Allelic Diversity of Acetyl Coenzyme A Carboxylase accD/bccp Genes Implicated in Nuclear-Cytoplasmic Conflict in the Wild and Domesticated Pea (Pisum sp.)

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results
2.1. Structure and Variation of accD Gene
2.2. Variation in Nuclear bccp Gene
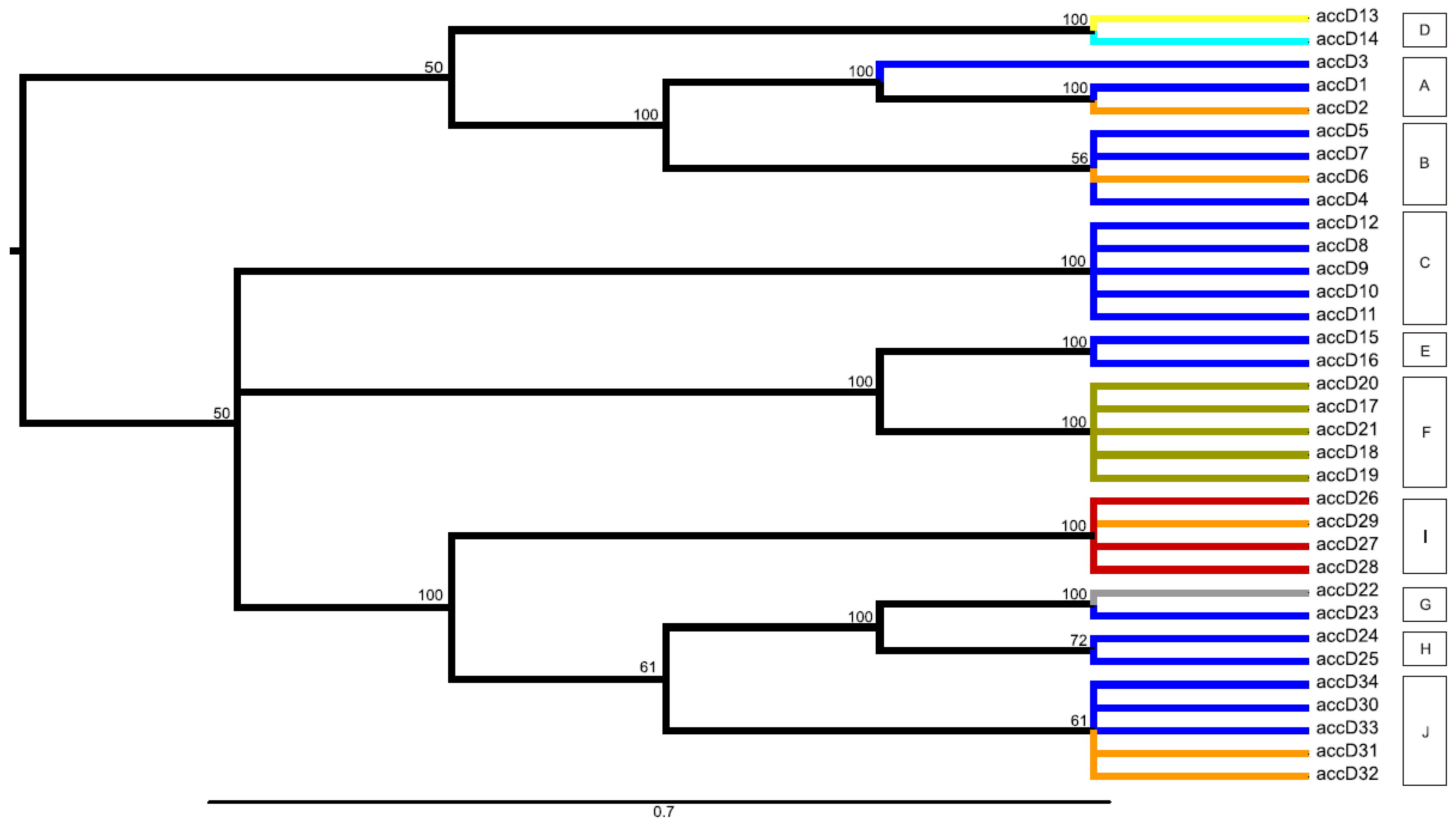
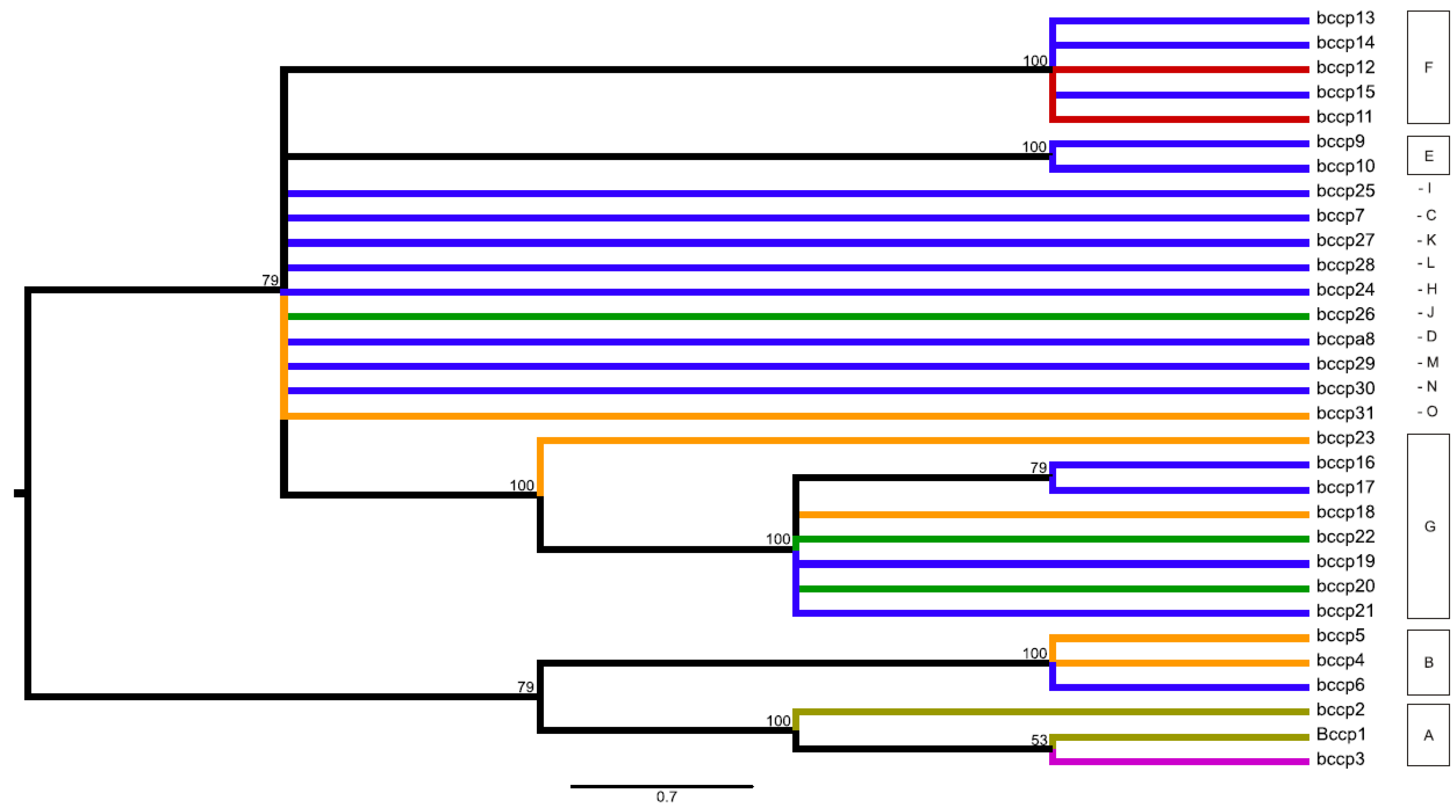
2.3. Network and Maximum Parsimony Analyses
2.4. Frequency of Amino Acid Substitutions and Their Distribution
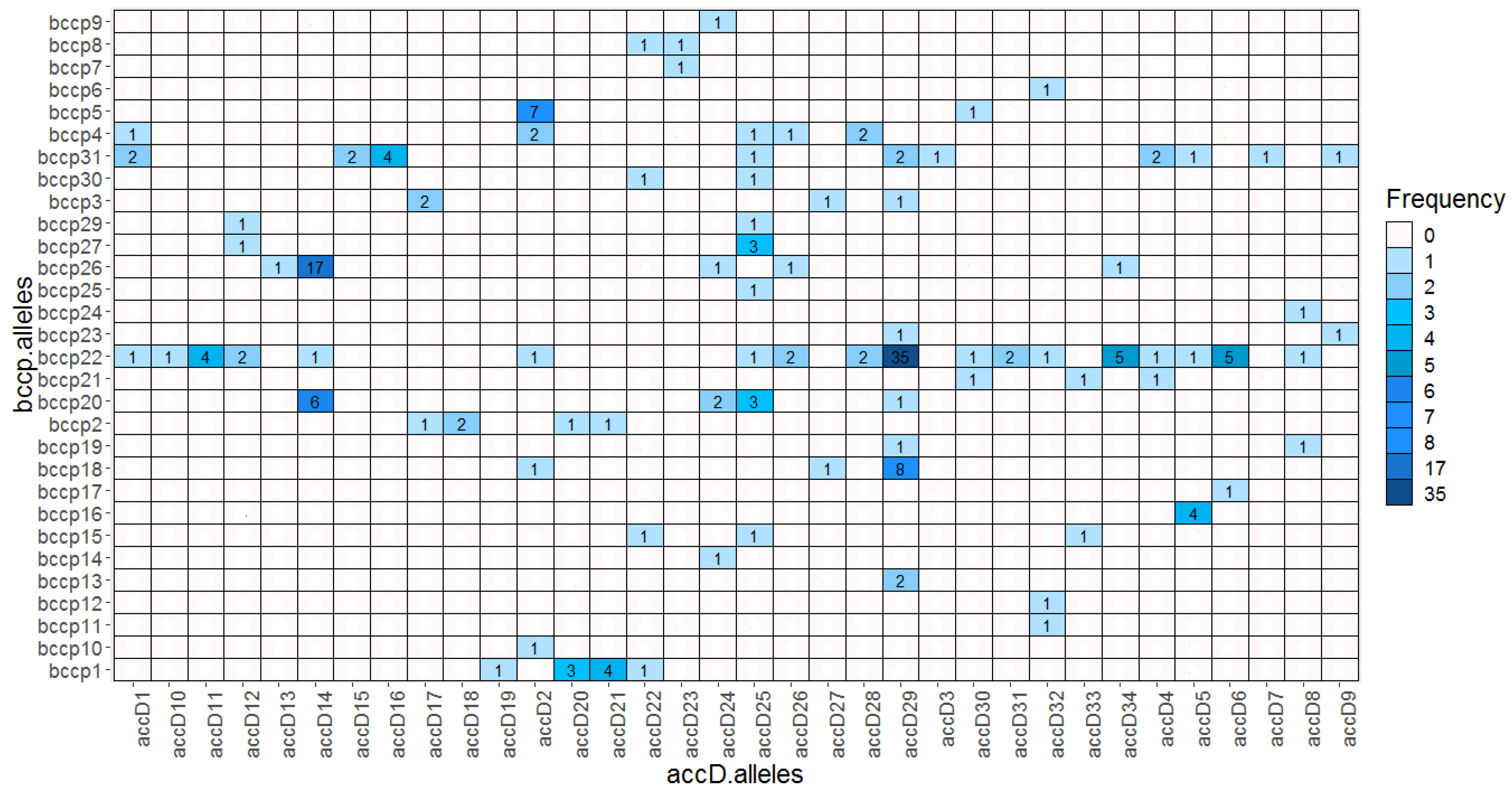
2.5. Allelic accD/bccp Combinations
2.6. Relationship to Pisum Genetic Diversity
2.7. Geographic Distribution of accD/bccp Alleles
3. Discussion
3.1. Hypervariability of the Chloroplast accD Gene
3.2. Allelic accD/bccp Combinations Found in Wild and Domesticated Peas
3.3. Domestication and Hybrid Incompatibility
4. Material and Methods
4.1. Plant Material
4.2. DNA and RNA Analysis
4.3. Sequence Analysis
4.4. Tandem Repeat Analysis
4.5. Protein Sequence Analysis and Structure Modelling
4.6. Mapping Protein Sequence Polymorphisms on Predicted Structure
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coyne, J.A. Genetics and speciation. Nature 1992, 355, 511–515. [Google Scholar] [CrossRef]
- Coyne, J.A.; Orr, H.A. Speciation. Sinauer, Sunderland; Oxford University Press: New York, NY, USA, 2004; pp. 1–545. [Google Scholar]
- Givnish, T.J. Ecology of plant speciation. Taxon 2010, 59, 1326–1366. [Google Scholar] [CrossRef]
- Case, A.L.; Finseth, F.R.; Barr, C.M.; Fishman, L. Selfish evolution of cytonuclear hybrid incompatibility in Mimulus. Proc. Biol. Sci. 2016, 283, 20161493. [Google Scholar] [CrossRef]
- Sambatti, J.B.M.; Ortiz-Barrientos, D.; Baack, E.J.; Rieseberg, L.H. Ecological selection maintains cytonuclear incompatibilities in hybridizing sunflowers. Ecol. Lett. 2008, 11, 1082–1091. [Google Scholar] [CrossRef]
- Bateson, W. Mendel’s Principles of Heredity; Cambridge University Press: Cambridge, UK, 1909. [Google Scholar]
- Dobzhansky, T. Genetics and the Origin of Species. In Columbia Biological Series; Columbia University Press: New York, NY, USA, 1937; Volume 9, pp. 1–364. [Google Scholar]
- Muller, H.J. Isolating mechanisms, evolution, and temperature. Biol. Symp. 1942, 6, 71–125. [Google Scholar]
- Fishman, L.; Sweigart, A.L. When Two Rights Make a Wrong: The Evolutionary Genetics of Plant Hybrid Incompatibilities. Annu. Rev. Plant Biol. 2018, 69, 707–731. [Google Scholar] [CrossRef]
