Future Aspects for Cannabinoids in Breast Cancer Therapy

 ,
, 

Abstract
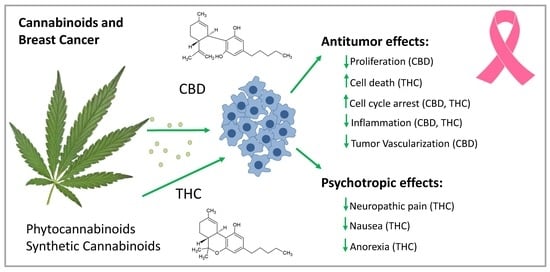
1. Introduction: Cannabis sativa and Cannabinoids
2. Mechanism of Cannabinoid Action
3. Cannabinoids from Cannabis sativa
3.1. Cannabidiol (CBD)
3.2. Delta-9-tetrahydrocannabinol (THC)
3.3. Minor Phytocannabinoids
Other CBs from C. sativa
3.4. Drugs Based on CBs from C. sativa
3.5. Synthetic Cannabinoid Analogues
4. Cannabinoids in Breast Cancer
4.1. Molecular Effects of CBs in Breast Cancer
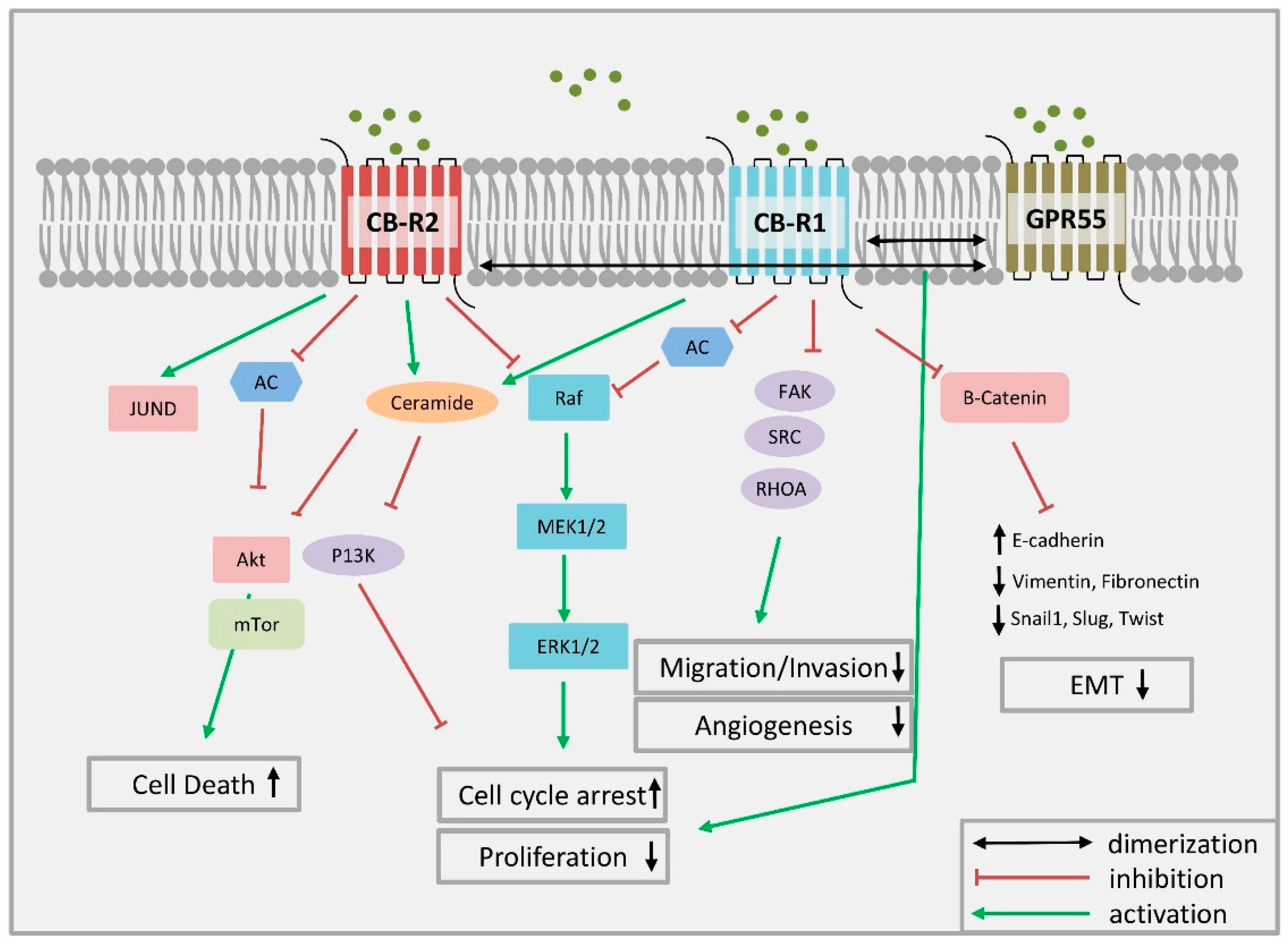
4.2. Cannabinoid Receptor Signaling
4.3. The Effect of Cannabinoids in Breast Cancer Cell Lines
4.3.1. Phytocannabinoids and Synthetic Analogues
4.3.2. Endocannabinoids
4.4. Preclinical Evidence of the Effects of CBs in Animal Models
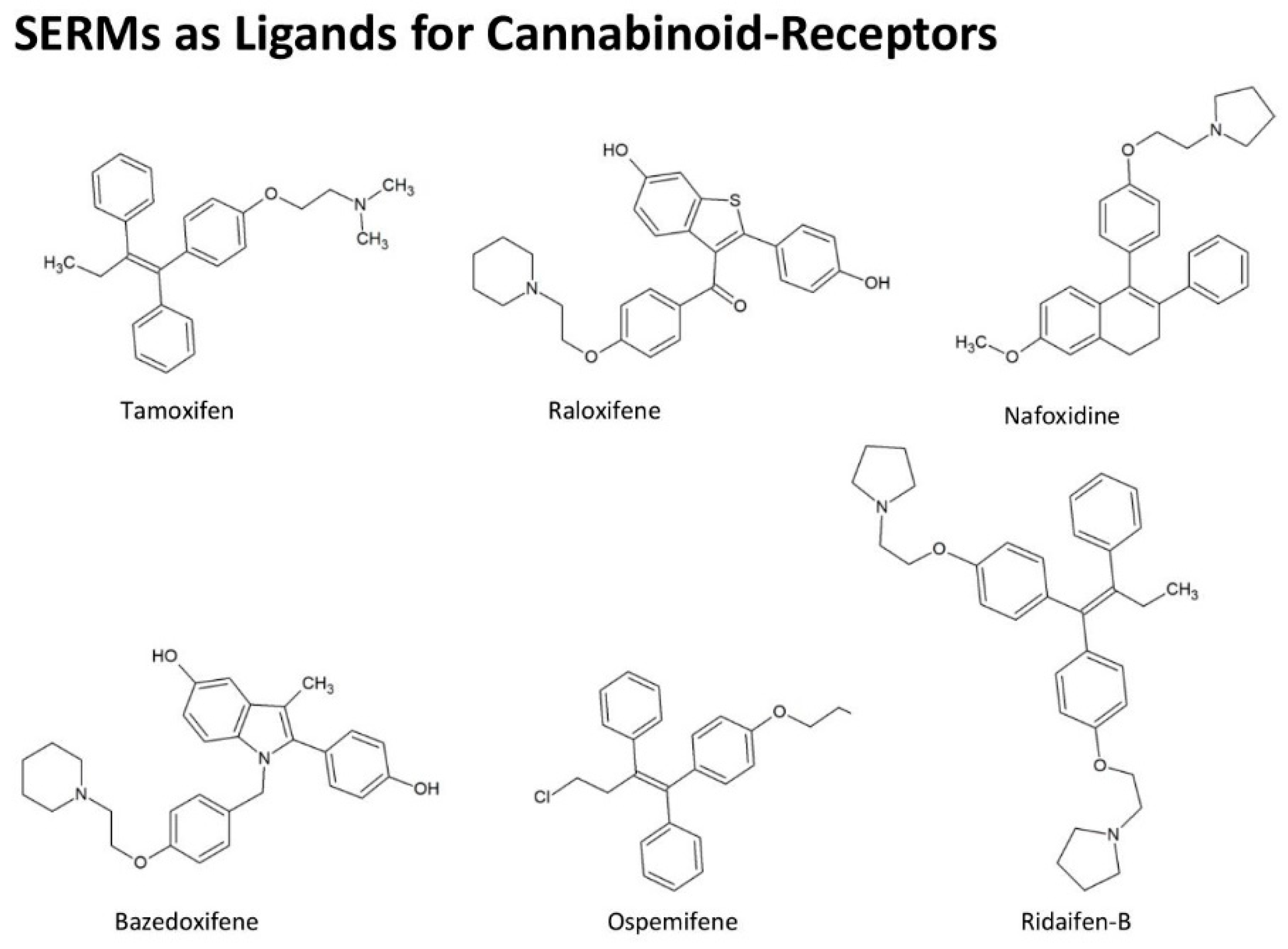
4.5. Effect of Cannabinoids Related to Estrogen
5. Current Therapeutic Application of Cannabinoids in Cancer Patients
6. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2-AG | 2-Arachidonoylglycerol |
| AA | Arachidonic acid |
| AEA | N-arachindonoylethanolamine (Anandamide) |
| AIDS | Acquired Immune Deficiency Syndrome |
| Akt | Protein kinase B |
| cAMP | cyclic adenosine monophosphate |
| CB | Cannabinoid |
| CBN | Cannabinol |
| CB1-R | Cannabinoid receptor-1 |
| CB2-R | Cannabinoid receptor-2 |
| CBD | Cannibidiol |
| CBDA | Cannabidiolic acid |
| CB-R | Cannabinoid receptor |
| Cdc | Cell division control |
| COX | Cyclooxygenase |
| DAG | Diacylglycerol |
| E2 | 17β-Estradiol |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| ER | Estrogen receptor |
| erb-B2 | Epidermal growth factor receptor |
| ERK | Extracellular-signal-regulated kinase |
| FAAH | Fatty acid amide hydrolase |
| GPR | G-protein coupled receptor |
| GPER | G-protein coupled for estrogen |
| HER2 | human epidermal growth factor receptor 2 |
| ID-1 | inhibitor of DNA binding 1 |
| IL | Interleukin |
| MAGL | Monoacylglycerol lipase |
| MAPK | Mitogen-activated protein kinase |
| MMP | Metalloproteinases |
| mTOR | Mammalian target of rapamycin |
| NAPE | N-acyl-phosphatidylethanolamine |
| NAT | N-acyltransferase |
| NGF | Nerve growth factor |
| PI3K | Phosphoinoside 3-kinase |
| PLC | Phospholipase C |
| PPAR | Peroxisome proliferator-activated receptor |
| Phyto-CBs | Phytocannabinoids |
| ROS | Reactive oxygen species |
| SERM | Selective estrogen receptor modulator |
| SRC | Proto-oncogene tyrosine-protein kinase Src |
| STAT | Signal transducer and activator of transcription |
| THC | Delta-9-Tetrahydrocannabinol |
| THCA | Delta-9-tetrahydrocannabinolic acid |
| TNF | Tumor necrosis factor |
| VEGF | Vascular endothelial growth factor receptor |
References
- Abrams, D.I. Integrating cannabis into clinical cancer care. Curr. Oncol. 2016, 23, S8–S14. [Google Scholar]
- Bonini, S.A.; Premoli, M.; Tambaro, S.; Kumar, A.; Maccarinelli, G.; Memo, M.; Mastinu, A. Cannabis sativa: A comprehensive ethnopharmacological review of a medicinal plant with a long history. J. Ethnopharmacol. 2018, 227, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Hanus, L.O.; Meyer, S.M.; Munoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef]
- Nuutinen, T. Medicinal properties of terpenes found in Cannabis sativa and Humulus lupulus. Eur. J. Med. Chem. 2018, 157, 198–228. [Google Scholar] [PubMed]
