Characterization of Microbiota Associated with Digesta and Mucosa in Different Regions of Gastrointestinal Tract of Nursery Pigs


Abstract
1. Introduction
2. Results
2.1. Animals
2.2. Summary of Microbiota Data
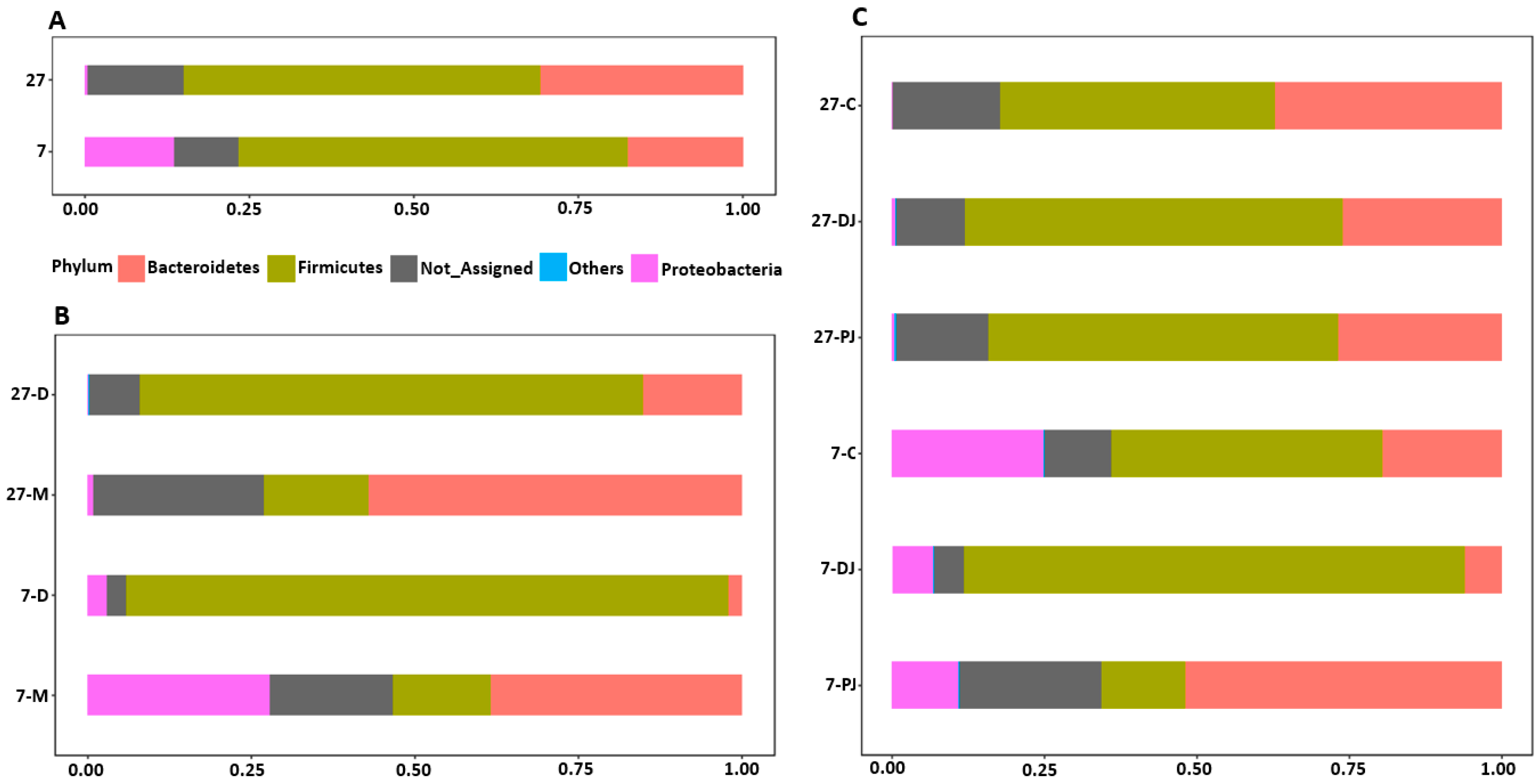
2.3. Taxonomy Assignment at Phylum Level
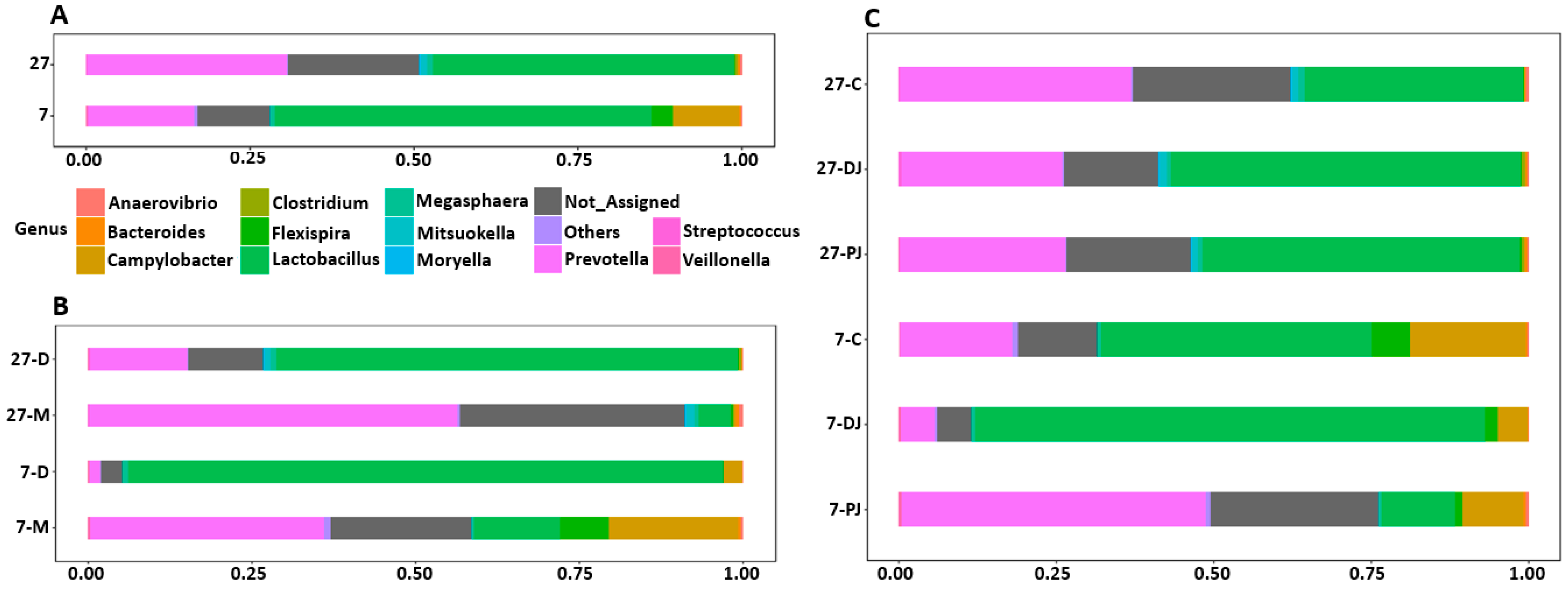
2.4. Genus Level
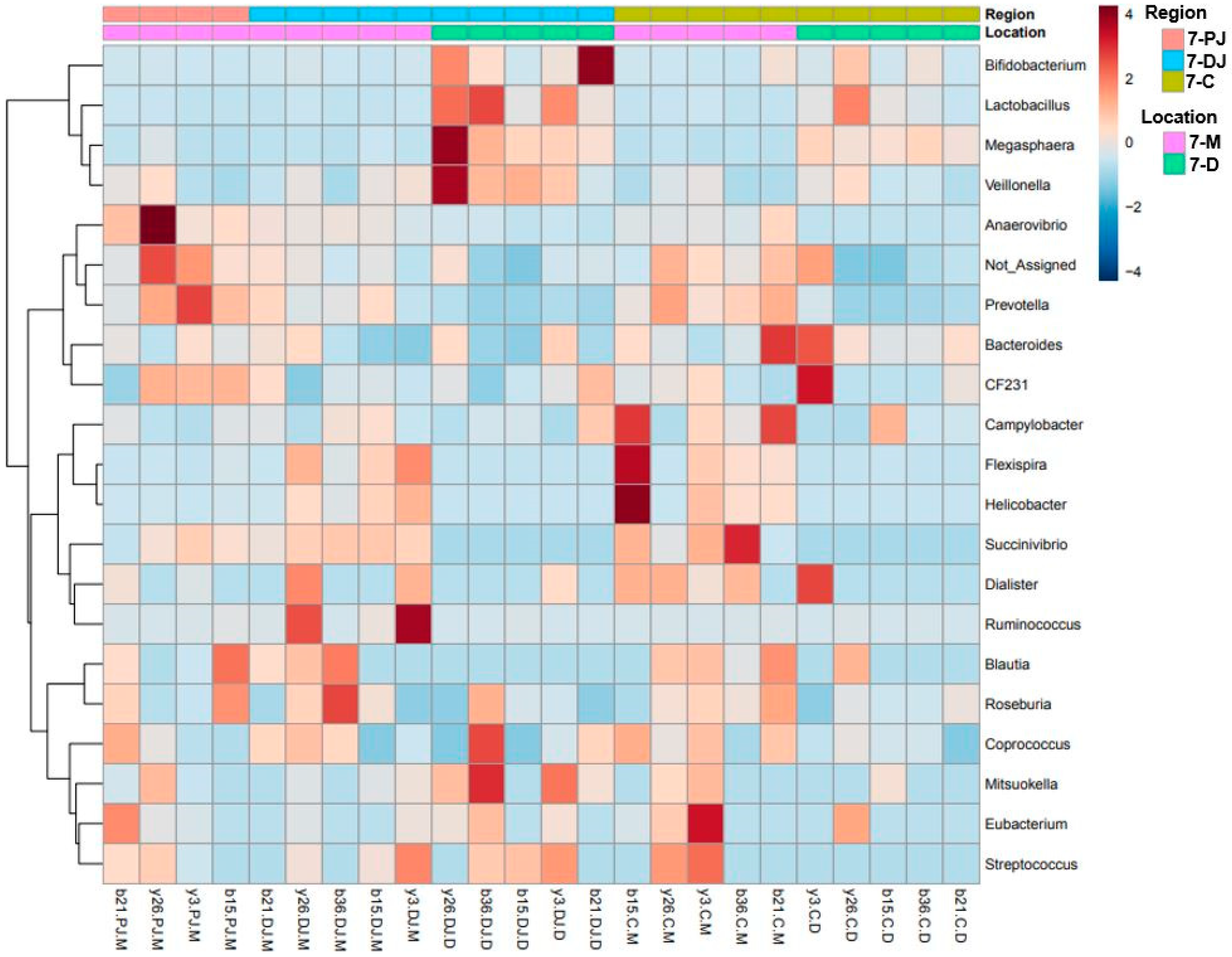
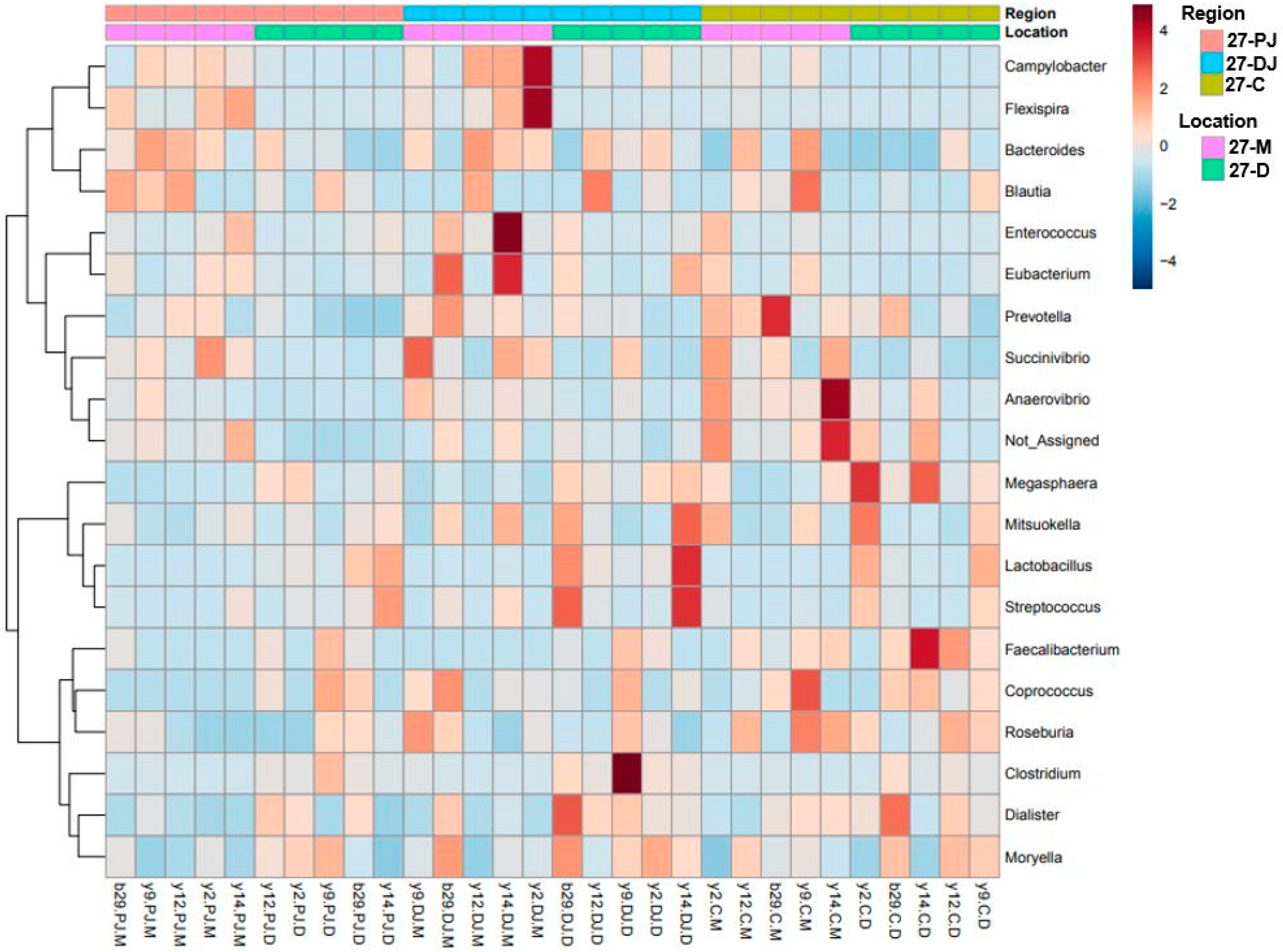
2.5. Differentially Abundant Taxa at Two Different Ages, Locations, and Regions of GIT
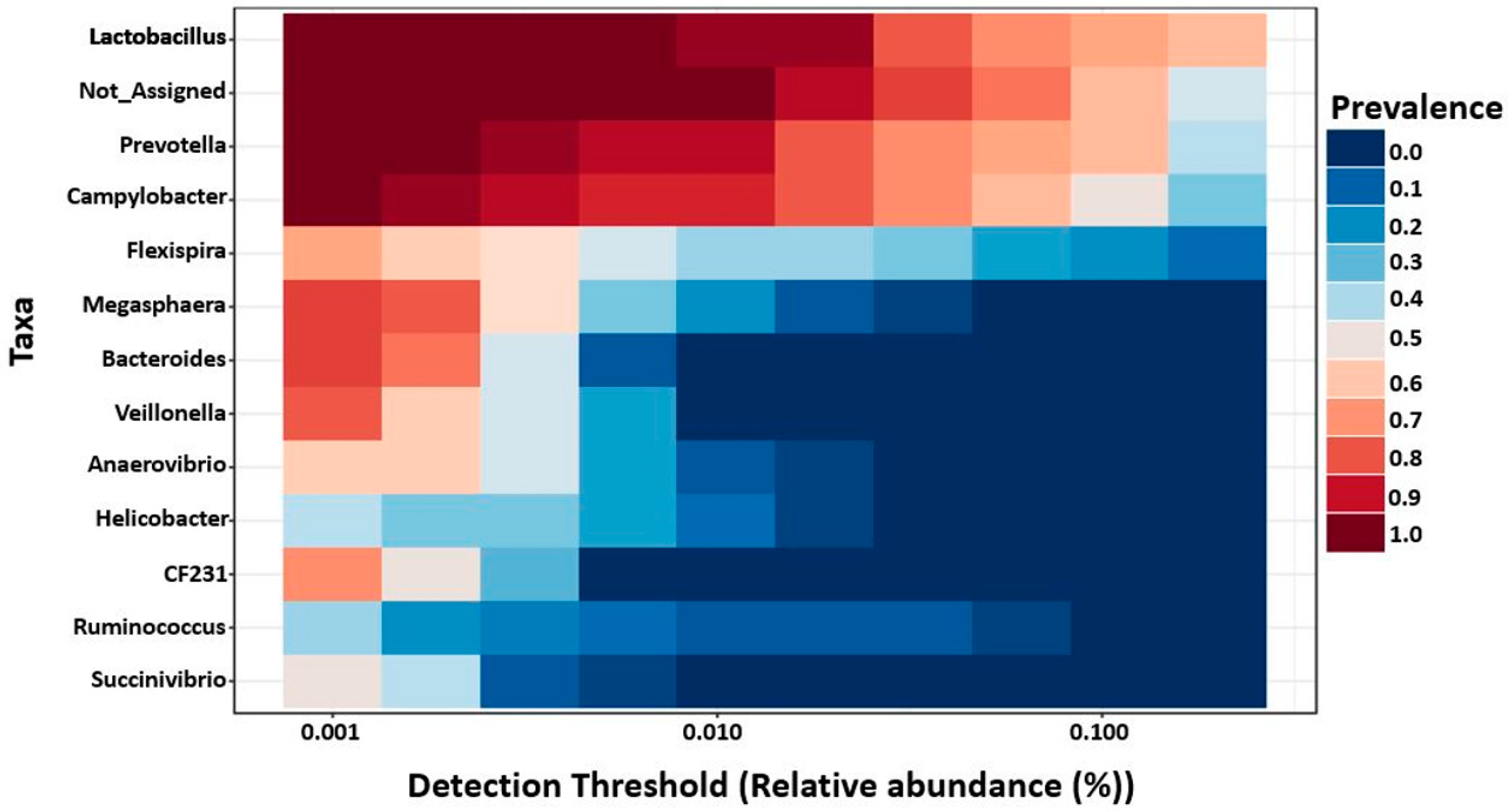
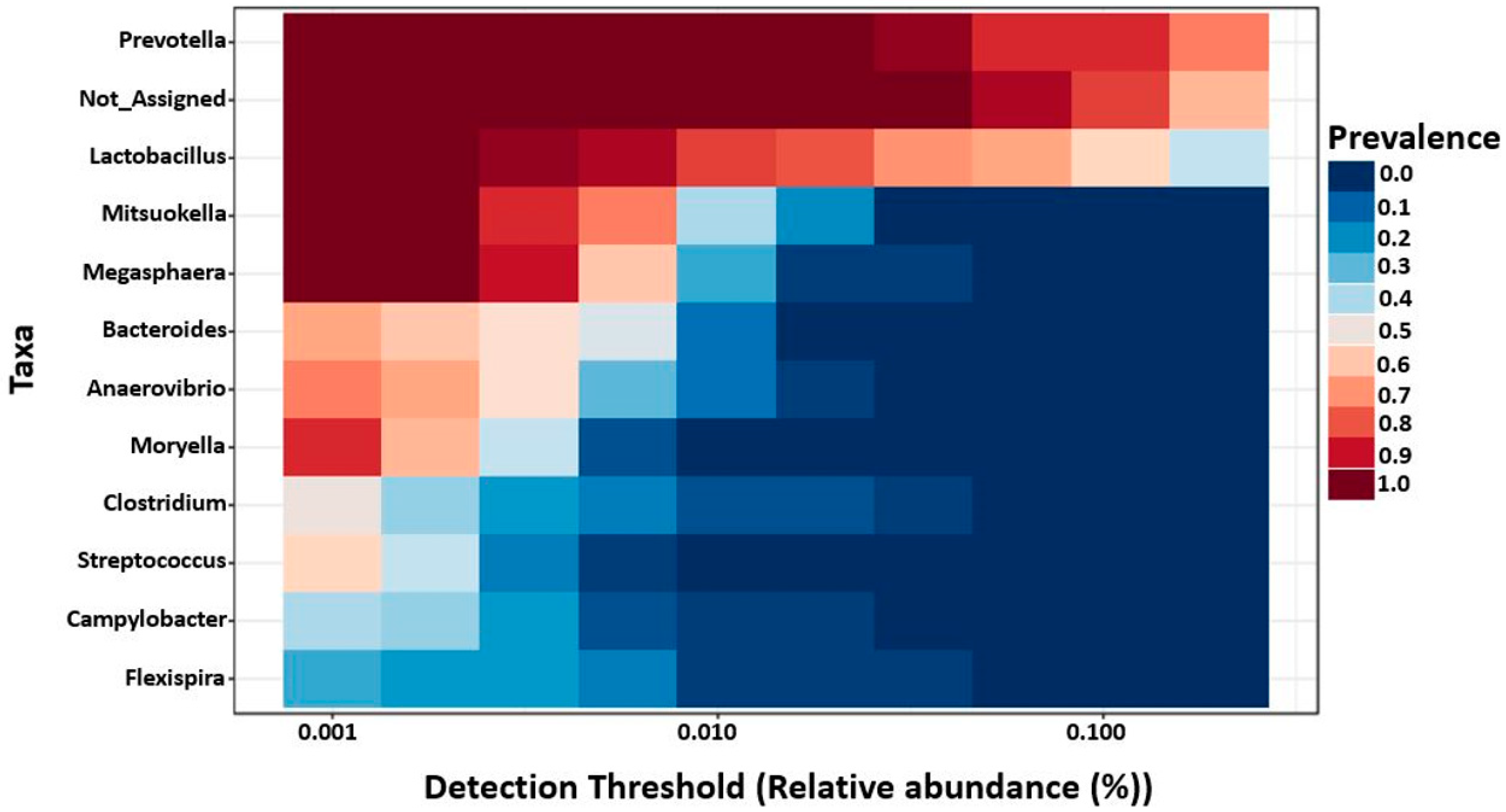
2.6. Core Microbiota on 7- and 27-Day Post-Weaned Pigs
2.7. Differentially Abundant Taxa in Piglets with Diarrhea
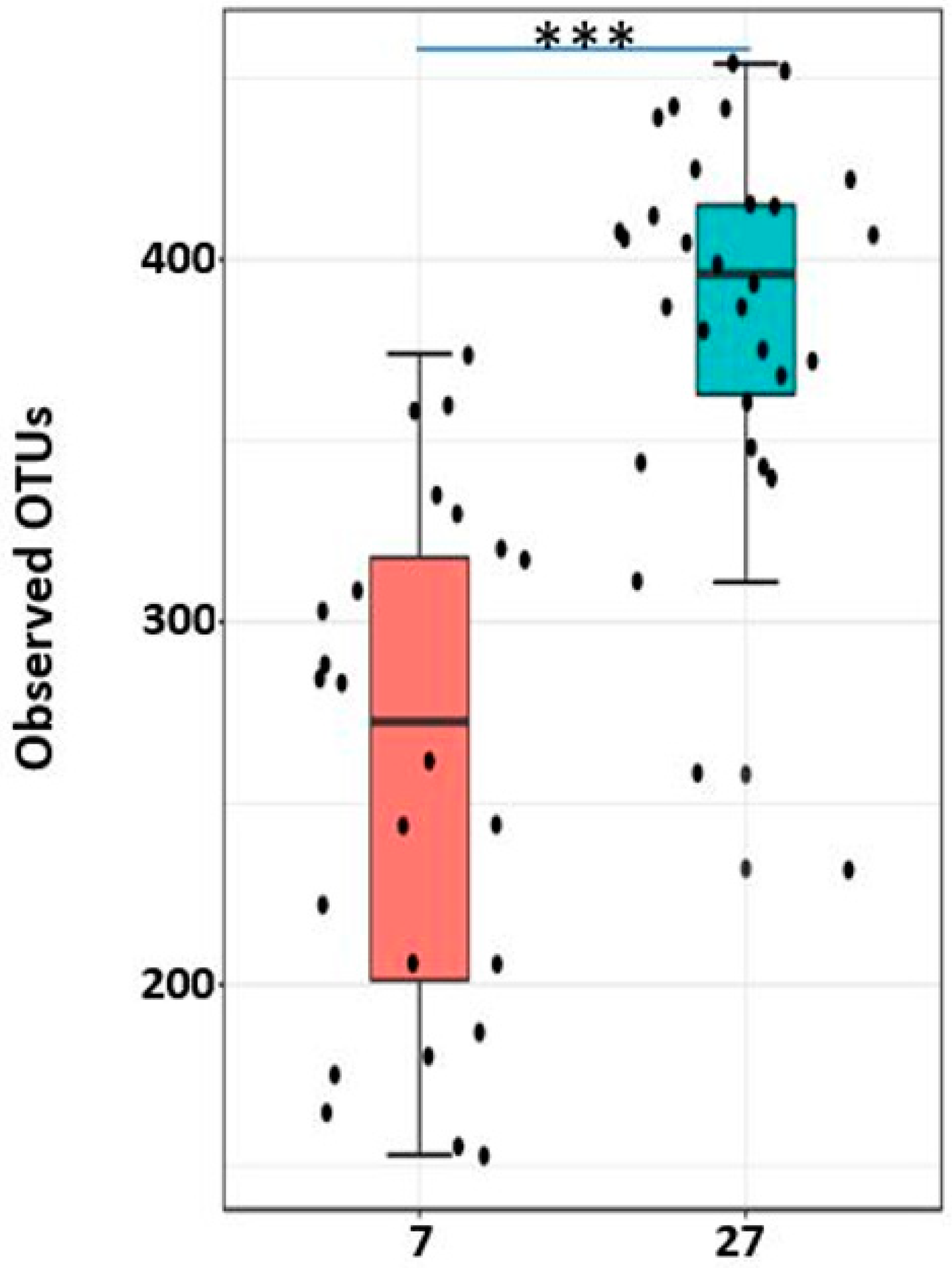
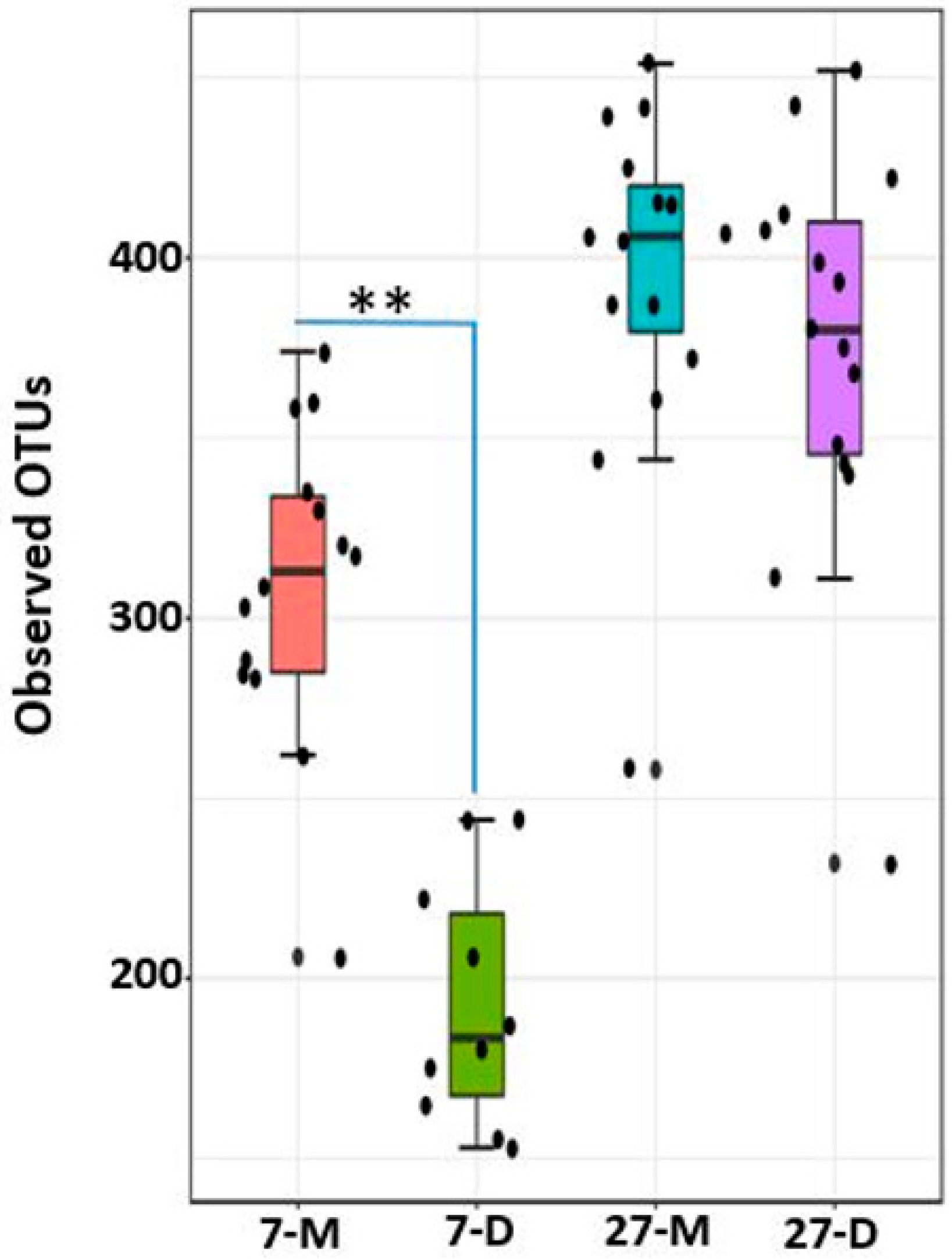
2.8. Alpha Diversity
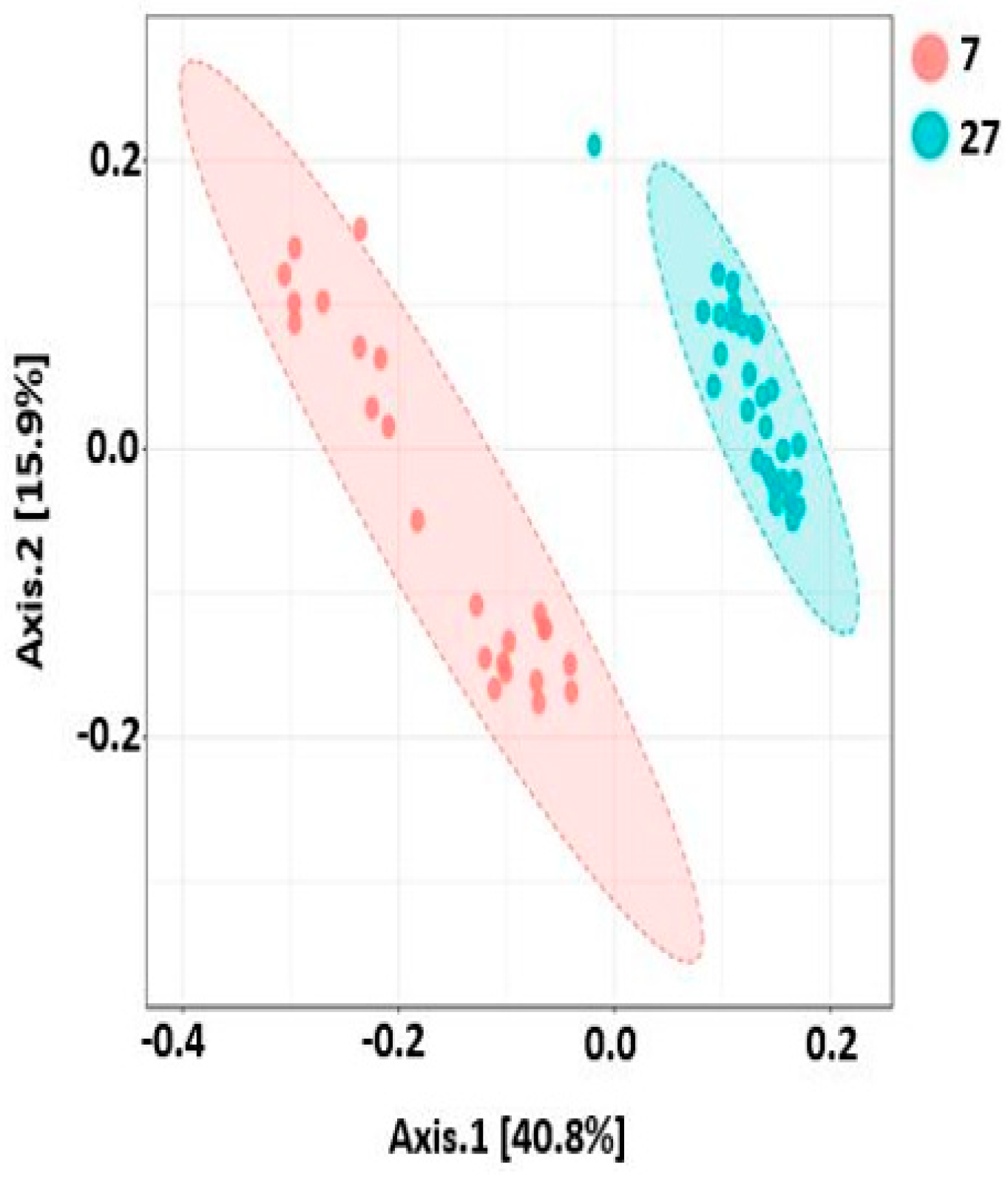
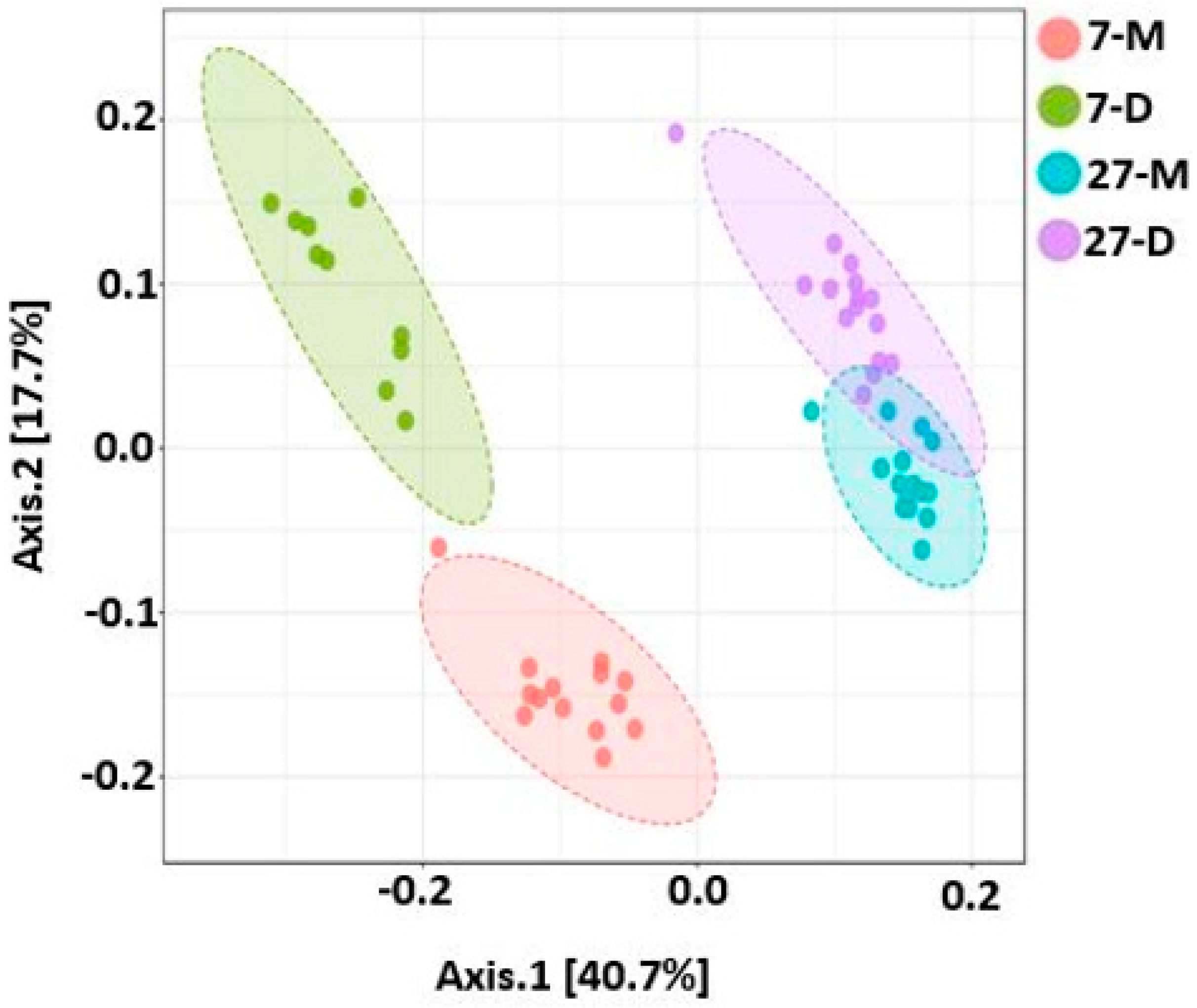
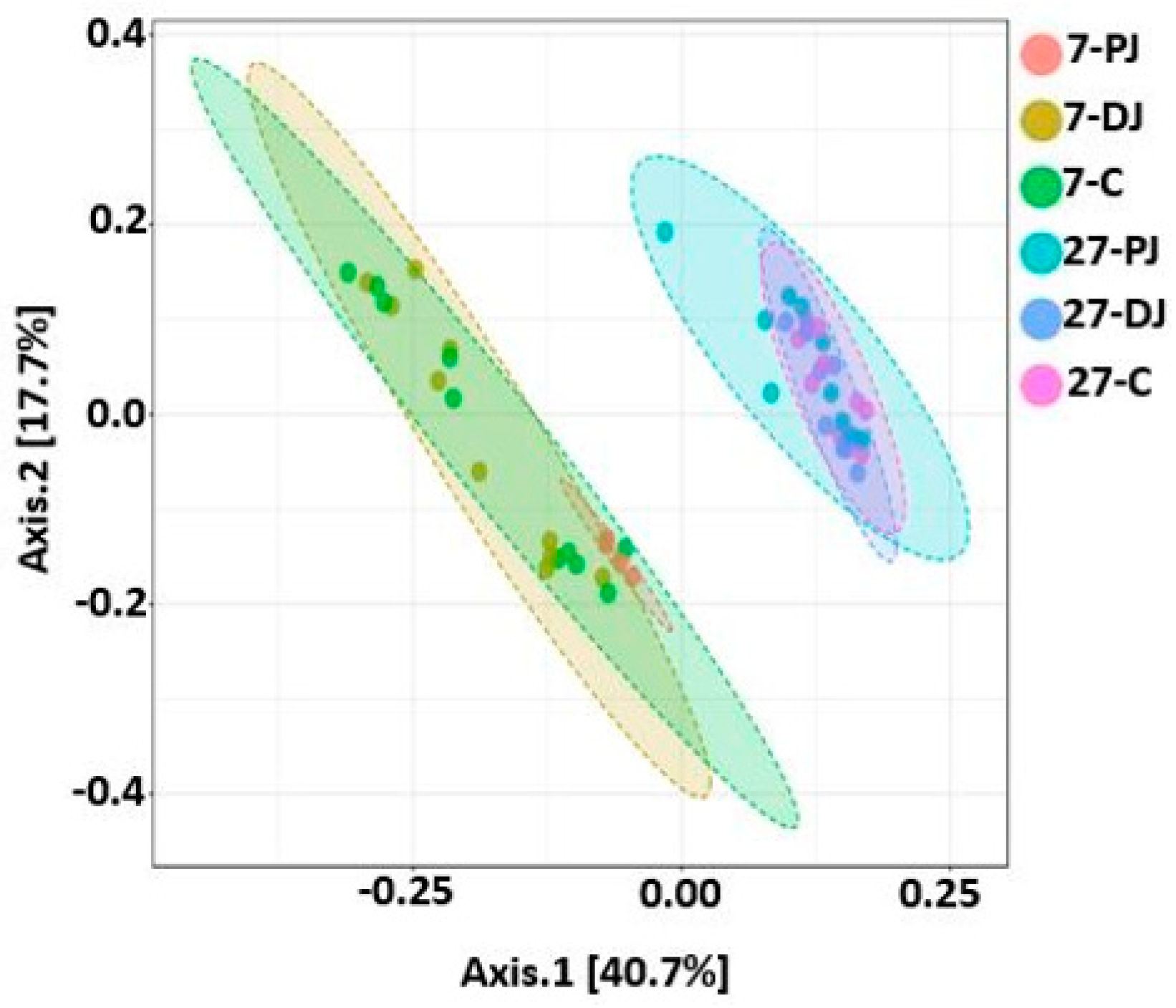
2.9. Beta Diversity
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Sample Collection and Processing
4.3. DNA Extraction and PCR
4.4. Data Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Looft, T.; Allen, H.K.; Cantarel, B.L.; Levine, U.Y.; Bayles, D.O.; Alt, D.P.; Henrissat, B.; Stanton, T.B. Bacteria, phages and pigs: The effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 2014, 8, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, B.; Kwon, Y.M. Characterization of the culturable subpopulations of lactobacillus in the chicken intestinal tract as a resource for probiotic development. Front. Microbiol. 2017, 8, 1389. [Google Scholar] [CrossRef] [PubMed]
- Morissette, B.; Talbot, G.; Beaulieu, C.; Lessard, M. Growth performance of piglets during the first two weeks of lactation affects the development of the intestinal microbiota. J. Anim. Physiol. Anim Nutr. 2018, 102, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Looft, T.; Johnson, T.A.; Allen, H.K.; Bayles, D.O.; Alt, D.P.; Stedtfeld, R.D.; Sul, W.J.; Stedtfeld, T.M.; Chai, B.; Hashsham, S.A.; et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. USA 2012, 109, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Frese, S.A.; Parker, K.; Calvert, C.C.; Mills, D.A. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 2015, 3, 28. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Li, S.; Gan, R.Y.; Zhou, T.; Xu, D.P.; Li, H.B. Impacts of gut bacteria on human health and diseases. Int. J. Mol. Sci. 2015, 16, 7493–7519. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Duffy, A. Factors influencing the gut microbiota, inflammation, and type 2 diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kwon, Y.M.; Kim, I.S.; Kim, J.A.; Yu, D.Y.; Adhikari, B.; Lee, S.S.; Choi, I.S.; Cho, K.K. Effects