholar] [CrossRef]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell. Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef]
- Waye, M.M.Y.; Cheng, H.Y. Genetics and epigenetics of autism: A Review. Psychiatry Clin. Neurosci. 2018, 72, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Poschmann, J.; Cruz-Herrera Del Rosario, R.; Parikshak, N.N.; Hajan, H.S.; Kumar, V.; Ramasamy, R.; Belgard, T.G.; Elanggovan, B.; Wong, C.C.Y.; et al. Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell 2016, 167, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Wajner, M.; Amaral, A.U. Mitochondrial dysfunction in fatty acid oxidation disorders, insights from human and animal studies. Biosci. Rep. 2016, 36, e00281. [Google Scholar] [CrossRef] [PubMed]
- Celestino-Soper, P.B.; Violante, S.; Crawford, E.L.; Luo, R.; Lionel, A.C.; Delaby, E.; Cai, G.; Sadikovic, B.; Lee, K.; Lo, C.; et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc. Natl. Acad. Sci. USA 2012, 109, 7974–7981. [Google Scholar] [CrossRef]
- Schönfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the central nervous system. Biomed. Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, J.N.; Bowman, C.E.; Wolfgang, M.J.; Scafidi, S. Developmental regulation and localization of carnitine palmitoyltransferases (CPTs) in rat brain. J. Neurochem. 2017, 142, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Jones, A.; Deeney, J.T.; Hur, S.K.; Bankaitis, V.A. Inborn errors of long-chain fatty acid β-oxidation link neural stem cell self-renewal to autism. Cell Rep. 2016, 14, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Pilz, G.A.; Ghesquière, B.; Kovacs, W.J.; Wegleiter, T.; Moore, D.L.; Hruzova, M.; Zamboni, N.; Carmeliet, P.; Jessberger, S. Fatty acid oxidation-dependent metabolic shift regulates adult neural stem cell activity. Cell Rep. 2017, 20, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont, J.P.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2, biochemical.; molecular and medical aspects. Mol. Asp. Med. 2004, 25, 495–520. [Google Scholar] [CrossRef] [PubMed]
- Tein, I. Impact of fatty acid oxidation disorders in child neurology, from Reye syndrome to Pandora’s box. Dev. Med. Child Neurol. 2015, 57, 304–306. [Google Scholar] [CrossRef]
- Ozonoff, S.; Williams, B.J.; Landa, R. Parental report of the early development of children with regressive autism. The delays-plus-regression phenotype. Autism 2005, 9, 461–486. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Yao, D.; Yamaguchi, M.; Chida, J.; Yao, D.; Kido, H. Bezafibrate upregulates carnitine palmitoyltransferase II expression and promotes mitochondrial energy crisis dissipation in fibroblasts of patients with influenza-associated encephalopathy. Mol. Genet. Metab. 2011, 104, 265–272. [Google Scholar] [CrossRef] [PubMed]
- El-Ansary, A.K.; Bacha, A.G.; Al-Ayahdi, L.Y. Plasma fatty acids as diagnostic markers in autistic patients from Saudi Arabia. Lipids Health Dis. 2011, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Pastural, E.; Ritchie, S.; Lu, Y.; Jin, W.; Kavianpour, A.; Khine Su-Myat, K.; Heath, D.; Wood, P.L.; Fisk, M.; Goodenowe, D.B. Novel plasma phospholipid biomarkers of autism, mitochondrial dysfunction as a putative causative mechanism. Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.C.; Liang, B.B.; Jarvi, E.J.; Cooper, A.J.; Lu, D.R. Differential effects of fatty acyl coenzyme A derivatives on citrate synthase and glutamate dehydrogenase. Res. Commun. Chem. Pathol. Pharmacol. 1993, 82, 331–338. [Google Scholar] [PubMed]
