Glial Cell Expression of PD-L1


Abstract
{kind=link}
{kind=link}
1. Introduction
2. Expression of PD-L1 within the Brain
3. PD-L1 and Pro-Inflammatory Cytokines
4. Role of PD-L1 in the Development of Brain-Resident Memory Cells (bTRM)
5. PD-L1: Mixed Blessings in CNS Disease
6. PD-L1, Glioma, and Checkpoint Blockade
7. Implications for PD-L1 Blockade Therapy in Clearing Viral Brain Reservoirs
8. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PD | Programmed Death |
| CNS | Central nervous system |
| TRM | Tissue resident memory cells |
| CTLA | Cytotoxic T-lymphocyte- associated protein |
| NO | Nitric oxide |
| iNOS | Inducible NO synthase |
| EAE | Experimental autoimmune encephalitis |
| MCMV | Murine cytomegalovirus |
| TMEV-IDD | Theiler’s murine encephalomyelitis virus- induced demyelinating disease |
| MS | Multiple sclerosis |
| HSK | Herpetic stromal keratitis |
| HSV | Herpes simplex virus |
| NSCLC | Non-small cell lung cancer |
| RCC | Renal cell carcinoma |
| irAEs | Immune-related adverse events |
| HAND | HIV-associated neurocognitive disorders |
| IFN | Interferon |
| MHC | Major histocompatibility complex |
| Ag | Antigen |
| TCR | T-cell receptor |
References
- Solomos, A.C.; Rall, G.F. Get It through Your Thick Head: Emerging Principles in Neuroimmunology and Neurovirology Redefine Central Nervous System “Immune Privilege”. ACS Chem. Neurosci. 2016, 7, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Mutnal, M.B.; Hu, S.; Little, M.R.; Lokensgard, J.R. Memory T cells persisting in the brain following MCMV infection induce long-term microglial activation via interferon-gamma. J. Neurovirol. 2011, 17, 424–437. [Google Scholar] [CrossRef]
- Mutnal, M.B.; Hu, S.; Lokensgard, J.R. Persistent humoral immune responses in the CNS limit recovery of reactivated murine cytomegalovirus. PLoS ONE 2012, 7, e33143. [Google Scholar] [CrossRef]
- Lokensgard, J.R.; Mutnal, M.B.; Prasad, S.; Sheng, W.; Hu, S. Glial cell activation, recruitment, and survival of B-lineage cells following MCMV brain infection. J. Neuroinflamm. 2016, 13, 114. [Google Scholar] [CrossRef]
- Cheeran, M.C.; Gekker, G.; Hu, S.; Palmquist, J.M.; Lokensgard, J.R. T cell-mediated restriction of intracerebral murine cytomegalovirus infection displays dependence upon perforin but not interferon-gamma. J. Neurovirol. 2005, 11, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.E.; Liang, S.C.; Wu, Y.; Chernova, T.; Sobel, R.A.; Klemm, M.; Kuchroo, V.K.; Freeman, G.J.; Sharpe, A.H. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10691–10696. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.Y.; Lee, Y.J.; Seo, S.K.; Lee, S.W.; Park, S.J.; Lee, J.N.; Sohn, H.S.; Yao, S.; Chen, L.; Choi, I. Blocking of monocyte-associated B7-H1 (CD274) enhances HCV-specific T cell immunity in chronic hepatitis C infection. J. Leukoc. Biol. 2008, 83, 755–764. [Google Scholar] [CrossRef]
- Magnus, T.; Schreiner, B.; Korn, T.; Jack, C.; Guo, H.; Antel, J.; Ifergan, I.; Chen, L.; Bischof, F.; Bar-Or, A.; et al. Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: Implications for immune responses and autoimmunity in the CNS. J. Neurosci. 2005, 25, 2537–2546. [Google Scholar] [CrossRef]