- Barnard-Kubow, K.B.; So, N.; Galloway, L.F. Cytonuclear incompatibility contributes to the early stages of speciation. Evolution 2016, 70, 2752–2766. [Google Scholar] [CrossRef] [PubMed]
- Mayr, E. Systematics and the Origin of Species; Columbia University Press: New York, NY, USA, 1942. [Google Scholar]
- Rieseberg, L.H.; Blackman, B.K. Speciation genes in plants. Ann. Bot. 2010, 106, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lin, H.-X. Evolution and Molecular Control of Hybrid Incompatibility in Plants. Front. Plant Sci. 2016, 7, 1208. [Google Scholar] [CrossRef]
- Orr, H.A.; Masly, J.P.; Presgraves, D.C. Speciation genes. Curr. Opin. Genet. Dev. 2004, 14, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Bomblies, K. Doomed lovers: Mechanisms of isolation and incompatibility in plants. Annu. Rev. Plant Biol. 2010, 61, 109–124. [Google Scholar] [CrossRef]
- Sweigart, A.L.; Willis, J.H. Molecular evolution and genetics of postzygotic reproductive isolation in plants. F1000 Biol. Rep. 2012, 4, 23. [Google Scholar] [CrossRef]
- Baack, E.; Melo, M.C.; Rieseberg, L.H.; Ortiz-Barrientos, D. The origins of reproductive isolation in plants. New Phytol. 2015, 207, 968–984. [Google Scholar] [CrossRef]
- Moison, M.; Roux, F.; Quadrado, M.; Duval, R.; Ekovich, M.; Lê, D.-H.; Verzaux, M.; Budar, F. Cytoplasmic phylogeny and evidence of cyto-nuclear co-adaptation in Arabidopsis thaliana. Plant J. 2010, 63, 728–738. [Google Scholar] [CrossRef]
- Levin, D.A. The cytoplasmic factor in plant speciation. Syst. Bot. 2003, 28, 5–11. [Google Scholar]
- Burton, R.S.; Pereira, R.J.; Barreto, F.S. Cytonuclear Genomic Interactions and Hybrid Breakdown. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 281–302. [Google Scholar] [CrossRef]
- Moyle, L.C.; Nakazato, T. Complex Epistasis for Dobzhansky–Muller Hybrid Incompatibility in Solanum. Genetics 2009, 181, 347–351. [Google Scholar] [CrossRef][Green Version]
- Ouyang, Y.; Liu, Y.-G.; Zhang, Q. Hybrid sterility in plant: Stories from rice. Curr. Opin. Plant Biol. 2010, 13, 186–192. [Google Scholar] [CrossRef]
- Barr, C.M.; Fishman, L. The Nuclear Component of a Cytonuclear Hybrid Incompatibility in Mimulus Maps to a Cluster of Pentatricopeptide Repeat Genes. Genetics 2010, 184, 455–465. [Google Scholar] [CrossRef]