- Ventura, F.V.; Ruiter, J.P.; Ijlst, L.; de Almeida, I.T.; Wanders, R.J. Inhibitory effect of 3-hydroxyacyl-CoAs and other long chain fatty acid beta-oxidation intermediates on mitochondrial oxidative phosphorylation. J. Inherit. Metab. Dis. 1996, 19, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Melnyk, S.; MacFabe, D.F. Unique acyl-carnitine profiles are potential biomarkers for acquired mitochondrial disease in autism spectrum disorder. Transl. Psychiatry 2013, 3, e220. [Google Scholar] [CrossRef]
- Barone, R.; Alaimo, S.; Messina, M.; Pulvirenti, A.; Bastin, J.; MIMIC-Autism group; Ferro, A.; Frye, R.E.; Rizzo, R. A subset of patients with Autism Spectrum Disorders show a distinctive metabolic profile by dried blood spot analyses. Front. Psychiatry 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, R.; Allen, K.J.; Koplin, J.J.; Roche, P.; Greaves, R.F. Advantages and Challenges of Dried Blood Spot Analysis by Mass Spectrometry Across the Total Testing Process. EJIFCC 2016, 27, 288–317. [Google Scholar]
- Shultz, S.R.; MacFabe, D.F.; Ossenkopp, K.P.; Scratch, S.; Whelan, J.; Taylor, R.; Cain, D.P. Intracerebroventricular injection of propionic acid, an enteric bacterial metabolic end-product, impairs social behavior in the rat, implications for an animal model of autism. Neuropharmacology 2008, 54, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.H.; Foley, K.A.; Mepham, J.R.; Tichenoff, L.J.; Possmayer, F.; MacFabe, D.F. Altered brain phospholipid and acylcarnitine profiles in propionic acid infused rodents, further development of a potential model of autism spectrum disorders. J. Neurochem. 2010, 113, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Bennuri, S.C.; Davis, J.E.; Wynne, R.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; MacFabe, D.F.; et al. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl. Psychiatry 2018, 2, 42. [Google Scholar] [CrossRef]
- Frye, R.E.; Rose, S.; Chacko, J.; Wynne, R.; Bennuri, S.C.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; et al. Modulation of mitochondrial function by the microbiome metabolite propionic acid in autism and control cell lines. Transl. Psychiatry 2016, 25, e927. [Google Scholar] [CrossRef] [PubMed]
- Witters, P.; Debbold, E.; Crivelly, K.; Vande Kerckhove, K.; Corthouts, K.; Debbold, B.; Andersson, H.; Vannieuwenborg, L.; Geuens, S.; Baumgartner, M.; et al. Autism in patients with propionic acidemia. Mol. Genet. Metab. 2016, 119, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, K.K.; Levy, R.J. Evidence of mitochondrial dysfunction in autism, biochemical links, genetic-based associations, and non-energy-related mechanisms. Oxid. Med. Cell. Longev. 2017, 2017, 4314025. [Google Scholar] [CrossRef] [PubMed]