of the Brown Seaweed Laminaria japonica Supplementation on Serum Concentrations of IgG, Triglycerides, and Cholesterol, and Intestinal Microbiota Composition in Rats. Front. Nutr. 2018, 5, 23. [Google Scholar] [CrossRef]
- Leser, T.D.; Amenuvor, J.Z.; Jensen, T.K.; Lindecrona, R.H.; Boye, M.; Møller, K. Culture-independent analysis of gut bacteria: The pig gastrointestinal tract microbiota revisited. Appl. Environ. Microbiol. 2002, 68, 673–690. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, Y.; Liu, S.; Huang, J.; Zhai, Z.; He, C.; Ding, J.; Wang, J.; Wang, H.; Zhao, J. The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS ONE 2015, 10, e0117441. [Google Scholar] [CrossRef]
- Burrough, E.R.; Arruda, B.L.; Plummer, P.J. Comparison of the luminal and mucosa-associated microbiota in the colon of pigs with and without swine dysentery. Front. Vet. Sci. 2017, 4, 139. [Google Scholar] [CrossRef]
- Holman, D.B.; Brunelle, B.W.; Trachsel, J.; Allen, H.K. Meta-analysis to define a core microbiota in the swine gut. mSystems 2017, 2, e00004-17. [Google Scholar] [CrossRef]
- Gresse, R.; Chaucheyras-Durand, F.; Fleury, M.A.; Van de Wiele, T.; Forano, E.; Blanquet-Diot, S. Gut microbiota dysbiosis in postweaning piglets: Understanding the keys to health. Trends Microbiol. 2017, 25, 851–873. [Google Scholar] [CrossRef]