- Greiner, S.; Rauwolf, U.; Meurer, J.; Herrmann, R.G. The role of plastids in plant speciation. Mol. Ecol. 2011, 20, 671–691. [Google Scholar] [CrossRef]
- Leppälä, J.; Savolainen, O. Nuclear-Cytoplasmic Interactions Reduce Male Fertility in Hybrids of Arabidopsis Lyrata Subspecies. Evolution 2011, 65, 2959–2972. [Google Scholar] [CrossRef]
- Törjék, O.; Witucka-Wall, H.; Meyer, R.C.; von Korff, M.; Kusterer, B.; Rautengarten, C.; Altmann, T. Segregation distortion in Arabidopsis C24/Col-0 and Col-0/C24 recombinant inbred line populations is due to reduced fertility caused by epistatic interaction of two loci. Theor. Appl. Genet. 2006, 113, 1551–1561. [Google Scholar] [CrossRef]
- Durand, S.; Bouché, N.; Perez Strand, E.; Loudet, O.; Camilleri, C. Rapid Establishment of Genetic Incompatibility through Natural Epigenetic Variation. Curr. Biol. 2012, 22, 326–331. [Google Scholar] [CrossRef]
- Birky, C.W. The Inheritance of Genes in Mitochondria and Chloroplasts: Laws, Mechanisms, and Models. Annu. Rev. Genet. 2001, 35, 125–148. [Google Scholar] [CrossRef]
- Fishman, L.; Willis, J.H. A cytonuclear incompatibility causes anther sterility in Mimulus hybrids. Evolution 2006, 60, 1372–1381. [Google Scholar] [CrossRef]
- Hanson, M.R.; Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 2004, 16 (Suppl. 1), S154–S169. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Y.-G. Male Sterility and Fertility Restoration in Crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef]
- Rhoades, M.M. The cytoplasmic inheritance of male sterility in Zea mays. J. Genet. 1931, 27, 71–93. [Google Scholar] [CrossRef]
- Renner, O. Die pflanzlichen Plastiden als selbstandige Elemente der genetischen Konstitution. Ber. Math. Phys. Kl. Sachs. Akad. 1934, 86, 241–266. [Google Scholar]
- Stebbins, G.L. Variation and Evolution in Plants; Columbia University Press: New York, NY, USA, 1950. [Google Scholar]
- Stubbe, W. The role of the plastome in evolution of the genus Oenothera. Genetica 1964, 35, 28–33. [Google Scholar] [CrossRef]
- Crosby, K.; Smith, D.R. Does the mode of plastid inheritance influence plastid genome architecture? PLoS ONE 2012, 7, e46260. [Google Scholar] [CrossRef]
- Roux, F.; Mary-Huard, T.; Barillot, E.; Wenes, E.; Botran, L.; Durand, S.; Villoutreix, R.; Martin-Magniette, M.-L.; Camilleri, C.; Budar, F. Cytonuclear interactions affect adaptive traits of the annual plant Arabidopsis thaliana in the field. Proc. Natl. Acad. Sci. USA 2016, 113, 3687–3692. [Google Scholar] [CrossRef]