- Fasinu, P.S.; Phillips, S.; ElSohly, M.A.; Walker, L.A. Current Status and Prospects for Cannabidiol Preparations as New Therapeutic Agents. Pharmacotherapy 2016, 36, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Jikomes, N.; Zoorob, M. The Cannabinoid Content of Legal Cannabis in Washington State Varies Systematically Across Testing Facilities and Popular Consumer Products. Sci. Rep. 2018, 8, 4519. [Google Scholar] [CrossRef]
- Chakravarti, B.; Ravi, J.; Ganju, R.K. Cannabinoids as therapeutic agents in cancer: Current status and future implications. Oncotarget 2014, 5, 5852–5872. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 23, S23–S32. [Google Scholar]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar]
- Pertwee, R.G. Targeting the endocannabinoid system with cannabinoid receptor agonists: Pharmacological strategies and therapeutic possibilities. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3353–3363. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; De Petrocellis, L.; Bisogno, T. The biosynthesis, fate and pharmacological properties of endocannabinoids. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2005; pp. 147–185. [Google Scholar]
- De Petrocellis, L.; Cascio, M.G.; Di Marzo, V. The endocannabinoid system: A general view and latest additions. Br. J. Pharmacol. 2004, 141, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Solinas, M.; Goldberg, S.R.; Piomelli, D. The endocannabinoid system in brain reward processes. Br. J. Pharmacol. 2008, 154, 369–383. [Google Scholar] [CrossRef]
- Mallet, C.; Dubray, C.; Duale, C. FAAH inhibitors in the limelight, but regrettably. Int. J. Clin. Pharm. 2016, 54, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Salort, G.; Alvaro-Bartolome, M.; Garcia-Sevilla, J.A. Regulation of cannabinoid CB2 receptor constitutive activity in vivo: Repeated treatments with inverse agonists reverse the acute activation of JNK and associated apoptotic signaling in mouse brain. Psychopharmacology 2017, 234, 925–941. [Google Scholar] [CrossRef] [PubMed]
- Preet, A.; Qamri, Z.; Nasser, M.W.; Prasad, A.; Shilo, K.; Zou, X.; Groopman, J.E.; Ganju, R.K. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev. Res. 2011, 4, 65–75. [Google Scholar] [CrossRef]
- Howlett, A.C. Cannabinoid receptor signaling. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2005; pp. 53–79. [Google Scholar]
- Galve-Roperh, I.; Sanchez, C.; Cortes, M.L.; Gomez del Pulgar, T.; Izquierdo, M.; Guzman, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef]
- Dalton, G.D.; Howlett, A.C. Cannabinoid CB1 receptors transactivate multiple receptor tyrosine kinases and regulate serine/threonine kinases to activate ERK in neuronal cells. Br. J. Pharmacol. 2012, 165, 2497–2511. [Google Scholar] [CrossRef]
- Leyva-Illades, D.; Demorrow, S. Orphan G protein receptor GPR55 as an emerging target in cancer therapy and management. Cancer Manag. Res. 2013, 5, 147–155. [Google Scholar]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB(1) and CB(2). Pharm. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef]
- Izzo, A.A.; Borrelli, F.; Capasso, R.; Di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 2009, 30, 515–527. [Google Scholar] [CrossRef]
- Casajuana Koguel, C.; Lopez-Pelayo, H.; Balcells-Olivero, M.M.; Colom, J.; Gual, A. Psychoactive constituents of cannabis and their clinical implications: A systematic review. Adicciones 2018, 30, 140–151. [Google Scholar]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef]
- Huestis, M.A. Human cannabinoid pharmacokinetics. Chem. Biodivers. 2007, 4, 1770–1804. [Google Scholar] [CrossRef] [PubMed]
