Proinsulin C-Peptide Enhances Cell Survival and Protects against Simvastatin-Induced Myotoxicity in L6 Rat Myoblasts


Abstract
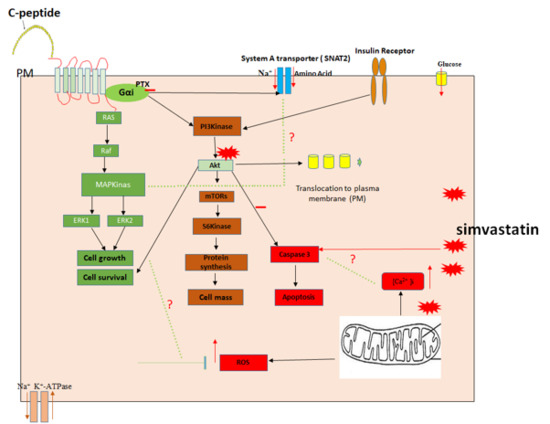
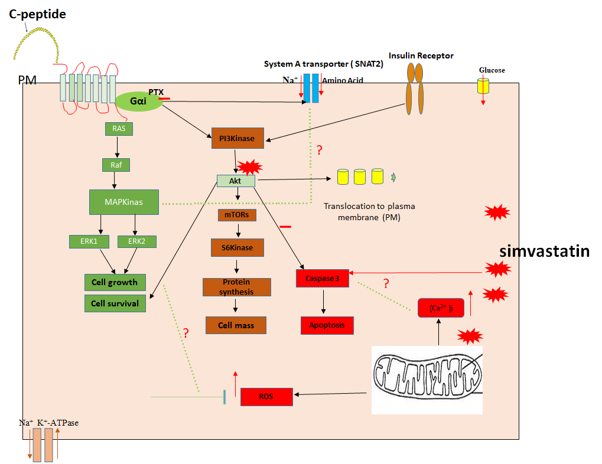{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