- Hollis, F.; Kanellopoulos, A.K.; Bagni, C. Mitochondrial dysfunction in autism spectrum disorder, clinical features and perspectives. Curr. Opin. Neurobiol. 2017, 45, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Wallace, D.C. Mitochondrial etiology of neuropsychiatric disorders. Biol. Psychiatry 2018, 83, 722–730. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Ene, H.M. Mitochondrial targeted therapies, where do we stand in mental disorders? Biol. Psychiatry 2018, 83, 770–779. [Google Scholar] [CrossRef]
- Mandard, S.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. Cell. Mol. Life Sci. 2004, 61, 393–416. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Nierenberg, A.A.; Ghaznavi, S.A.; Sande Mathias, I.; Ellard, K.K.; Janos, J.A.; Sylvia, L.G. peroxisome proliferator-activated receptor γ coactivator-1 α as a novel target for bipolar disorder and other neuropsychiatric disorders. Biol. Psychiatry. 2018, 83, 761–769. [Google Scholar] [CrossRef]
- Corona, J.C.; Duchen, M.R. PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic. Biol. Med. 2016, 100, 153–163. [Google Scholar] [CrossRef]
- Bugge, A.; Holst, D. PPAR agonists, - Could tissue targeting pave the way? Biochimie 2017, 136, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Djouadi, F.; Bonnefont, J.P.; Thuillier, L.; Droin, V.; Khadom, N.; Munnich, A.; Bastin, J. Correction of fatty acid oxidation in carnitine palmitoyl transferase II deficient cultured skin fibroblasts by bezafibrate. Pediatr. Res. 2003, 54, 446–451. [Google Scholar] [CrossRef]
- Djouadi, F.; Aubey, F.; Schlemmer, D.; Ruiter, J.P.; Wanders, R.J.; Strauss, A.W.; Bastin, J. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum. Mol. Genet. 2005, 14, 2695–2703. [Google Scholar] [CrossRef]
- Bastin, J.; Aubey, F.; Rötig, A.; Munnich, A.; Djouadi, F. Activation of peroxisome proliferator activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients’ cells lacking its components. J. Clin. Endocrinol. Metab. 2008, 93, 1433–1441. [Google Scholar] [CrossRef]
- Gobin-Limballe, F.; Djouadi, F.; Aubey, S.; Olpin, B.S.; Andresen, S.; Yamaguchi, H.; Mandel, H.; Fukao, T.; Ruiter, J.P.; Wanders, R.J.; et al. Genetic basis for correction of very long chain acyl-CoA dehydrogenase deficiency by bezafibrate in patient fibroblasts, towards a genotype-based therapy. Am. J. Hum. Genet. 2007, 81, 1133–1143. [Google Scholar] [CrossRef]
- Yamada, K.; Kobayashi, H.; Bo, R.; Purevsuren, J.; Mushimoto, Y.; Takahashi, T.; Hasegawa, Y.; Taketani, T.; Fukuda, S.; Yamaguchi, S. Efficacy of bezafibrate on fibroblasts of glutaric acidemia type II patients evaluated using an in vitro probe acylcarnitine assay. Brain Dev. 2017, 39, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont, J.P.; Bastin, J.; Behin, A.; Djouadi, F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N. Engl. J. Med. 2009, 360, 838–840. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont, J.P.; Bastin, J.; Laforêt, P.; Aubey, F.; Mogenet, A.; Romano, S.; Ricquier, D.; Gobin-Limballe, S.; Vassault, A.; Behin, A.; et al. Long term follow-up of bezafibrate treatment in the myopathic form of Carnitine-PalmitoylTransferase 2 deficiency. Clin. Pharm. Ther. 2010, 88, 101–108. [Google Scholar] [CrossRef]
- Newcastle University; Newcastle-upon-Tyne Hospitals NHS Trust. A Study of Bezafibrate in Mitochondrial Myopathy. Available online: https://clinicaltrials.gov/ct2/show/NCT02398201 (accessed on 16 February 2019).