- Duncan, D.S.; Miller, S.D. CNS expression of B7-H1 regulates pro-inflammatory cytokine production and alters severity of Theiler’s virus-induced demyelinating disease. PLoS ONE 2011, 6, e18548. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, B.; Bailey, S.L.; Shin, T.; Chen, L.; Miller, S.D. PD-1 ligands expressed on myeloid-derived APC in the CNS regulate T-cell responses in EAE. Eur. J. Immunol. 2008, 38, 2706–2717. [Google Scholar] [CrossRef]
- Phares, T.W.; Ramakrishna, C.; Parra, G.I.; Epstein, A.; Chen, L.; Atkinson, R.; Stohlman, S.A.; Bergmann, C.C. Target-dependent B7-H1 regulation contributes to clearance of central nervous system infection and dampens morbidity. J. Immunol. 2009, 182, 5430–5438. [Google Scholar] [CrossRef] [PubMed]
- Lafon, M.; Megret, F.; Meuth, S.G.; Simon, O.; Velandia Romero, M.L.; Lafage, M.; Chen, L.; Alexopoulou, L.; Flavell, R.A.; Prehaud, C.; et al. Detrimental contribution of the immuno-inhibitor B7-H1 to rabies virus encephalitis. J. Immunol. 2008, 180, 7506–7515. [Google Scholar] [CrossRef] [PubMed]
- Lipp, M.; Brandt, C.; Dehghani, F.; Kwidzinski, E.; Bechmann, I. PD-L1 (B7-H1) regulation in zones of axonal degeneration. Neurosci. Lett. 2007, 425, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef] [PubMed]
- Usui, Y.; Okunuki, Y.; Hattori, T.; Kezuka, T.; Keino, H.; Ebihara, N.; Sugita, S.; Usui, M.; Goto, H.; Takeuchi, M. Functional expression of B7H1 on retinal pigment epithelial cells. Exp. Eye Res. 2008, 86, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Rodig, N.; Ryan, T.; Allen, J.A.; Pang, H.; Grabie, N.; Chernova, T.; Greenfield, E.A.; Liang, S.C.; Sharpe, A.H.; Lichtman, A.H.; et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur. J. Immunol. 2003, 33, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Eppihimer, M.J.; Gunn, J.; Freeman, G.J.; Greenfield, E.A.; Chernova, T.; Erickson, J.; Leonard, J.P. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation 2002, 9, 133–145. [Google Scholar] [CrossRef]
- Wiendl, H.; Mitsdoerffer, M.; Schneider, D.; Chen, L.; Lochmuller, H.; Melms, A.; Weller, M. Human muscle cells express a B7-related molecule, B7-H1, with strong negative immune regulatory potential: A novel mechanism of counterbalancing the immune attack in idiopathic inflammatory myopathies. FASEB J. 2003, 17, 1892–1894. [Google Scholar] [CrossRef]
- Wintterle, S.; Schreiner, B.; Mitsdoerffer, M.; Schneider, D.; Chen, L.; Meyermann, R.; Weller, M.; Wiendl, H. Expression of the B7-related molecule B7-H1 by glioma cells: A potential mechanism of immune paralysis. Cancer Res. 2003, 63, 7462–7467. [Google Scholar]
- Hori, J.; Wang, M.; Miyashita, M.; Tanemoto, K.; Takahashi, H.; Takemori, T.; Okumura, K.; Yagita, H.; Azuma, M. B7-H1-induced apoptosis as a mechanism of immune privilege of corneal allografts. J. Immunol. 2006, 177, 5928–5935. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Holscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Liu, B.; Hong, J.S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Hu, S.; Sheng, W.S.; Prasad, S.; Lokensgard, J.R. Modulation of Microglial Cell Fcgamma Receptor Expression Following Viral Brain Infection. Sci. Rep. 2017, 7, 41889. [Google Scholar] [CrossRef]
- Bailey, S.L.; Carpentier, P.A.; McMahon, E.J.; Begolka, W.S.; Miller, S.D. Innate and adaptive immune responses of the central nervous system. Crit. Rev. Immunol. 2006, 26, 149–188. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef]