- McPartland, J.M. Cannabis and Eicosanoids: A Review of Molecular Pharmacology. J. Cannabis Ther. 2001, 1, 71–83. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer 2011, 10, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Hegde, V.L.; Singh, U.P.; Nagarkatti, P.S.; Nagarkatti, M. Critical Role of Mast Cells and Peroxisome Proliferator-Activated Receptor gamma in the Induction of Myeloid-Derived Suppressor Cells by Marijuana Cannabidiol In Vivo. J. Immunol. 2015, 194, 5211–5222. [Google Scholar] [CrossRef]
- Kogan, N.M. Cannabinoids and cancer. Mini Rev. Med. Chem. 2005, 5, 941–952. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lu, Y.; Anderson, H.D. Cannabinoid signaling in health and disease. Can. J. Physiol. Pharmacol. 2017, 95, 311–327. [Google Scholar] [CrossRef]
- Solinas, M.; Massi, P.; Cantelmo, A.R.; Cattaneo, M.G.; Cammarota, R.; Bartolini, D.; Cinquina, V.; Valenti, M.; Vicentini, L.M.; Noonan, D.M.; et al. Cannabidiol inhibits angiogenesis by multiple mechanisms. Br. J. Pharmacol. 2012, 167, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Pellati, F.; Borgonetti, V.; Brighenti, V.; Biagi, M.; Benvenuti, S.; Corsi, L. Cannabis sativa L. and Nonpsychoactive Cannabinoids: Their Chemistry and Role against Oxidative Stress, Inflammation, and Cancer. BioMed Res. Int. 2018, 2018, 1–15. [Google Scholar] [CrossRef]
- Nahtigal, I.; Blake, A.; Hand, A.; Florentinus-Mefailoski, A.; Hashemi, H.; Friedberg, J. The Pharmacological Properties of Cannabis; Nova Science: New York, NY, USA, 2016; Volume 9, pp. 481–491. [Google Scholar]
- Borrelli, F.; Fasolino, I.; Romano, B.; Capasso, R.; Maiello, F.; Coppola, D.; Orlando, P.; Battista, G.; Pagano, E.; Di Marzo, V.; et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem. Pharmacol. 2013, 85, 1306–1316. [Google Scholar] [CrossRef]
- Bab, I.; Zimmer, A.; Melamed, E. Cannabinoids and the skeleton: From marijuana to reversal of bone loss. Ann. Med. 2009, 41, 560–567. [Google Scholar] [CrossRef]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131. [Google Scholar]
- Castaneto, M.S.; Gorelick, D.A.; Desrosiers, N.A.; Hartman, R.L.; Pirard, S.; Huestis, M.A. Synthetic cannabinoids: Epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend. 2014, 144, 12–41. [Google Scholar] [CrossRef]
- Turgeman, I.; Bar-Sela, G. Cannabis for cancer—Illusion or the tip of an eceberg: A review of the evidence for the use of Cannabis and synthetic cannabinoids in oncology. Expert Opin. Investig. Drugs 2019, 28, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Fraguas-Sanchez, A.I.; Fernandez-Carballido, A.; Torres-Suarez, A.I. Phyto-, endo- and synthetic cannabinoids: Promising chemotherapeutic agents in the treatment of breast and prostate carcinomas. Expert Opin. Investig. Drugs 2016, 25, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Pokrywka, M.; Goralska, J.; Solnica, B. Cannabinoids—A new weapon against cancer? Postepy Higieny i Medycyny Doswiadczalnej (Online) 2016, 70, 1309–1320. [Google Scholar] [PubMed]
- Ramer, R.; Hinz, B. Cannabinoids as Anticancer Drugs. Adv. Pharmacol. 2017, 80, 397–436. [Google Scholar] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Prat, A.; Pineda, E.; Adamo, B.; Galvan, P.; Fernandez, A.; Gaba, L.; Diez, M.; Viladot, M.; Arance, A.; Munoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. 2), S26–S35. [Google Scholar] [CrossRef] [PubMed]