- Dempewolf, H.; Hodgins, K.A.; Rummell, S.E.; Ellstrand, N.C.; Rieseberg, L.H. Reproductive isolation during domestication. Plant Cell 2012, 24, 2710–2717. [Google Scholar] [CrossRef]
- Bogdanova, V.S.; Zaytseva, O.O.; Mglinets, A.V.; Shatskaya, N.V.; Kosterin, O.E.; Vasiliev, G.V. Nuclear-cytoplasmic conflict in pea (Pisum sativum L.) is associated with nuclear and plastidic candidate genes encoding acetyl-CoA carboxylase subunits. PLoS ONE 2015, 10, e0119835. [Google Scholar] [CrossRef]
- Sasaki, Y.; Nagano, Y. Plant acetyl-CoA carboxylase: Structure, biosynthesis, regulation, and gene manipulation for plant breeding. Biosci. Biotechnol. Biochem. 2004, 68, 1175–1184. [Google Scholar] [CrossRef]
- Nikolau, B.J.; Ohlrogge, J.B.; Wurtele, E.S. Plant biotin-containing carboxylases. Arch. Biochem. Biophys. 2003, 414, 211–222. [Google Scholar] [CrossRef]
- Szczepaniak, A.; Książkiewicz, M.; Podkowiński, J.; Czyż, K.B.; Figlerowicz, M.; Naganowska, B. Legume Cytosolic and Plastid Acetyl-Coenzyme—A Carboxylase Genes Differ by Evolutionary Patterns and Selection Pressure Schemes Acting before and after Whole-Genome Duplications. Genes 2018, 9, 563. [Google Scholar] [CrossRef]
- Asaf, S.; Khan, A.L.; Aaqil Khan, M.; Muhammad Imran, Q.; Kang, S.-M.; Al-Hosni, K.; Jeong, E.J.; Lee, K.E.; Lee, I.-J. Comparative analysis of complete plastid genomes from wild soybean (Glycine soja) and nine other Glycine species. PLoS ONE 2017, 12, e0182281. [Google Scholar] [CrossRef]
- Gurdon, C.; Maliga, P. Two Distinct Plastid Genome Configurations and Unprecedented Intraspecies Length Variation in the accD Coding Region in Medicago truncatula. DNA Res. 2014, 21, 417–427. [Google Scholar] [CrossRef]
- Magee, A.M.; Aspinall, S.; Rice, D.W.; Cusack, B.P.; Sémon, M.; Perry, A.S.; Stefanović, S.; Milbourne, D.; Barth, S.; Palmer, J.D.; et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010, 20, 1700–1710. [Google Scholar] [CrossRef]
- Ha, Y.-H.; Kim, C.; Choi, K.; Kim, J.-H. Molecular Phylogeny and Dating of Forsythieae (Oleaceae) Provide Insight into the Miocene History of Eurasian Temperate Shrubs. Front. Plant Sci. 2018, 9, 99. [Google Scholar] [CrossRef]
- Rockenbach, K.; Havird, J.C.; Monroe, J.G.; Triant, D.A.; Taylor, D.R.; Sloan, D.B. Positive Selection in Rapidly Evolving Plastid-Nuclear Enzyme Complexes. Genetics 2016, 204, 1507–1522. [Google Scholar] [CrossRef]