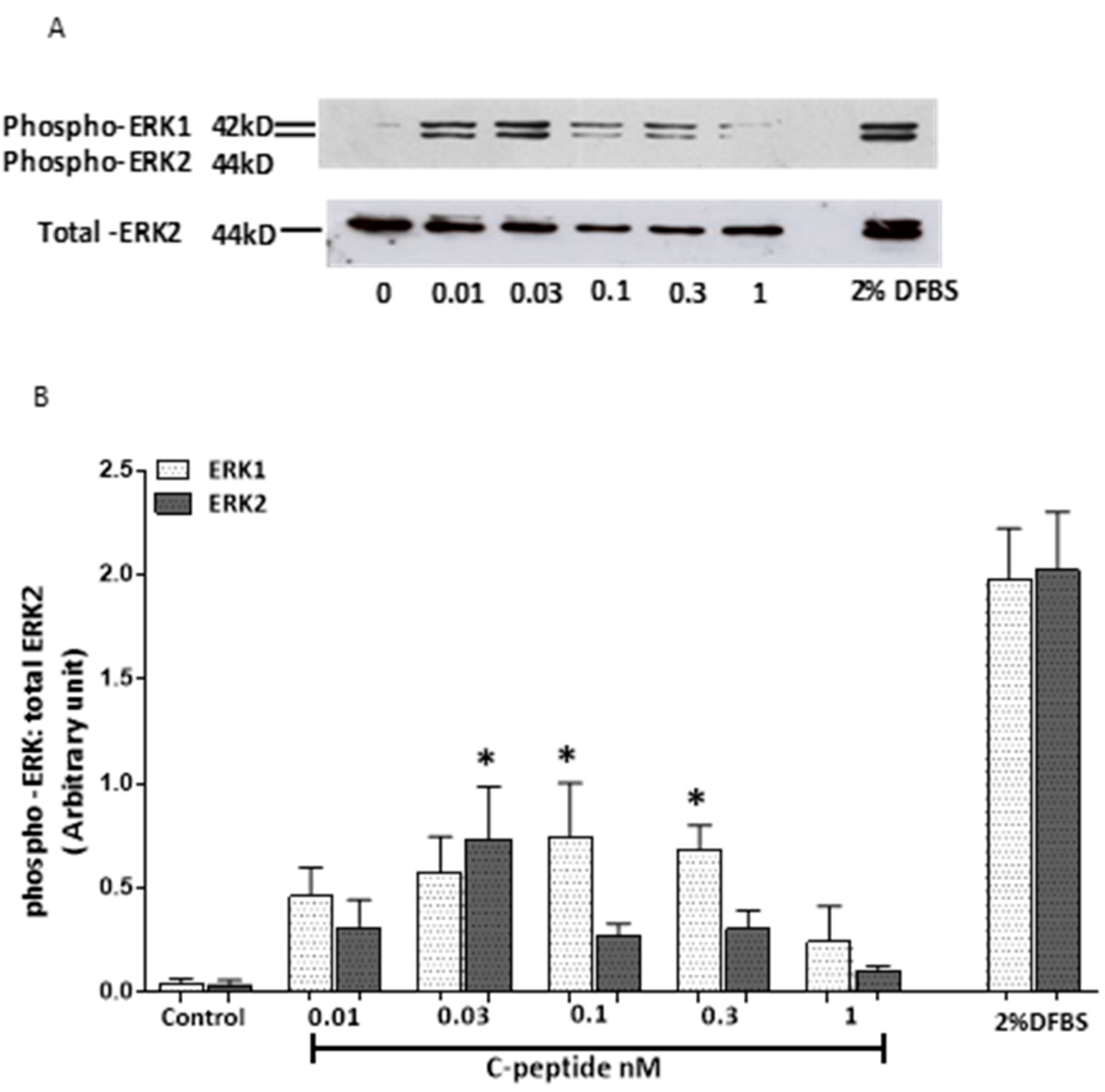
2.1. Effect of Rat C-Peptide on ERK1/2 Activation in L6 Cells
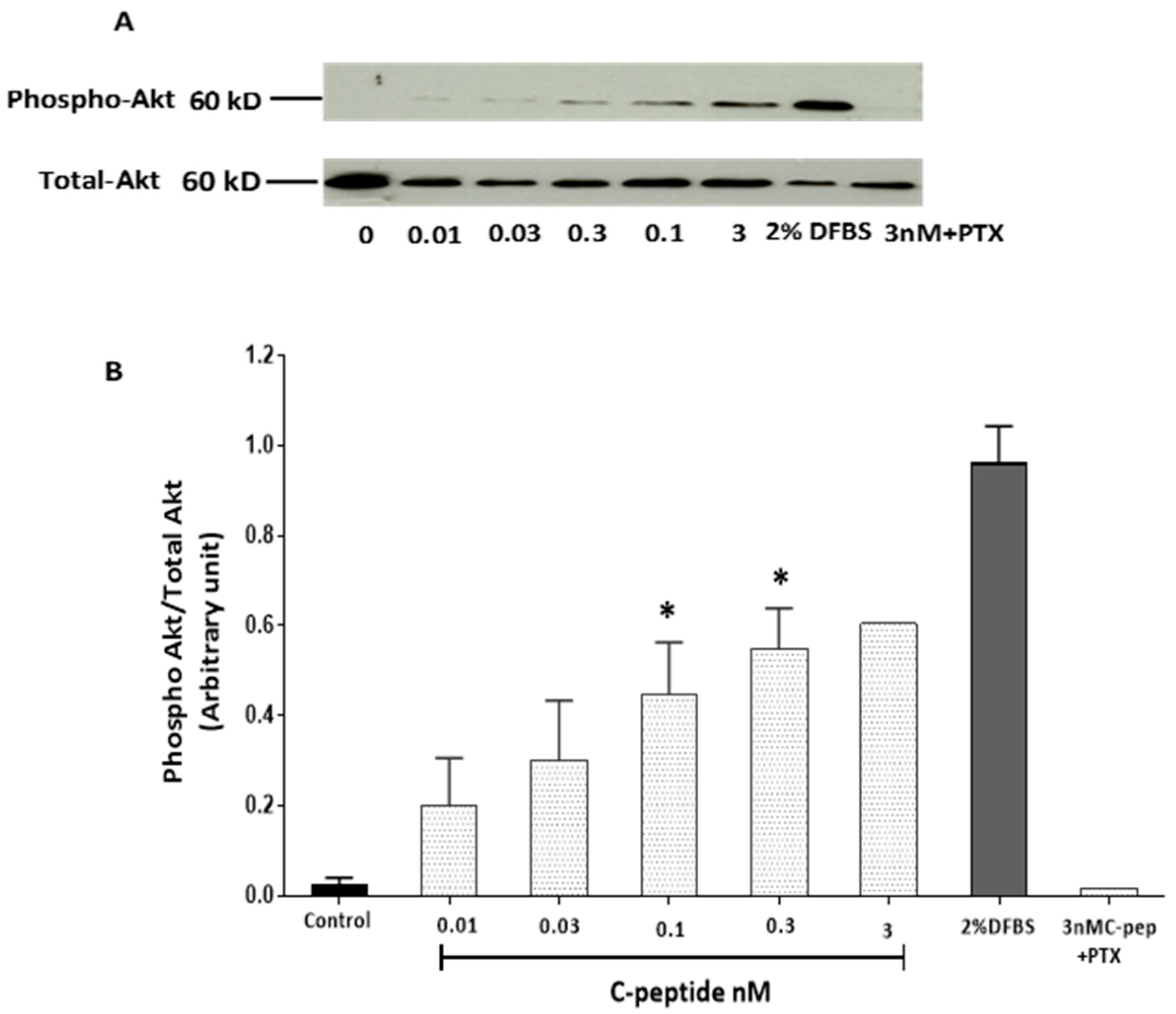
2.2. C-Peptide Stimulates Akt in L6 Myoblasts
2.3. C-Peptide Suppresses Simvastatin Induced Death Signaling in Skeletal Muscle Cells
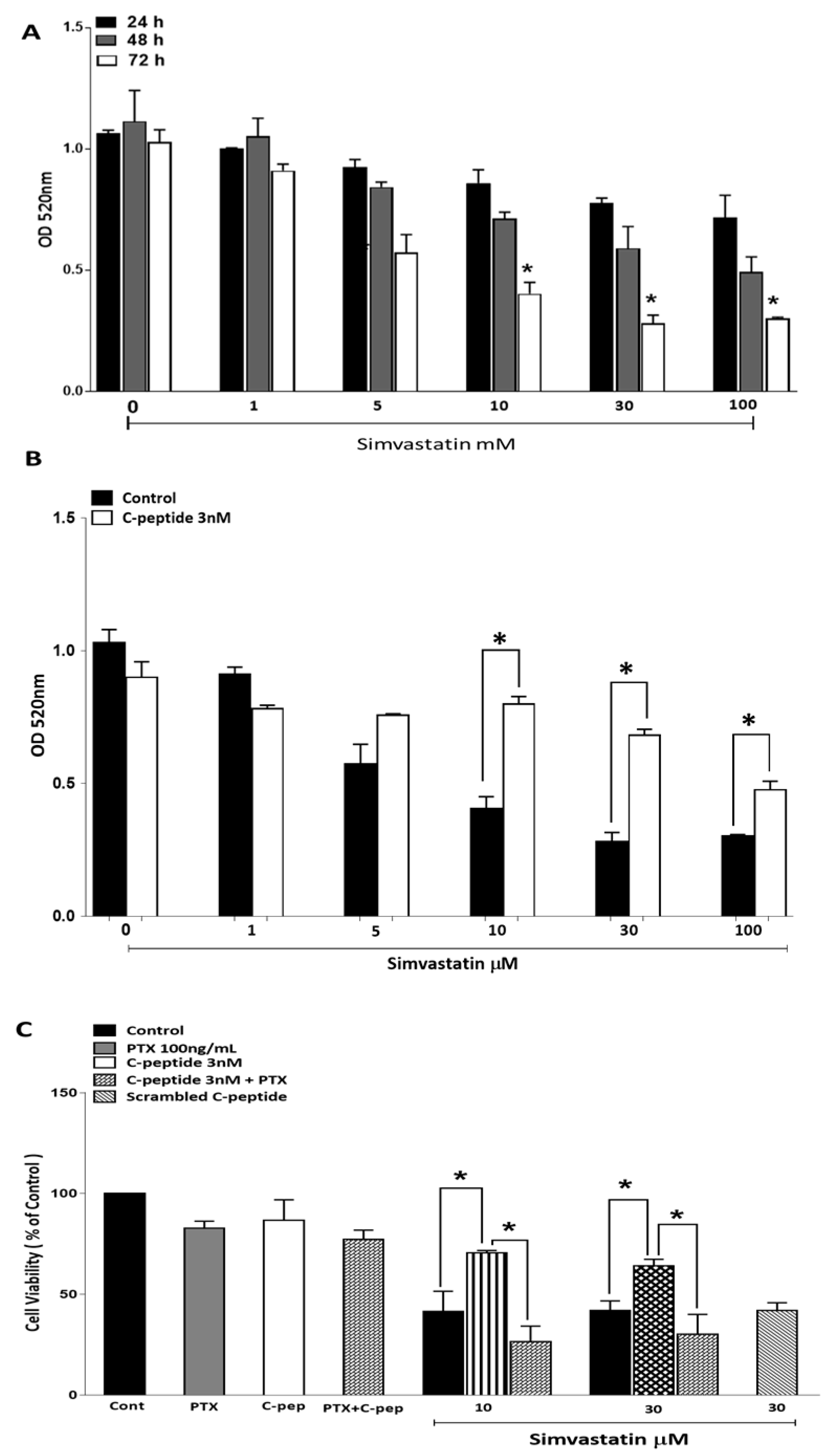
2.3.1. Simvastatin Effects on Myoblast Viability
2.3.2. C-Peptide Protects against Simvastatin-Induced Cytotoxicity
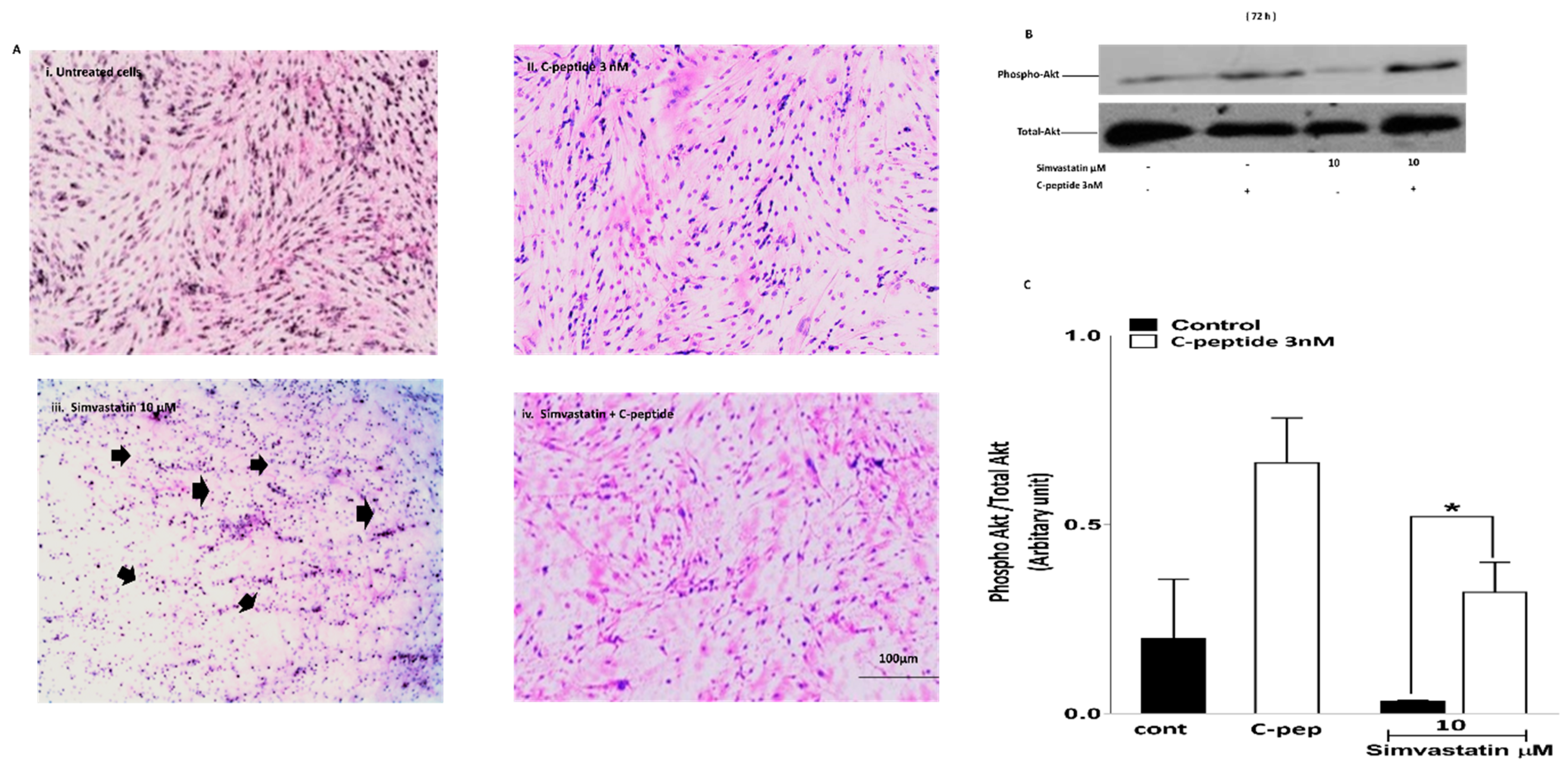
2.3.3. Morphological Visualization