- Jensen, P. Observations on the maternal behaviour of free-ranging domestic pigs. Appl. Anim. Behav. Sci. 1986, 16, 131–142. [Google Scholar] [CrossRef]
- Lalles, J.; Bosi, P.; Smidt, H.; Stokes, C.R. Nutritional management of gut health in pigs around weaning. Proc. Nutr. Soc. 2007, 66, 260–268. [Google Scholar] [CrossRef]
- Campbell, J.M.; Crenshaw, J.D.; Polo, J. The biological stress of early weaned piglets. J. Anim. Sci. Biotechnol. 2013, 4, 19. [Google Scholar] [CrossRef]
- Barton, M.D. Impact of antibiotic use in the swine industry. Curr. Opin. Microbiol. 2014, 19, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Umu, Ö.C.; Frank, J.A.; Fangel, J.U.; Oostindjer, M.; Da Silva, C.S.; Bolhuis, E.J.; Bosch, G.; Willats, W.G.T.; Pope, P.B.; Diep, D.B. Resistant starch diet induces change in the swine microbiome and a predominance of beneficial bacterial populations. Microbiome 2015, 3, 16. [Google Scholar] [CrossRef]
- Li, P.; Niu, Q.; Wei, Q.; Zhang, Y.; Ma, X.; Kim, S.W.; Lin, M.; Huang, R. Microbial shifts in the porcine distal gut in response to diets supplemented with Enterococcus faecalis as alternatives to antibiotics. Sci. Rep. 2017, 7, 41395. [Google Scholar] [CrossRef]