- Krüger, J.; Thomas, C.M.; Golstein, C.; Dixon, M.S.; Smoker, M.; Tang, S.; Mulder, L.; Jones, J.D.G. A tomato cysteine protease required for Cf-2-dependent disease resistance and suppression of autonecrosis. Science 2002, 296, 744–747. [Google Scholar] [CrossRef]
- Rooney, H.C.; Van’t Klooster, J.W.; van der Hoorn, R.A.; Joosten, M.H.; Jones, J.D.; de Wit, P.J. Cladosporium Avr2 inhibits tomato Rcr3 protease required for Cf-2-dependent disease resistance. Science 2005, 308, 1783–1786. [Google Scholar] [CrossRef]
- Bomblies, K.; Lempe, J.; Epple, P.; Warthmann, N.; Lanz, C.; Dangl, J.L.; Weigel, D. Autoimmune response as a mechanism for a Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol. 2007, 5, e236. [Google Scholar] [CrossRef]
- Bomblies, K.; Weigel, D. Hybrid necrosis: Autoimmunity as a potential gene-flow barrier in plant species. Nat. Rev. Genet. 2007, 8, 382–393. [Google Scholar] [CrossRef]
- Smýkal, P.; Hradilová, I.; Trněný, O.; Brus, J.; Rathore, A.; Bariotakis, M.; Das, R.R.; Bhattacharyya, D.; Richards, C.; Coyne, C.J.; et al. Genomic diversity and macroecology of the crop wild relatives of domesticated pea. Sci. Rep. 2017, 7, 17384. [Google Scholar] [CrossRef]
- Trněný, O.; Brus, J.; Hradilová, I.; Rathore, A.; Das, R.R.; Kopecký, P.; Coyne, C.J.; Reeves, P.; Richards, C.; Smýkal, P. Molecular Evidence for Two Domestication Events in the Pea Crop. Genes (Basel) 2018, 9, 535. [Google Scholar] [CrossRef]
- D’Agostino, N.; Tamburino, R.; Cantarella, C.; De Carluccio, V.; Sannino, L.; Cozzolino, S.; Cardi, T.; Scotti, N. The Complete Plastome Sequences of Eleven Capsicum Genotypes: Insights into DNA Variation and Molecular Evolution. Genes 2018, 9, 503. [Google Scholar] [CrossRef]
- Greiner, S.; Wang, X.; Herrmann, R.G.; Rauwolf, U.; Mayer, K.; Haberer, G.; Meurer, J. The complete nucleotide sequences of the 5 genetically distinct plastid genomes of Oenothera, subsection Oenothera: II. A microevolutionary view using bioinformatics and formal genetic data. Mol. Biol. Evol. 2008, 25, 2019–2030. [Google Scholar] [CrossRef]
- Sobanski, J.; Giavalisco, P.; Fischer, A.; Kreiner, J.; Walther, D.; Schoettler, M.A.; Pellizzer, T.; Golczyk, H.; Obata, T.; Bock, R.; et al. Chloroplast competition is controlled by lipid biosynthesis in evening primroses. Proc. Natl. Acad. Sci. USA 2019, 116, 5665–5674. [Google Scholar] [CrossRef]