- Caffarel, M.M.; Sarrió, D.; Palacios, J.; Guzmán, M.; Sánchez, C. Δ9-Tetrahydrocannabinol Inhibits Cell Cycle Progression in Human Breast Cancer Cells through Cdc2 Regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef] [PubMed]
- McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Delta-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J. Immunol. 2005, 174, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.D.; Christian, R.T.; Horowitz, M.P.; Garcia, A.; Desprez, P.Y. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol. Cancer 2007, 6, 2921–2927. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, A.; Moriello, A.S.; Starowicz, K.; Matias, I.; Pisanti, S.; De Petrocellis, L.; Laezza, C.; Portella, G.; Bifulco, M.; Di Marzo, V. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J. Pharm. Exp. 2006, 318, 1375–1387. [Google Scholar] [CrossRef]
- Sultan, A.S.; Marie, M.A.; Sheweita, S.A. Novel mechanism of cannabidiol-induced apoptosis in breast cancer cell lines. Breast 2018, 41, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S. Medicinal chemistry and pharmacology focused on cannabidiol, a major component of the fiber-type cannabis. Yakugaku Zasshi 2013, 133, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Misawa, K.; Yamamoto, I.; Watanabe, K. Cannabidiolic acid as a selective cyclooxygenase-2 inhibitory component in cannabis. Drug Metab. Dispos. 2008, 36, 1917–1921. [Google Scholar] [CrossRef]
- Takeda, S.; Okajima, S.; Miyoshi, H.; Yoshida, K.; Okamoto, Y.; Okada, T.; Amamoto, T.; Watanabe, K.; Omiecinski, C.J.; Aramaki, H. Cannabidiolic acid, a major cannabinoid in fiber-type cannabis, is an inhibitor of MDA-MB-231 breast cancer cell migration. Toxicol. Lett. 2012, 214, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Okazaki, H.; Ikeda, E.; Abe, S.; Yoshioka, Y.; Watanabe, K.; Aramaki, H. Down-regulation of cyclooxygenase-2 (COX-2) by cannabidiolic acid in human breast cancer cells. J. Toxicol. Sci. 2014, 39, 711–716. [Google Scholar] [CrossRef]
- Grimaldi, C.; Pisanti, S.; Laezza, C.; Malfitano, A.M.; Santoro, A.; Vitale, M.; Caruso, M.G.; Notarnicola, M.; Iacuzzo, I.; Portella, G.; et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp. Cell Res. 2006, 312, 363–373. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Melck, D.; Palmisano, A.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. USA 1998, 95, 8375–8380. [Google Scholar] [CrossRef]
- Melck, D.; Rueda, D.; Galve-Roperh, I.; De Petrocellis, L.; Guzman, M.; Di Marzo, V. Involvement of the cAMP/protein kinase A pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999, 463, 235–240. [Google Scholar] [CrossRef]
- Melck, D.; De Petrocellis, L.; Orlando, P.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. Suppression of nerve growth factor Trk receptors and prolactin receptors by endocannabinoids leads to inhibition of human breast and prostate cancer cell proliferation. Endocrinology 2000, 141, 118–126. [Google Scholar] [CrossRef]
- Sarnataro, D.; Grimaldi, C.; Pisanti, S.; Gazzerro, P.; Laezza, C.; Zurzolo, C.; Bifulco, M. Plasma membrane and lysosomal localization of CB1 cannabinoid receptor are dependent on lipid rafts and regulated by anandamide in human breast cancer cells. FEBS Lett. 2005, 579, 6343–6349. [Google Scholar] [CrossRef] [PubMed]
- Laezza, C.; D’Alessandro, A.; Paladino, S.; Maria Malfitano, A.; Chiara Proto, M.; Gazzerro, P.; Pisanti, S.; Santoro, A.; Ciaglia, E.; Bifulco, M. Anandamide inhibits the Wnt/beta-catenin signalling pathway in human breast cancer MDA MB 231 cells. Eur. J. Cancer 2012, 48, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Picardi, P.; Ciaglia, E.; Proto, M.; Pisanti, S. Anandamide inhibits breast tumor-induced angiogenesis. Transl. Med. UniSa 2014, 10, 8–12. [Google Scholar]