- Wei, H.K.; Xue, H.X.; Zhou, Z.X.; Peng, J. A carvacrol–thymol blend decreased intestinal oxidative stress and influenced selected microbes without changing the messenger RNA levels of tight junction proteins in jejunal mucosa of weaning piglets. Animal 2017, 11, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, Y.; Chen, X.; Fang, C.; Zhao, L.; Chen, F. The maturing development of gut microbiota in commercial piglets during the weaning transition. Front. Microbiol. 2017, 8, 1688. [Google Scholar] [CrossRef]
- Kim, H.B.; Borewicz, K.; White, B.A.; Singer, R.S.; Sreevatsan, S.; Tu, Z.J.; Isaacson, R.E. Microbial shifts in the swine distal gut in response to the treatment with antimicrobial growth promoter, tylosin. Proc. Natl. Acad. Sci. USA 2012, 109, 15485–15490. [Google Scholar] [CrossRef]
- Pajarillo, E.A.B.; Chae, J.; Balolong, M.P.; Kim, H.B.; Kang, D. Assessment of fecal bacterial diversity among healthy piglets during the weaning transition. J. Gen. Appl. Microbiol. 2014, 60, 140–146. [Google Scholar] [CrossRef]
- Shin, N.; Whon, T.W.; Bae, J. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Yang, Y.; Su, Y.; Zoetendal, E.G.; Zhu, W. Differences in microbiota membership along the gastrointestinal tract of piglets and their differential alterations following an early-life antibiotic intervention. Front. Microbiol. 2017, 8, 797. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef]