- Li, J.; Su, Y.; Wang, T. The Repeat Sequences and Elevated Substitution Rates of the Chloroplast accD Gene in Cupressophytes. Front. Plant Sci. 2018, 9, 533. [Google Scholar] [CrossRef]
- Ujihara, T.; Hayashi, N.; Kohata, K.; Matsushita, S.; Kitajima, S. Intraspecific Sequence Variation in the rbcL-accD Region of the Chloroplast Genome in Tea (Camellia sinensis). Tea Res. J. 2007, 104, 15–23. [Google Scholar] [CrossRef][Green Version]
- Smýkal, P.; Trněný, O.; Brus, J.; Hanáček, P.; Rathore, A.; Roma, R.D.; Pechanec, V.; Duchoslav, M.; Bhattacharyya, D.; Bariotakis, M.; et al. Genetic structure of wild pea (Pisum sativum subsp. elatius) populations in the northern part of the Fertile Crescent reflects moderate cross-pollination and strong effect of geographic but not environmental distance. PLoS ONE 2018, 13, e0194056. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Light, S.; Sagit, R.; Sachenkova, O.; Ekman, D.; Elofsson, A. Protein expansion is primarily due to indels in intrinsically disordered regions. Mol. Biol. Evol. 2013, 30, 2645–2653. [Google Scholar] [CrossRef]
- Simon, M.; Hancock, J.M. Tandem and cryptic amino acid repeats accumulate in disordered regions of proteins. Genome Biol. 2009, 10, R59. [Google Scholar] [CrossRef]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef]
- Corriveau, J.L.; Coleman, A.W. Rapid Screening Method to Detect Potential Biparental Inheritance of Plastid DNA and Results for Over 200 Angiosperm Species. Am. J. Bot. 1988, 75, 1443–1458. [Google Scholar] [CrossRef]
- Zhang, Q.; Sodmergen. Why does biparental plastid inheritance revive in angiosperms? J. Plant Res. 2010, 123, 201–206. [Google Scholar] [CrossRef]
- Greiner, S.; Sobanski, J.; Bock, R. Why are most organelle genomes transmitted maternally? Bioessays 2015, 37, 80–94. [Google Scholar] [CrossRef]
- Christie, J.R.; Beekman, M. Uniparental Inheritance Promotes Adaptive Evolution in Cytoplasmic Genomes. Mol. Biol. Evol. 2017, 34, 677–691. [Google Scholar] [CrossRef]
- Bogdanova, V.S.; Kosterin, O.E.; Yadrikhinskiy, A.K. Wild peas vary in their cross-compatibility with cultivated pea (Pisum sativum subsp. sativum L.) depending on alleles of a nuclear-cytoplasmic incompatibility locus. Theor. Appl. Genet. 2014, 127, 1163–1172. [Google Scholar] [CrossRef]