- Qamri, Z.; Preet, A.; Nasser, M.W.; Bass, C.E.; Leone, G.; Barsky, S.H.; Ganju, R.K. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol. Cancer 2009, 8, 3117–3129. [Google Scholar] [CrossRef] [PubMed]
- Caffarel, M.M.; Andradas, C.; Mira, E.; Perez-Gomez, E.; Cerutti, C.; Moreno-Bueno, G.; Flores, J.M.; Garcia-Real, I.; Palacios, J.; Manes, S.; et al. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol. Cancer 2010, 9, 196. [Google Scholar] [CrossRef]
- Perez-Gomez, E.; Andradas, C.; Blasco-Benito, S.; Caffarel, M.M.; Garcia-Taboada, E.; Villa-Morales, M.; Moreno, E.; Hamann, S.; Martin-Villar, E.; Flores, J.M.; et al. Role of cannabinoid receptor CB2 in HER2 pro-oncogenic signaling in breast cancer. J. Natl. Cancer Inst. 2015, 107, djv077. [Google Scholar] [CrossRef]
- Ursini-Siegel, J.; Schade, B.; Cardiff, R.D.; Muller, W.J. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat. Rev. Cancer 2007, 7, 389–397. [Google Scholar] [CrossRef]
- Ford, L.A.; Roelofs, A.J.; Anavi-Goffer, S.; Mowat, L.; Simpson, D.G.; Irving, A.J.; Rogers, M.J.; Rajnicek, A.M.; Ross, R.A. A role for L-alpha-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br. J. Pharmacol. 2010, 160, 762–771. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, S.; Liu, Y.; Su, M.; Ling, X.; Liu, F.; Ge, Y.; Bai, M. Combined CB2 Receptor Agonist and Photodynamic Therapy Synergistically Inhibit Tumor Growth in Triple Negative Breast Cancer. Photodiagn. Photodyn. 2018, 24, 185–191. [Google Scholar] [CrossRef]
- Wu, H.Y.; Huang, C.H.; Lin, Y.H.; Wang, C.C.; Jan, T.R. Cannabidiol induced apoptosis in human monocytes through mitochondrial permeability transition pore-mediated ROS production. Free Radic. Biol. Med. 2018, 124, 311–318. [Google Scholar] [CrossRef]
- Bouquie, R.; Deslandes, G.; Mazare, H.; Cogne, M.; Mahe, J.; Gregoire, M.; Jolliet, P. Cannabis and anticancer drugs: Societal usage and expected pharmacological interactions—A review. Fundam. Clin. Pharm. 2018, 32, 462–484. [Google Scholar] [CrossRef]
- Murase, R.; Kawamura, R.; Singer, E.; Pakdel, A.; Sarma, P.; Judkins, J.; Elwakeel, E.; Dayal, S.; Martinez-Martinez, E.; Amere, M.; et al. Targeting multiple cannabinoid anti-tumour pathways with a resorcinol derivative leads to inhibition of advanced stages of breast cancer. Br. J. Pharmacol. 2014, 171, 4464–4477. [Google Scholar] [CrossRef]
- Scott, K.A.; Dalgleish, A.G.; Liu, W.M. Anticancer effects of phytocannabinoids used with chemotherapy in leukaemia cells can be improved by altering the sequence of their administration. Int. J. Oncol. 2017, 51, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Yasmin-Karim, S.; Moreau, M.; Mueller, R.; Sinha, N.; Dabney, R.; Herman, A.; Ngwa, W. Enhancing the Therapeutic Efficacy of Cancer Treatment With Cannabinoids. Front. Oncol. 2018, 8, 114. [Google Scholar] [CrossRef]
- Radin, D.P.; Patel, P. Delineating the molecular mechanisms of tamoxifen’s oncolytic actions in estrogen receptor-negative cancers. Eur. J. Pharmacol. 2016, 781, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.D.; Migliaccio, I.; Malorni, L.; Guarducci, C.; Biganzoli, L.; Di Leo, A. Challenges in the management of advanced, ER-positive, HER2-negative breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, M.; Nasser, M.W.; Ravi, J.; Wani, N.A.; Ahirwar, D.K.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E., III; et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef]