- Schachtele, S.J.; Hu, S.; Sheng, W.S.; Mutnal, M.B.; Lokensgard, J.R. Glial cells suppress postencephalitic CD8+ T lymphocytes through PD-L1. Glia 2014, 62, 1582–1594. [Google Scholar] [CrossRef] [PubMed]
- Lokensgard, J.R.; Schachtele, S.J.; Mutnal, M.B.; Sheng, W.S.; Prasad, S.; Hu, S. Chronic reactive gliosis following regulatory T cell depletion during acute MCMV encephalitis. Glia 2015, 63, 1982–1996. [Google Scholar] [CrossRef] [PubMed]
- Pittet, C.L.; Newcombe, J.; Antel, J.P.; Arbour, N. The majority of infiltrating CD8 T lymphocytes in multiple sclerosis lesions is insensitive to enhanced PD-L1 levels on CNS cells. Glia 2011, 59, 841–856. [Google Scholar] [CrossRef]
- Vardjan, N.; Gabrijel, M.; Potokar, M.; Svajger, U.; Kreft, M.; Jeras, M.; de Pablo, Y.; Faiz, M.; Pekny, M.; Zorec, R. IFN-gamma-induced increase in the mobility of MHC class II compartments in astrocytes depends on intermediate filaments. J. Neuroinflamm. 2012, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Armien, A.G.; Hu, S.; Little, M.R.; Robinson, N.; Lokensgard, J.R.; Low, W.C.; Cheeran, M.C. Chronic cortical and subcortical pathology with associated neurological deficits ensuing experimental herpes encephalitis. Brain Pathol. 2010, 20, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.P.; Cheeran, M.C.; Palmquist, J.M.; Hu, S.; Urban, S.L.; Lokensgard, J.R. Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J. Immunol. 2008, 181, 6417–6426. [Google Scholar] [CrossRef]
- El Annan, J.; Goyal, S.; Zhang, Q.; Freeman, G.J.; Sharpe, A.H.; Dana, R. Regulation of T-cell chemotaxis by programmed death-ligand 1 (PD-L1) in dry eye-associated corneal inflammation. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3418–3423. [Google Scholar] [CrossRef] [PubMed]
- Blais, V.; Rivest, S. Effects of TNF-alpha and IFN-gamma on nitric oxide-induced neurotoxicity in the mouse brain. J. Immunol. 2004, 172, 7043–7052. [Google Scholar] [CrossRef]
- Prasad, S.; Lokensgard, J.R. Brain-Resident T Cells Following Viral Infection. Viral. Immunol. 2019, 32, 48–54. [Google Scholar] [CrossRef]
- Shwetank; Abdelsamed, H.A.; Frost, E.L.; Schmitz, H.M.; Mockus, T.E.; Youngblood, B.A.; Lukacher, A.E. Maintenance of PD-1 on brain-resident memory CD8 T cells is antigen independent. Immunol. Cell Biol. 2017, 95, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Smolders, J.; Heutinck, K.M.; Fransen, N.L.; Remmerswaal, E.B.M.; Hombrink, P.; Ten Berge, I.J.M.; van Lier, R.A.W.; Huitinga, I.; Hamann, J. Tissue-resident memory T cells populate the human brain. Nat. Commun. 2018, 9, 4593. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef]
- Prasad, S.; Hu, S.; Sheng, W.S.; Chauhan, P.; Singh, A.; Lokensgard, J.R. The PD-1: PD-L1 pathway promotes development of brain-resident memory T cells following acute viral encephalitis. J. Neuroinflamm. 2017, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Hu, S.; Sheng, W.S.; Chauhan, P.; Lokensgard, J.R. Reactive glia promote development of CD103(+) CD69(+) CD8(+) T-cells through programmed cell death-ligand 1 (PD-L1). Immun. Inflamm. Dis. 2018, 6, 332–344. [Google Scholar] [CrossRef]
- Pavelko, K.D.; Bell, M.P.; Harrington, S.M.; Dong, H. B7-H1 Influences the Accumulation of Virus-Specific Tissue Resident Memory T Cells in the Central Nervous System. Front. Immunol. 2017, 8, 1532. [Google Scholar] [CrossRef]