- North, N.; Casey, R.; Domoney, C. Inheritance and mapping of seed lipoxygenase polypeptides in Pisum. Theor. Appl. Genet. 1998, 77, 805–808. [Google Scholar] [CrossRef]
- Hradilová, I.; Trněný, O.; Válková, M.; Cechová, M.; Janská, A.; Prokešová, L.; Aamir, K.; Krezdorn, N.; Rotter, B.; Winter, P.; et al. A Combined Comparative Transcriptomic, Metabolomic, and Anatomical Analyses of Two Key Domestication Traits: Pod Dehiscence and Seed Dormancy in Pea (Pisum sp.). Front. Plant Sci. 2017, 8, 542. [Google Scholar] [CrossRef]
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef]
- Ben-Ze’Ev, N.; Zohary, D. Species relationships in the genus Pisum L. Isr. J. Bot. 1973, 22, 73–91. [Google Scholar]
- Errico, A.; Conicella, C.; Taliercio, U. Cytological and Morphological Characterization of Pisum sativum and Pisum fulvum Tetraploids. Plant Breed. 1991, 106, 141–148. [Google Scholar] [CrossRef]
- Lu, Y.; Kermicle, J.L.; Evans, M.M.S. Genetic and cellular analysis of cross-incompatibility in Zea mays. Plant Reprod. 2014, 27, 19–29. [Google Scholar] [CrossRef]
- Saitoh, K.; Onishi, K.; Mikami, I.; Thidar, K.; Sano, Y. Allelic diversification at the C (OsC1) locus of wild and cultivated rice: Nucleotide changes associated with phenotypes. Genetics 2004, 168, 997–1007. [Google Scholar] [CrossRef]
- Kubo, T. Genetic mechanisms of postzygotic reproductive isolation: An epistatic network in rice. Breed. Sci. 2013, 63, 359–366. [Google Scholar] [CrossRef]
- Kumari, M.; Clarke, H.J.; Small, I.; Siddique, K.H.M. Albinism in Plants: A Major Bottleneck in Wide Hybridization, Androgenesis and Doubled Haploid Culture. Crit. Rev. Plant Sci. 2009, 28, 393–409. [Google Scholar] [CrossRef]
- Kumari, M.; Clarke, H.J.; des Francs-Small, C.C.; Small, I.; Khan, T.N.; Siddique, K.H.M. Albinism does not correlate with biparental inheritance of plastid DNA in interspecific hybrids in Cicer species. Plant Sci. 2011, 180, 628–633. [Google Scholar] [CrossRef]
- Bohra, A.; Jha, U.C.; Adhimoolam, P. Cytoplasmic male sterility (CMS) in hybrid breeding in field crops. Plant Cell Rep. 2016, 35, 967–993. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Zhang, D. Molecular Control of Male Fertility for Crop Hybrid Breeding. Trends Plant Sci. 2018, 23, 53–65. [Google Scholar] [CrossRef]