- Blasco-Benito, S.; Seijo-Vila, M.; Caro-Villalobos, M.; Tundidor, I.; Andradas, C.; Garcia-Taboada, E.; Wade, J.; Smith, S.; Guzman, M.; Perez-Gomez, E.; et al. Appraising the “entourage effect”: Antitumor action of a pure cannabinoid versus a botanical drug preparation in preclinical models of breast cancer. Biochem. Pharmacol. 2018, 157, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Mohammadpour, F.; Ostad, S.N.; Aliebrahimi, S.; Daman, Z. Anti-invasion Effects of Cannabinoids Agonist and Antagonist on Human Breast Cancer Stem Cells. Iran. J. Pharm. Res. 2017, 16, 1479–1486. [Google Scholar] [PubMed]
- Pan, H.; Mukhopadhyay, P.; Rajesh, M.; Patel, V.; Mukhopadhyay, B.; Gao, B.; Haskó, G.; Pacher, P. Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J. Pharmacol. Exp. Ther. 2009, 328, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef]
- Portella, G.; Laezza, C.; Laccetti, P.; De Petrocellis, L.; Di Marzo, V.; Bifulco, M. Inhibitory effects of cannabinoid CB1 receptor stimulation on tumor growth and metastatic spreading: Actions on signals involved in angiogenesis and metastasis. FASEB J. 2003, 17, 1771–1773. [Google Scholar] [CrossRef]
- Chen, S.-H.; Cheung, C.H.A. Challenges in Treating Estrogen Receptor-Positive Breast Cancer. IntechOpen 2018. [Google Scholar] [CrossRef]
- Kogan, N.M.; Schlesinger, M.; Priel, E.; Rabinowitz, R.; Berenshtein, E.; Chevion, M.; Mechoulam, R. HU-331, a novel cannabinoid-based anticancer topoisomerase II inhibitor. Mol. Cancer 2007, 6, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef]
- Hsu, L.H.; Chu, N.M.; Lin, Y.F.; Kao, S.H. G-Protein Coupled Estrogen Receptor in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 306. [Google Scholar] [CrossRef]
- Dobovisek, L.; Hojnik, M.; Ferk, P. Overlapping molecular pathways between cannabinoid receptors type 1 and 2 and estrogens/androgens on the periphery and their involvement in the pathogenesis of common diseases (Review). Int. J. Mol. Med. 2016, 38, 1642–1651. [Google Scholar] [CrossRef]
- Sharma, D.; Kumar, S.; Narasimhan, B. Estrogen alpha receptor antagonists for the treatment of breast cancer: A review. Chem. Cent. J. 2018, 12, 107. [Google Scholar] [CrossRef]
- Wang, M.; Chen, H.; Wu, K.; Ding, A.; Zhang, M.; Zhang, P. Evaluation of the prognostic stage in the 8th edition of the American Joint Committee on Cancer in locally advanced breast cancer: An analysis based on SEER 18 database. Breast 2018, 37, 56–63. [Google Scholar] [CrossRef]
- Tan, W.; Li, Q.; Chen, K.; Su, F.; Song, E.; Gong, C. Estrogen receptor beta as a prognostic factor in breast cancer patients: A systematic review and meta-analysis. Oncotarget 2016, 7, 10373–10385. [Google Scholar] [CrossRef] [PubMed]
- Ruh, M.F.; Taylor, J.A.; Howlett, A.C.; Welshons, W.V. Failure of cannabinoid compounds to stimulate estrogen receptors. Biochem. Pharmacol. 1997, 53, 35–41. [Google Scholar] [CrossRef]
- Takeda, S.; Yoshida, K.; Nishimura, H.; Harada, M.; Okajima, S.; Miyoshi, H.; Okamoto, Y.; Amamoto, T.; Watanabe, K.; Omiecinski, C.J.; et al. Delta(9)-Tetrahydrocannabinol disrupts estrogen-signaling through up-regulation of estrogen receptor beta (ERbeta). Chem. Res. Toxicol. 2013, 26, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Mungenast, F.; Thalhammer, T. Estrogen biosynthesis and action in ovarian cancer. Front. Endocrinol. 2014, 5, 192. [Google Scholar] [CrossRef]