2.3.4. C-Peptide Activates Akt and Suppresses the Inhibitory Effect of Simvastatin on Akt Activation in Myoblasts.
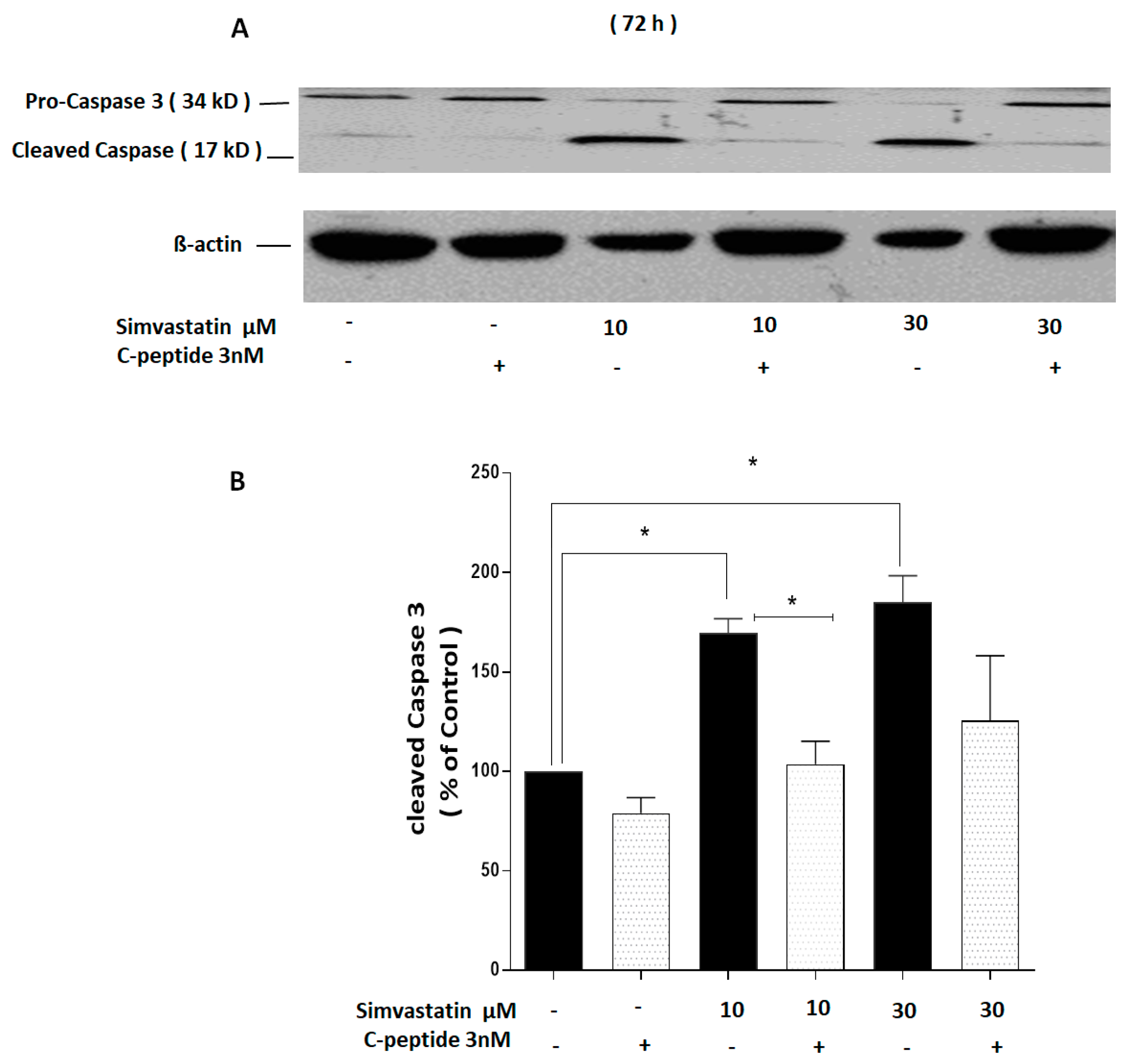
2.3.5. C-Peptide Suppresses Simvastatin-Induced Caspase-3 Cleavage
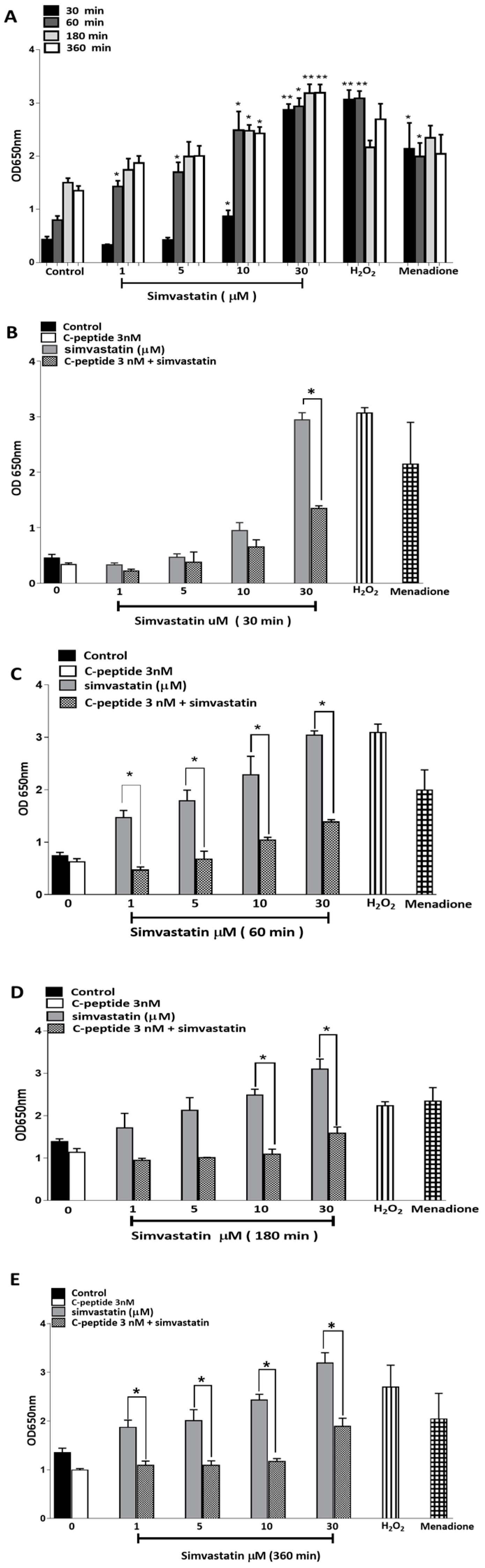
2.3.6. Simvastatin-Induced Reactive Oxygen Species (ROS) Generation in L6 Myoblasts
2.3.7. C-Peptide Reduces Simvastatin-Induced ROS Generation in L6 Myoblasts.
3. Discussion
4. Material and Methods
4.1. Chemical and Reagents
4.2. Cell Culture
4.3. Immunoblotting
4.4. Cell Viability and Growth