- Villesen, P. FaBox: An online toolbox for fasta sequences. Mol. Ecol. Notes 2007, 7, 965–968. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Kalendar, R.; Khassenov, B.; Ramanculov, E.; Samuilova, O.; Ivanov, K.I. FastPCR: An in silico tool for fast primer and probe design and advanced sequence analysis. Genomics 2017, 109, 312–319. [Google Scholar] [CrossRef]
- Heger, A.; Holm, L. Rapid automatic detection and alignment of repeats in protein sequences. Proteins 2000, 41, 224–237. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012, 13, 173. [Google Scholar] [CrossRef]
- Hooft, R.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in protein structures. Nature 1996, 381, 272. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Substitutions/Alignment Length | Indels/Alignment Length | ||
|---|---|---|---|---|
| Modelled | Not Modelled | Modelled | Not Modelled | |
| accD | 36/299 | 50/256* | 2/299 | 17/256** |
| bccp | 17/134 | 19/138 | 0/134 | 1/138 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nováková, E.; Zablatzká, L.; Brus, J.; Nesrstová, V.; Hanáček, P.; Kalendar, R.; Cvrčková, F.; Majeský, Ľ.; Smýkal, P. Allelic Diversity of Acetyl Coenzyme A Carboxylase accD/bccp Genes Implicated in Nuclear-Cytoplasmic Conflict in the Wild and Domesticated Pea (Pisum sp.). Int. J. Mol. Sci. 2019, 20, 1773. https://doi.org/10.3390/ijms20071773
Nováková E, Zablatzká L, Brus J, Nesrstová V, Hanáček P, Kalendar R, Cvrčková F, Majeský Ľ, Smýkal P. Allelic Diversity of Acetyl Coenzyme A Carboxylase accD/bccp Genes Implicated in Nuclear-Cytoplasmic Conflict in the Wild and Domesticated Pea (Pisum sp.). International Journal of Molecular Sciences. 2019; 20(7):1773. https://doi.org/10.3390/ijms20071773
Chicago/Turabian StyleNováková, Eliška, Lenka Zablatzká, Jan Brus, Viktorie Nesrstová, Pavel Hanáček, Ruslan Kalendar, Fatima Cvrčková, Ľuboš Majeský, and Petr Smýkal. 2019. "Allelic Diversity of Acetyl Coenzyme A Carboxylase accD/bccp Genes Implicated in Nuclear-Cytoplasmic Conflict in the Wild and Domesticated Pea (Pisum sp.)" International Journal of Molecular Sciences 20, no. 7: 1773. https://doi.org/10.3390/ijms20071773
APA StyleNováková, E., Zablatzká, L., Brus, J., Nesrstová, V., Hanáček, P., Kalendar, R., Cvrčková, F., Majeský, Ľ., & Smýkal, P. (2019). Allelic Diversity of Acetyl Coenzyme A Carboxylase accD/bccp Genes Implicated in Nuclear-Cytoplasmic Conflict in the Wild and Domesticated Pea (Pisum sp.). International Journal of Molecular Sciences, 20(7), 1773. https://doi.org/10.3390/ijms20071773