- Prather, P.L.; FrancisDevaraj, F.; Dates, C.R.; Greer, A.K.; Bratton, S.M.; Ford, B.M.; Franks, L.N.; Radominska-Pandya, A. CB1 and CB2 receptors are novel molecular targets for Tamoxifen and 4OH-Tamoxifen. Biochem. Biophys. Res. Commun. 2013, 441, 339–343. [Google Scholar] [CrossRef]
- Franks, L.N.; Ford, B.M.; Fujiwara, T.; Zhao, H.; Prather, P.L. The tamoxifen derivative ridaifen-B is a high affinity selective CB2 receptor inverse agonist exhibiting anti-inflammatory and anti-osteoclastogenic effects. Toxicol. Appl. Pharmacol. 2018, 353, 31–42. [Google Scholar] [CrossRef]
- Kumar, P.; Song, Z.H. CB2 cannabinoid receptor is a novel target for third-generation selective estrogen receptor modulators bazedoxifene and lasofoxifene. Biochem. Biophys. Res. Commun. 2014, 443, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Franks, L.N.; Ford, B.M.; Prather, P.L. Selective Estrogen Receptor Modulators: Cannabinoid Receptor Inverse Agonists with Differential CB1 and CB2 Selectivity. Front. Pharmacol. 2016, 7, 503. [Google Scholar] [CrossRef]
- Ma, H.; Yan, D.; Wang, Y.; Shi, W.; Liu, T.; Zhao, C.; Huo, S.; Duan, J.; Tao, J.; Zhai, M.; et al. Bazedoxifene exhibits growth suppressive activity by Targeting IL-6/GP130/STAT3 Signaling in Hepatocellular Carcinoma. Cancer Sci. 2019, 110, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Cyr, C.; Arboleda, M.F.; Aggarwal, S.K.; Balneaves, L.G.; Daeninck, P.; Neron, A.; Prosk, E.; Vigano, A. Cannabis in palliative care: Current challenges and practical recommendations. Ann. Palliat. Med. 2018, 7, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.P. Cannabinoids for Symptom Management and Cancer Therapy: The Evidence. J. Natl. Compr. Cancer Netw. 2016, 14, 915–922. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CB | Cell Line | IC50 | Antitumoral Activity | Receptor Mechanism | Citation |
|---|---|---|---|---|---|
| THC | MDA-MB-231 MDA-MB-468 SKBR-3 MCF-7 EVSA-T T-47D | 5.0 ± 1.2 µM 4.4 ± 0.3 µM 4.5 ± 0.4 µM 10.2 ± 0.7 µM 4.0 ± 0.1 µM 6.7 ± 0.2 µM | Induction of apoptosis Cell cycle arrest, Inhibition of G2-M transition via downregulation of Cdc2 | CB2-R | [44] |
| MDA-MB-231 MCF-7 4T1 | n.d. | Increased production of IL-4 and IL-10 Suppression of the cell-mediated Th1 response and enhancement of the Th2-response | CB1-R CB2-R | [45] | |
| MDA-MB-231 MDA-MB-468 | 1.2 µmol/L 2.5 µmol/L | Antiproliferative activity Reduction of invasiveness via ID-1 | n.d. | [46] | |
| MCF-7 MDA-MB-231 | 14.2 ± 2.1 µM 24.3 ± 4.2 µM | Inhibition of cell growth | CB2-R | [47] | |
| THCA | MCF-7 MDA-MB-231 | 9.8 ± 0.4 µM 18.2 ± 5.3 µM | Inhibition of cell growth | CB2-R | [47] |
| CBD | MDA-MB-231 MCF-7 SK-BR-3 ZR-75-1 MCF-10A (n.m.) | n.d. | Inhibition of cell viability Induction of apoptosis/autophagy No influence on cell viability | CB1-R CB2-R TRPV | [27] |
| MDA-MB-231 MCF-7 | 8.2 ± 0.3 µM 10.6 ± 1.8 µM | Inhibition of cell viability Cell cycle arrest at the G1/S transition Induction of apoptosis via pro-caspase-3 cleavage to caspase-3, induction of endoplasmic reticulum stress, inhibition of mTOR and Akt | CB2-R | [47] | |
| MDA-MB-231 T-47D | 2.2 µM 5.0 µM | Induction of apoptosis, inhibition of mTOR, upregulation of PPARγ | n.d. | [48] | |
| MDA-MB-231 MDA-MB-436 | 1.3 µmol/L 1.6 µmol/L | Antiproliferative activity Invasiveness reduction via ID-1 | n.d. | [46] | |