4.5. Wright Stain
4.6. Determination of ROS Generation
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- D’Souza, D.M.; Al-Sajee, D.; Hawke, T.J. Diabetic myopathy: Impact of diabetes mellitus on skeletal muscle progenitor cells. Front. Physiol. 2013, 4, 379. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J. The effect of insulin on the disposal of intravenous glucose: Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.P.; Riddell, M.C.; Hawke, T.J. Effects of type 1 diabetes mellitus on skeletal muscle: Clinical observations and physiological mechanisms. Pediatr. Diabetes 2011, 12, 345–364. [Google Scholar] [CrossRef]
- Charlton, M.; Nair, K.S. Protein metabolism in insulin-dependent diabetes mellitus. J. Nutr. 1998, 128, 323S–327S. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Dirks, A.J.; Jones, K.M. Statin-induced apoptosis and skeletal myopathy. Am. J. Physiol.-Cell Physiol. 2006, 291, C1208–C1212. [Google Scholar] [CrossRef] [PubMed]
- Owczarek, J.; Jasiñska, M.; Orszulak-Michalak, D. Drug-induced myopathies. An overview of the possible mechanisms. Pharmacol. Rep. 2005, 57, 23–34. [Google Scholar] [PubMed]
- Porter, K.E.; Turner, N.A. Statins and myocardial remodelling: Cell and molecular pathways. Expert Rev. Mol. Med. 2011, 13, e22. [Google Scholar] [CrossRef]
- Hoogwerf, B.J.; Huang, J.C. Lipid-Lowering Strategies and Reduction of Coronary Heart Disease Risk. Physician’s Guide to Cardiovascular Disease Prevention, 2012. Cleveland Clinic. Available online: https://commed.vcu.edu/Chronic_Disease/Heart/2013/CLevClinPrevRX.pdf (accessed on 10 February 2019).