- Park, S.L.; Mackay, L.K. PD-1: Always on my mind. Immunol. Cell Biol. 2017, 95, 857–858. [Google Scholar] [CrossRef] [PubMed]
- Marban, C.; Forouzanfar, F.; Ait-Ammar, A.; Fahmi, F.; El Mekdad, H.; Daouad, F.; Rohr, O.; Schwartz, C. Targeting the Brain Reservoirs: Toward an HIV Cure. Front. Immunol. 2016, 7, 397. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Sheng, W.S.; Hu, S.; Prasad, S.; Lokensgard, J.R. Nitrosative damage during retrovirus infection-induced neuropathic pain. J. Neuroinflamm. 2018, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Tripodis, Y.; Alvarez, V.E.; Huber, B.; Kiernan, P.T.; Daneshvar, D.H.; Mez, J.; Montenigro, P.H.; Solomon, T.M.; Alosco, M.L.; et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol. Commun. 2016, 4, 112. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; He, H.; Yang, Z.; Zhu, G.; Kang, L.; Jing, X.; Lu, H.; Song, W.; Bai, B.; Tang, H. Programmed Death Ligand-1 on Microglia Regulates Th1 Differentiation via Nitric Oxide in Experimental Autoimmune Encephalomyelitis. Neurosci. Bull. 2016, 32, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Tsirka, S.E. Animal Models of MS Reveal Multiple Roles of Microglia in Disease Pathogenesis. Neurol. Res. Int. 2011, 2011, 383087. [Google Scholar] [CrossRef] [PubMed]
- Phares, T.W.; Stohlman, S.A.; Hinton, D.R.; Bergmann, C.C. Enhanced CD8 T-cell anti-viral function and clinical disease in B7-H1-deficient mice requires CD4 T cells during encephalomyelitis. J. Neuroinflamm. 2012, 9, 269. [Google Scholar] [CrossRef]
- Carter, L.L.; Leach, M.W.; Azoitei, M.L.; Cui, J.; Pelker, J.W.; Jussif, J.; Benoit, S.; Ireland, G.; Luxenberg, D.; Askew, G.R.; et al. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2007, 182, 124–134. [Google Scholar] [CrossRef]
- Ortler, S.; Leder, C.; Mittelbronn, M.; Zozulya, A.L.; Knolle, P.A.; Chen, L.; Kroner, A.; Wiendl, H. B7-H1 restricts neuroantigen-specific T cell responses and confines inflammatory CNS damage: Implications for the lesion pathogenesis of multiple sclerosis. Eur. J. Immunol. 2008, 38, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Lund, H.; Mia, S.; Parsa, R.; Harris, R.A. Adoptive transfer of cytokine-induced immunomodulatory adult microglia attenuates experimental autoimmune encephalomyelitis in DBA/1 mice. Glia 2014, 62, 804–817. [Google Scholar] [CrossRef]
- Phares, T.W.; Stohlman, S.A.; Hinton, D.R.; Atkinson, R.; Bergmann, C.C. Enhanced antiviral T cell function in the absence of B7-H1 is insufficient to prevent persistence but exacerbates axonal bystander damage during viral encephalomyelitis. J. Immunol. 2010, 185, 5607–5618. [Google Scholar] [CrossRef] [PubMed]
- Gibbons Johnson, R.M.; Dong, H. Functional Expression of Programmed Death-Ligand 1 (B7-H1) by Immune Cells and Tumor Cells. Front. Immunol. 2017, 8, 961. [Google Scholar] [CrossRef]
- Miyasato, Y.; Takashima, Y.; Takeya, H.; Yano, H.; Hayano, A.; Nakagawa, T.; Makino, K.; Takeya, M.; Yamanaka, R.; Komohara, Y. The expression of PD-1 ligands and IDO1 by macrophage/microglia in primary central nervous system lymphoma. J. Clin. Exp. Hematop. 2018, 58, 95–101. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e5. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514. [Google Scholar] [CrossRef]