- Bian, G.; Ma, S.; Zhu, Z.; Su, Y.; Zoetendal, E.G.; Mackie, R.; Liu, J.; Mu, C.; Huang, R.; Smidt, H.; et al. Age, introduction of solid feed and weaning are more important determinants of gut bacterial succession in piglets than breed and nursing mother as revealed by a reciprocal cross-fostering model. Environ. Microbiol. 2016, 18, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Xiao, Y.; Chen, J.; Chen, J.; He, X.; Yang, H. Microbial composition in different gut locations of weaning piglets receiving antibiotics. Asian-Australas. J. Anim. Sci. 2017, 30, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Janssens, Y.; Nielandt, J.; Bronselaer, A.; Debunne, N.; Verbeke, F.; Wynendaele, E.; Van Immerseel, F.; Vandewynckel, Y.P.; De Tré, G.; De Spiegeleer, B. Disbiome database: Linking the microbiome to disease. BMC Microbiol. 2018, 18, 50. [Google Scholar] [CrossRef]
- Hao, W.L.; Lee, Y.K. Microflora of the gastrointestinal tract: A review. Methods Mol. Biol. 2004, 268, 491–502. [Google Scholar] [PubMed]
- Crespo-Piazuelo, D.; Estellé, J.; Revilla, M.; Criado-Mesas, L.; Ramayo-Caldas, Y.; Óvilo, C.; Fernández, A.I.; Ballester, M.; Folch, J.M. Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci. Rep. 2018, 8, 12727. [Google Scholar] [CrossRef] [PubMed]
- Rhouma, M.; Fairbrother, J.M.; Beaudry, F.; Letellier, A. Post weaning diarrhea in pigs: Risk factors and non-colistin-based control strategies. Acta Vet. Scand. 2017, 59, 31. [Google Scholar] [CrossRef]