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: A randomised placebo-controlled trial. Lancet 2003, 361, 2005–2016. [Google Scholar] [CrossRef]
- Parker, B.A.; Thompson, P.D. Effect of statins on skeletal muscle: Exercise, myopathy, and muscle outcomes. Exerc. Sport Sci. Rev. 2012, 40, 188–194. [Google Scholar] [CrossRef]
- Sirvent, P.; Mercier, J.; Lacampagne, A. New insights into mechanisms of statin-associated myotoxicity. Curr. Opin. Pharmacol. 2008, 8, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Bouitbir, J.; Charles, A.-L.; Echaniz-Laguna, A.; Kindo, M.; Daussin, F.; Auwerx, J.; Piquard, F.; Geny, B.; Zoll, J. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: A ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur. Heart J. 2012, 33, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Wahren, J.; Larsson, C. C-peptide: New Findings and Therapeutic Possibilities. Diabetes Res. Clin. Pract. 2015, 107, 309–319. [Google Scholar] [CrossRef]
- Brunskill, N. C-peptide and kidney disease. J. Intern. Med. 2017, 281, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Cifarelli, V.; Geng, X.; Styche, A.; Lakomy, R.; Trucco, M.; Luppi, P. C-peptide reduces high-glucose-induced apoptosis of endothelial cells and decreases NAD (P) H-oxidase reactive oxygen species generation in human aortic endothelial cells. Diabetologia 2011, 54, 2702–2712. [Google Scholar] [CrossRef]
- Bhatt, M.P.; Lim, Y.-C.; Kim, Y.-M.; Ha, K.-S. C-Peptide activates AMPKα and prevents ROS-mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes 2013, 62, 3851–3862. [Google Scholar] [CrossRef]
- Grunberger, G.; Qiang, X.; Li, Z.; Mathews, S.; Sbrissa, D.; Shisheva, A.; Sima, A. Molecular basis for the insulinomimetic effects of C-peptide. Diabetologia 2001, 44, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Wahren, J.; Ekberg, K.; Johansson, J.; Henriksson, M.; Pramanik, A.; Johansson, B.-L.; Rigler, R.; Jörnvall, H. Role of C-peptide in human physiology. Am. J. Physiol.-Endocrinol. Metab. 2000, 278, E759–E768. [Google Scholar] [CrossRef] [PubMed]
- Mullen, P.J.; Lüscher, B.; Scharnagl, H.; Krähenbühl, S.; Brecht, K. Effect of simvastatin on cholesterol metabolism in C2C12 myotubes and HepG2 cells, and consequences for statin-induced myopathy. Biochem. Pharmacol. 2010, 79, 1200–1209. [Google Scholar] [CrossRef]
- Rigler, R.; Pramanik, A.; Jonasson, P.; Kratz, G.; Jansson, O.; Nygren, P.-Å.; Ståhl, S.; Ekberg, K.; Johansson, B.-L.; Uhlen, S. Specific binding of proinsulin C-peptide to human cell membranes. Proc. Natl. Acad. Sci. USA 1999, 96, 13318–13323. [Google Scholar] [CrossRef]
- Waldhäusl, W. C-peptide as a remedy for diabetic microangiopathy? Diabetes 2013, 62, 39–40. [Google Scholar] [CrossRef]
- Al-Rasheed, N.M. Proinsulin C-peptide: Activation of intracellular signalling pathways and modulation of transcription factors in opossum kidney proximal tubular cells. Ph.D. Thesis, University of Leicester, Leicester, UK, 2006. [Google Scholar]
- Zhong, Z.; Davidescu, A.; Ehren, I.; Ekberg, K.; Jörnvall, H.; Wahren, J.; Chibalin, A. C-peptide stimulates ERK1/2 and JNK MAP kinases via activation of protein kinase C in human renal tubular cells. Diabetologia 2005, 48, 187–197. [Google Scholar] [CrossRef]