- Pardoll, D. Cancer and the Immune System: Basic Concepts and Targets for Intervention. Semin. Oncol. 2015, 42, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kroemer, G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology 2012, 1, 1223–1225. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef]
- Cuzzubbo, S.; Javeri, F.; Tissier, M.; Roumi, A.; Barlog, C.; Doridam, J.; Lebbe, C.; Belin, C.; Ursu, R.; Carpentier, A.F. Neurological adverse events associated with immune checkpoint inhibitors: Review of the literature. Eur. J. Cancer 2017, 73, 1–8. [Google Scholar] [CrossRef]
- Anderson, E.S.; Postow, M.A.; Wolchok, J.D.; Young, R.J.; Ballangrud, A.; Chan, T.A.; Yamada, Y.; Beal, K. Melanoma brain metastases treated with stereotactic radiosurgery and concurrent pembrolizumab display marked regression; efficacy and safety of combined treatment. J. Immunother. Cancer 2017, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Grauer, O.M.; Reichelt, D.; Gruneberg, U.; Lohmann, H.; Schneider-Hohendorf, T.; Schulte-Mecklenbeck, A.; Gross, C.C.; Meuth, S.G.; Wiendl, H.; Husstedt, I.W. Neurocognitive decline in HIV patients is associated with ongoing T-cell activation in the cerebrospinal fluid. Ann. Clin. Transl. Neurol. 2015, 2, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Wilson, M.; Pruckner, C.; Kirkwood, J.M. PD-1 Blockade in Advanced Melanoma in Patients with Hepatitis C and/or HIV. Case Rep. Oncol. Med. 2015, 2015, 737389. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: Meta-analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A. Immune Regulation of Antibody Access to Neuronal Tissues. Trends Mol. Med. 2017, 23, 227–245. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Zinn, R.; Vissel, B. Microglia: A new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol. Learn. Mem. 2013, 105, 40–53. [Google Scholar] [CrossRef]
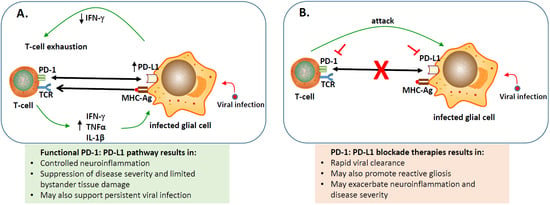
 represents blocking either PD-1 or PD-L1 using inhibitors;
represents blocking either PD-1 or PD-L1 using inhibitors;  indicates blocking the PD-1: PD-L1 pathway;
indicates blocking the PD-1: PD-L1 pathway;  engagement of PD-1 to PD-L1;
engagement of PD-1 to PD-L1;  binding of major histocompatibility complex-antigen (MHC-Ag) to T-cell receptor (TCR).
represents blocking either PD-1 or PD-L1 using inhibitors; indicates blocking the PD-1: PD-L1 pathway; engagement of PD-1 to PD-L1; binding of major histocompatibility complex-antigen (MHC-Ag) to T-cell receptor (TCR).
binding of major histocompatibility complex-antigen (MHC-Ag) to T-cell receptor (TCR).
represents blocking either PD-1 or PD-L1 using inhibitors; indicates blocking the PD-1: PD-L1 pathway; engagement of PD-1 to PD-L1; binding of major histocompatibility complex-antigen (MHC-Ag) to T-cell receptor (TCR).
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauhan, P.; Lokensgard, J.R. Glial Cell Expression of PD-L1. Int. J. Mol. Sci. 2019, 20, 1677. https://doi.org/10.3390/ijms20071677
Chauhan P, Lokensgard JR. Glial Cell Expression of PD-L1. International Journal of Molecular Sciences. 2019; 20(7):1677. https://doi.org/10.3390/ijms20071677
Chicago/Turabian StyleChauhan, Priyanka, and James R. Lokensgard. 2019. "Glial Cell Expression of PD-L1" International Journal of Molecular Sciences 20, no. 7: 1677. https://doi.org/10.3390/ijms20071677
APA StyleChauhan, P., & Lokensgard, J. R. (2019). Glial Cell Expression of PD-L1. International Journal of Molecular Sciences, 20(7), 1677. https://doi.org/10.3390/ijms20071677