- Morishima, T.; Togashi, T.; Yokota, S.; Okuno, Y.; Miyazaki, C.; Tashiro, M.; Okabe, N. Encephalitis and encephalopathy associated with an influenza epidemic in Japan. Clin. Infect. Dis. 2002, 35, 512–517. [Google Scholar] [CrossRef]
- Torres-Espínola, F.J.; Altmäe, S.; Segura, M.T.; Jerez, A.; Anjos, T.; Chisaguano, M.; Carmen López-Sabater, M.; Entrala, C.; Alvarez, J.C.; Agil, A.; et al. Maternal PPARG Pro12Ala polymorphism is associated with infant’s neurodevelopmental outcomes at 18 months of age. Early Hum. Dev. 2015, 91, 457–462. [Google Scholar] [CrossRef]
- Racke, M.K.; Drew, P.D. PPARs in Neuroinflammation. PPAR Res. 2008, 2008, 638356. [Google Scholar] [CrossRef]
- Chandra, A.; Sharma, A.; Calingasan, N.Y.; White, J.M.; Shurubor, Y.; Yang, X.W.; Beal, M.F.; Johri, A. Enhanced mitochondrial biogenesis ameliorates disease phenotype in a full-length mouse model of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 2269–2282. [Google Scholar] [CrossRef]
- Hondares, E.; Pineda-Torra, I.; Iglesias, R.; Staels, B.; Villarroya, F.; Giralt, M. PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem. Biophys. Res. Commun. 2007, 354, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, S.; Reiser, G. Role of the peroxisome proliferator-activated receptors (PPAR)-α,β/δ and γ triad in regulation of reactive oxygen species signaling in brain. Biol. Chem. 2013, 394, 1553–1570. [Google Scholar] [CrossRef]
- Cirnigliaro, M.; Barbagallo, C.; Gulisano, M.; Domini, C.N.; Barone, R.; Barbagallo, D.; Ragusa, M.; Di Pietro, C.; Rizzo, R.; Purrello, M. Expression and Regulatory Network Analysis of miR-140-3p, a New Potential Serum Biomarker for Autism Spectrum Disorder. Front. Mol. Neurosci. 2017, 10, 250. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A.; Alzahrani, M.Z.; Alshammari, M.A.; Alanazi, W.A.; Alasmari, A.F.; Attia, S.M. Resveratrol attenuates pro-inflammatory cytokines and activation of JAK1-STAT3 in BTBR T+ Itpr3tf/J autistic mice. Eur. J. Pharmacol. 2018, 829, 70–78. [Google Scholar] [CrossRef]
- Bastin, J.; Lopes-Costa, A.; Djouadi, F. Exposure to resveratrol triggers pharmacological correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum. Mol. Genet. 2011, 20, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Aires, V.; Delmas, D.; Le Bachelier, C.; Latruffe, N.; Schlemmer, D.; Benoist, J.F.; Djouadi, F.; Bastin, J. Stilbenes and resveratrol metabolites improve mitochondrial fatty acid oxidation defects in human fibroblasts. Orphanet J. Rare Dis. 2014, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, K.E.; Hill, A.P.; Guion, K.; Voltolina, L.; Fombonne, E. Overweight and obesity, prevalence and correlates in a large clinical sample of children with autism spectrum disorder. J. Autism Dev. Disord. 2014, 44, 1708–1719. [Google Scholar] [CrossRef]
- Kerr, D.M.; Downey, L.; Conboy, M.; Finn, D.P.; Roche, M. Alterations in the endocannabinoid system in the rat valproic acid model of autism. Behav. Brain Res. 2013, 249, 124–132. [Google Scholar] [CrossRef]
- Bertolino, B.; Crupi, R.; Impellizzeri, D.; Bruschetta, G.; Cordaro, M.; Siracusa, R.; Esposito, E.; Cuzzocrea, S. Beneficial effects of co-ultramicronized palmitoylethanolamide/ luteolin in a mouse model of autism and in a case report of autism. CNS Neurosci. Ther. 2017, 23, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, C.; Pirozzi, C.; Coretti, L.; Cavaliere, G.; Lama, A.; Russo, R.; Lembo, F.; Mollica, M.P.; Meli, R.; Calignano, A.; et al. Palmitoylethanolamide counteracts autistic-like behaviours in BTBR T+tf/J mice, contribution of central and peripheral mechanisms. Brain Behav. Immun. 2018, 74, 166–175. [Google Scholar] [CrossRef]