- Sjöberg, S.; Johansson, B.-L.; Östman, J.; Wahren, J. Renal and splanchnic exchange of human biosynthetic C-peptide in type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1991, 34, 423–428. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochimica et Biophysica Acta (BBA)-Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Jeong, S.-J.; Dasgupta, A.; Jung, K.-J.; Um, J.-H.; Burke, A.; Park, H.U.; Brady, J.N. PI3K/AKT inhibition induces caspase-dependent apoptosis in HTLV-1-transformed cells. Virology 2008, 370, 264–272. [Google Scholar] [CrossRef]
- Raz, L.; Zhang, Q.-G.; Zhou, C.-F.; Han, D.; Gulati, P.; Yang, L.C.; Yang, F.; Wang, R.M.; Brann, D.W. Role of Rac1 GTPase in NADPH oxidase activation and cognitive impairment following cerebral ischemia in the rat. PLoS ONE 2010, 5, e12606. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, D. Retention of differentiation potentialities during prolonged cultivation of myogenic cells. Proc. Natl. Acad. Sci. USA 1968, 61, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Chana, R.S.; Lewington, A.J.; Brunskill, N.J. Differential effects of peroxisome proliferator activated receptor-gamma (PPAR gamma) ligands in proximal tubular cells: Thiazolidinediones are partial PPAR gamma agonists. Kidney Int. 2004, 65, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.H. A rapid method for the differential staining of blood films and malarial parasites. J. Med. Res. 1902, 7, 138–144. [Google Scholar] [PubMed]
- Shuto, E.; Taketani, Y.; Tanaka, R.; Harada, N.; Isshiki, M.; Sato, M. Dietary Phosphorus Acutely Impairs Endothelial Function. Nephrology 2009, 20, 1504–1512. [Google Scholar] [CrossRef]
- Misirkic, M.; Janjetovic, K.; Vucicevic, L.; Tovilovic, G.; Ristic, B.; Vilimanovich, U.; Harhaji-Trajkovic, L.; Sumarac-Dumanovic, M.; Micic, D.; Bumbasirevic, V. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacol. Res. 2012, 65, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Gorham, L.M.; Ma, B.; Liu, L.; Yu, X.; Zhao, J.J.; Slaughter, D.E.; Arison, B.H.; Vyas, K.P. In vitro metabolism of simvastatin in humans [SBT] identification of metabolizing enzymes and effect of the drug on hepatic P450s. Drug Metab. Dispos. 1997, 25, 1191–1199. [Google Scholar] [PubMed]
- Peric, D.; Barragan, I.; Giraud-Triboult, K.; Egesipe, A.L.; Meyniel-Schicklin, L.; Cousin, C.; Lotteau, V.; Petit, V.; Touhami, J.; Battini, J.L.; et al. Cytostatic Effect of Repeated Exposure to Simvastatin: A Mechanism for Chronic Myotoxicity Revealed by the Use of Mesodermal Progenitors Derived from Human Pluripotent Stem Cells. Stem Cells 2015, 33, 2936–2948. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Essid, S.M.; Bevington, A.; Brunskill, N.J. Proinsulin C-Peptide Enhances Cell Survival and Protects against Simvastatin-Induced Myotoxicity in L6 Rat Myoblasts. Int. J. Mol. Sci. 2019, 20, 1654. https://doi.org/10.3390/ijms20071654
Essid SM, Bevington A, Brunskill NJ. Proinsulin C-Peptide Enhances Cell Survival and Protects against Simvastatin-Induced Myotoxicity in L6 Rat Myoblasts. International Journal of Molecular Sciences. 2019; 20(7):1654. https://doi.org/10.3390/ijms20071654
Chicago/Turabian StyleEssid, Sumia Mohamed, Alan Bevington, and Nigel J. Brunskill. 2019. "Proinsulin C-Peptide Enhances Cell Survival and Protects against Simvastatin-Induced Myotoxicity in L6 Rat Myoblasts" International Journal of Molecular Sciences 20, no. 7: 1654. https://doi.org/10.3390/ijms20071654
APA StyleEssid, S. M., Bevington, A., & Brunskill, N. J. (2019). Proinsulin C-Peptide Enhances Cell Survival and Protects against Simvastatin-Induced Myotoxicity in L6 Rat Myoblasts. International Journal of Molecular Sciences, 20(7), 1654. https://doi.org/10.3390/ijms20071654