| CBDA | MDA-MB-231 | >100 μM | Inhibition of cell migration by modulating the activity and expression of COX-2 | CB1-R CB2-R | [49,50,51,52] |
| MDA-MB-231 | 25 μM | Inhibition of cAMP-dependent protein kinase A via activation of the small GTPase, RhoA | CB1-R CB2-R | [51] | |
| MDA-MB-231 | >25 µM | Invasiveness reduction via ID-1 and SHARP1 | n.d. | [52] | |
| MCF-7 MDA-MB-231 | 21.7 ± 3.2 µM >25 µM | Inhibition of cell growth | CB2-R | [47] | |
| CBN | MDA-MB-231 MDA-MB-468 | 1.2 µmol/L 2.6 µmol/L | Antiproliferative activity Invasiveness reduction via ID-1 | n.d. | [46] |
| CBG | MDA-MB-231 MDA-MB-468 | 2.3 µmol/L 2.1 µmol/L | Antiproliferative activity Invasiveness reduction via ID-1 | n.d. | [46] |
| MCF-7 MDA-MB-231 | 9.8 ± 3.4 µM 20.4 ± 2.6 µM | Inhibition of cell growth | CB2-R | [47] | |
| CBC | MCF-7 MDA-MB-231 | 14.2 ± 1.4 µM >25 µM | Inhibition of cell growth | CB2-R | [47] |
| AEA | MDA-MB-231 | n.d. | No growth inhibition <10 µM | CB1-R | [53] |
| MCF-7 EFM-19 BT-474 T-47D | 0.5 µM 1.5 ± 0.3 µM 1.9 µM 1.9 µM | Cell cycle arrest, inhibition of G1/S transition | CB1-R | [54] | |
| MCF-7 EFM-19 | 1.4 ± 0.9 µM 1.5 ± 0.3 µM | Inhibition of adenylyl cyclase and activation of MAPK, thereby exerting a downregulation of PRLr and trk | n.d. | [55] | |
| MCF-7 T-47D | 1.4 ± 0.9 µM 1.9 ± 0.2 µM | Inhibition of proliferation, inhibition of forskolin-induced cAMP formation, stimulation of RAF1 translocation and MAPK activity | CB1-R CB2-R TRPV | [56] | |
| MDA-MB-231 | n.d. | Regulation of lipid rafts | CB1-R | [57] | |
| 2-AG | EFM-19 | n.d. | Cell cycle arrest, inhibition of G1/S transition | CB1-R | [54] |
| MCF-7 T-47D | 1.4 ± 0.3 µM 5.0 ± 1.1 µM | Inhibition of proliferation, inhibition of forskolin-induced cAMP formation, stimulation of RAF1 translocation and MAPK activity | CB1-R CB2-R TRPV | [56] | |
| Met-F-AEA | MDA-MB-231 T-47D | n.d. | Inhibition of adhesion and migration on type IV collagen without modifying integrin expression | CB1-R | [53] |
| MDA-MB-231 | n.d. | Inhibition of proliferation by degradation of b-catenin and decrease in cyclin D1, c-Myc and MMP-2 Cell cycle arrest, inhibition of G1/S transition Upregulation of E-cadherin accompanied by the reduction of vimentin, fibronectin and N-cadherin | CB1-R | [58] | |
| MDA-MB-231 | n.d. | Inhibition of angiogenesis by the reduction of pro-angiogenic factors VEGF Reduction of metalloproteinases, TIMP1 and TIMP2 | n.d. | [59] | |
| (R)-Met-AEA | EFM-19 | 0.8 µM | Cell cycle arrest, inhibition of G1/S transition | CB1-R | [54] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kisková, T.; Mungenast, F.; Suváková, M.; Jäger, W.; Thalhammer, T. Future Aspects for Cannabinoids in Breast Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 1673. https://doi.org/10.3390/ijms20071673
Kisková T, Mungenast F, Suváková M, Jäger W, Thalhammer T. Future Aspects for Cannabinoids in Breast Cancer Therapy. International Journal of Molecular Sciences. 2019; 20(7):1673. https://doi.org/10.3390/ijms20071673
Chicago/Turabian StyleKisková, Terézia, Felicitas Mungenast, Mária Suváková, Walter Jäger, and Theresia Thalhammer. 2019. "Future Aspects for Cannabinoids in Breast Cancer Therapy" International Journal of Molecular Sciences 20, no. 7: 1673. https://doi.org/10.3390/ijms20071673
APA StyleKisková, T., Mungenast, F., Suváková, M., Jäger, W., & Thalhammer, T. (2019). Future Aspects for Cannabinoids in Breast Cancer Therapy. International Journal of Molecular Sciences, 20(7), 1673. https://doi.org/10.3390/ijms20071673