- Modolo, J.R.; Margato, L.F.F.; Gottschalk, A.F.; Lopes, C.A.D.M. Incidence of Campylobacter in pigs with and without diarrhea. Rev. Microbiol. 1999, 30, 19–21. [Google Scholar] [CrossRef]
- Burrough, E.; Terhorst, S.; Sahin, O.; Zhang, Q. Prevalence of Campylobacter spp. relative to other enteric pathogens in grow-finish pigs with diarrhea. Anaerobe 2013, 22, 111–114. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Nutrient Requirements of Swine, 11th ed.; The National Academies Press: Washington, DC, USA, 2012. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 2 April 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Aronesty, E. ea-utils: Command-Line Tools for Processing Biological Sequencing Data. 2011. Available online: https://expressionanalysis.github.io/ea-utils/ (accessed on 2 April 2019).
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Location | No of Samples or Average Reads/Samples (Mean ± SE) | Days 7 | Days 27 |
|---|---|---|---|---|
| Proximate Jejunum | Mucosa | No. samples | 4 | 5 |
| Reads/sample | 10,064 ± 1781.96 | 5548.6 ± 715.70 | ||
| Digesta | No. samples | NA (missing) | 5 | |
| Reads/sample | NA (missing) | 8588.8 ± 2275.10 | ||
| Distal Jejunum | Mucosa | No. samples | 5 | 5 |
| Reads/sample | 4200.6 ± 458.17 | 10,382.6 ± 2622.45 | ||
| Digesta | No. samples | 5 | 5 | |