- Khalaj, M.; Saghazadeh, A.; Shirazi, E.; Shalbafan, M.R.; Alavi, K.; Shooshtari, M.H.; Laksari, F.Y.; Hosseini, M.; Mohammadi, M.R.; Akhondzadeh, S. Palmitoylethanolamide as adjunctive therapy for autism: Efficacy and safety results from a randomized controlled trial. J. Psychiatr. Res. 2018, 103, 104–111. [Google Scholar] [CrossRef]
- Zdravkovic, V.; Hamilton, J.K.; Daneman, D.; Cummings, E.A. Pioglitazone as adjunctive therapy in adolescents with type 1 diabetes. J. Pediatr. 2006, 149, 845–849. [Google Scholar] [CrossRef]
- Pilipović, K.; Župan, Ž.; Dolenec, P.; Mršić-Pelčić, J.; Župan, G. A single dose of PPARγ agonist pioglitazone reduces cortical oxidative damage and microglial reaction following lateral fluid percussion brain injury in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 59, 8–20. [Google Scholar] [CrossRef]
- Noriega, D.B.; Savelkoul, H.F. Immune dysregulation in autism spectrum disorder. Eur. J. Pediatr. 2014, 173, 33–43. [Google Scholar] [CrossRef]
- Edmiston, E.; Ashwood, P.; Van De Water, J. Autoimmunity, autoantibodies, and autism spectrum disorders (ASD). Biol. Psychiatry 2017, 81, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.N.; Van de Water, J. Altered T cell responses in children with autism. Brain Behav. Immun. 2011, 25, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Gładysz, D.; Krzywdzińska, A.; Hozyasz, K.K. Immune abnormalities in autism spectrum disorder-could they hold promise for causative treatment? Mol. Neurobiol. 2018, 55, 6387–6435. [Google Scholar] [CrossRef]
- Emanuele, E.; Lossano, C.; Politi, P.; Barale, F. Pioglitazone as a therapeutic agent in autistic spectrum disorder. Med. Hypotheses 2007, 69, 699. [Google Scholar] [CrossRef] [PubMed]
- Dembic, M.; Andersen, H.S.; Bastin, J.; Doktor, T.K.; Corydon, T.J.; Sass, J.O.; Costa, A.L.; Djouadi, F.; Andresen, B.S. Next generation sequencing of RNA reveals novel targets of resveratrol with possible implications for Canavan disease. Mol. Genet. Metab. 2018, 126, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.F. Social Skills Deficits in Autism Spectrum Disorder: Potential Biological Origins and Progress in Developing Therapeutic Agents. CNS Drugs 2018, 32, 713–734. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Scott, E.; Brown, V.A.; Gescher, A.J.; Steward, P.W.; Brown, K. Clinical trials of resveratrol. Ann. R. Y. Acad. Sci. 2011, 1215, 161–169. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barone, R.; Rizzo, R.; Tabbì, G.; Malaguarnera, M.; Frye, R.E.; Bastin, J. Nuclear Peroxisome Proliferator-Activated Receptors (PPARs) as Therapeutic Targets of Resveratrol for Autism Spectrum Disorder. Int. J. Mol. Sci. 2019, 20, 1878. https://doi.org/10.3390/ijms20081878
Barone R, Rizzo R, Tabbì G, Malaguarnera M, Frye RE, Bastin J. Nuclear Peroxisome Proliferator-Activated Receptors (PPARs) as Therapeutic Targets of Resveratrol for Autism Spectrum Disorder. International Journal of Molecular Sciences. 2019; 20(8):1878. https://doi.org/10.3390/ijms20081878
Chicago/Turabian StyleBarone, Rita, Renata Rizzo, Giovanni Tabbì, Michele Malaguarnera, Richard E. Frye, and Jean Bastin. 2019. "Nuclear Peroxisome Proliferator-Activated Receptors (PPARs) as Therapeutic Targets of Resveratrol for Autism Spectrum Disorder" International Journal of Molecular Sciences 20, no. 8: 1878. https://doi.org/10.3390/ijms20081878
APA StyleBarone, R., Rizzo, R., Tabbì, G., Malaguarnera, M., Frye, R. E., & Bastin, J. (2019). Nuclear Peroxisome Proliferator-Activated Receptors (PPARs) as Therapeutic Targets of Resveratrol for Autism Spectrum Disorder. International Journal of Molecular Sciences, 20(8), 1878. https://doi.org/10.3390/ijms20081878