| Reads/sample | 16,912.8 ± 5330.91 | 20,574.8 ± 8276.75 | ||
| Mid Colon | Mucosa | No. samples | 5 | 5 |
| Reads/sample | 9156.6 ± 1178.52 | 15,440 ± 2789.66 | ||
| Digesta | No. samples | 5 | 5 | |
| Reads/sample | 5975 ± 1928.96 | 17,698 ± 4778.06 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adhikari, B.; Kim, S.W.; Kwon, Y.M. Characterization of Microbiota Associated with Digesta and Mucosa in Different Regions of Gastrointestinal Tract of Nursery Pigs. Int. J. Mol. Sci. 2019, 20, 1630. https://doi.org/10.3390/ijms20071630
Adhikari B, Kim SW, Kwon YM. Characterization of Microbiota Associated with Digesta and Mucosa in Different Regions of Gastrointestinal Tract of Nursery Pigs. International Journal of Molecular Sciences. 2019; 20(7):1630. https://doi.org/10.3390/ijms20071630
Chicago/Turabian StyleAdhikari, Bishnu, Sung Woo Kim, and Young Min Kwon. 2019. "Characterization of Microbiota Associated with Digesta and Mucosa in Different Regions of Gastrointestinal Tract of Nursery Pigs" International Journal of Molecular Sciences 20, no. 7: 1630. https://doi.org/10.3390/ijms20071630
APA StyleAdhikari, B., Kim, S. W., & Kwon, Y. M. (2019). Characterization of Microbiota Associated with Digesta and Mucosa in Different Regions of Gastrointestinal Tract of Nursery Pigs. International Journal of Molecular Sciences, 20(7), 1630. https://doi.org/10.3390/ijms20071630