Electrically-Induced Ventricular Fibrillation Alters Cardiovascular Function and Expression of Apoptotic and Autophagic Proteins in Rat Hearts

Abstract
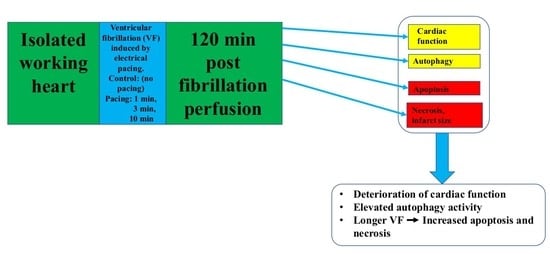
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effects of Pacing Induced Ventricular Fibrillation on Cardiac Recovery
2.2. Induced Ventricular Fibrillation Effects on the Infarcted Zone Magnitude
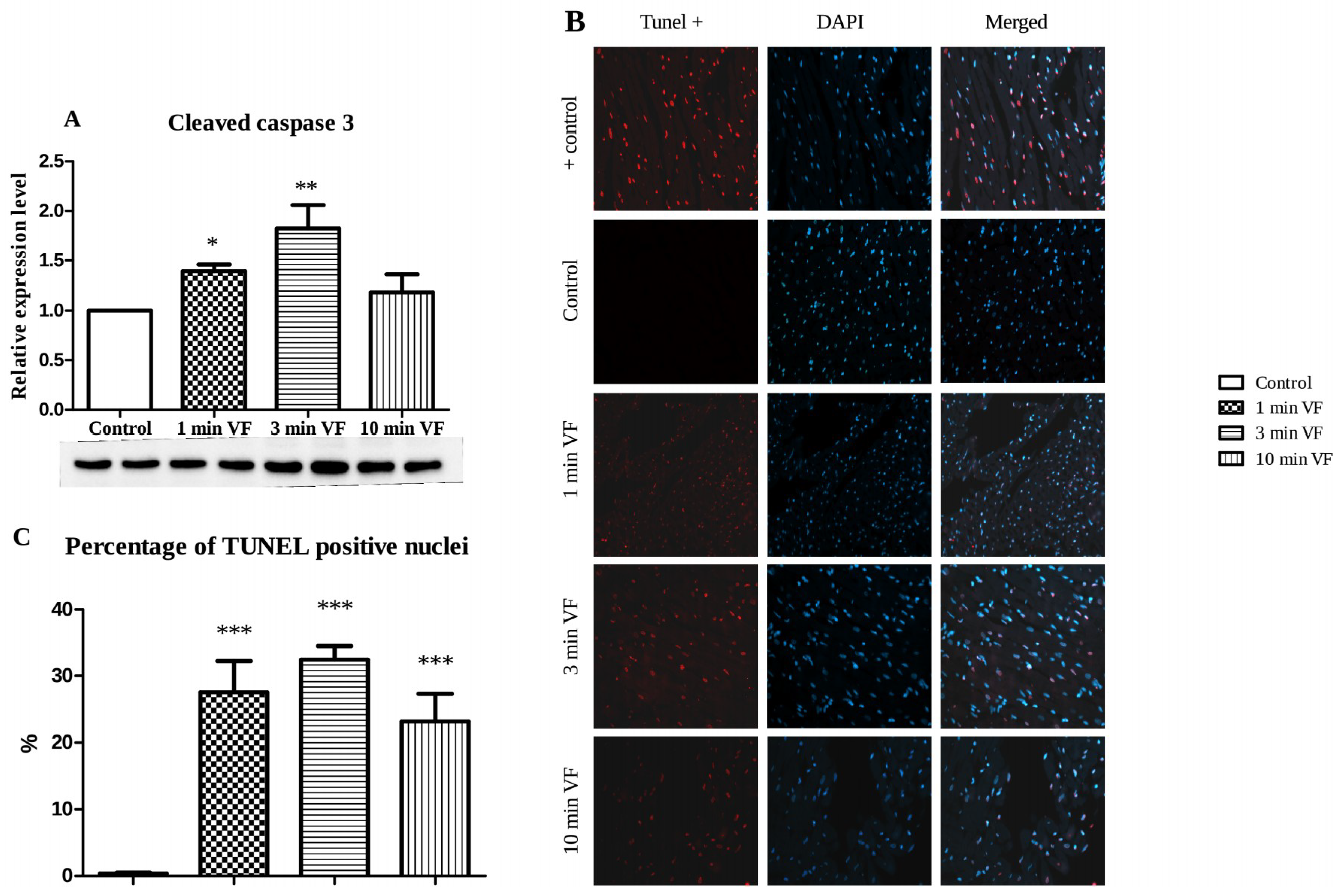
2.3. Induced Fibrillation on Caspase-3 Expression and TUNEL Positivity
3. Discussion
3.1. Pacing-Induced VF on Infarct Size and Caspase-3 Activation
3.2. VF Influence on Apoptosis- and Autophagy-Associated Proteins: Western Blot Studies
3.3. Usefulness and Limitations on Use of LC3BII/LC3BI and Atg5-12 Complex Proteins as Autophagy Indicators in Fibrillated Hearts
3.4. Conclusions
4. Materials and Methods
4.1. Animals
4.2. Chemical Reagents
4.3. Isolated Working Heart Preparation and Perfusion Protocol
4.4. Test Groups
4.5. Infarction Zone Magnitude in VF-Treated Isolated Hearts
4.6. Westerns Blot Analysis
4.7. Fluorescens Cell Death Detection
4.8. Data Analysis and Statistics
5. Limitation of the Study
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bolli, R.; McCay, P.B. Use of spin traps in intact animals undergoing myocardial ischemia/reperfusion: A new approach to assessing the role of oxygen radicals in myocardial “stunning”. Free Radic. Res. Commun. 1990, 9, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Tosaki, A.; Engelman, D.T.; Engelman, R.M.; Das, D.K. The evolution of diabetic response to ischemia/reperfusion and preconditioning in isolated working rat hearts. Cardiovasc. Res. 1996, 31, 526–536. [Google Scholar] [CrossRef]
- Curtis, M.J.; Hearse, D.J. Reperfusion-induced arrhythmias are critically dependent upon occluded zone size: Relevance to the mechanism of arrhythmogenesis. J. Mol. Cell. Cardiol. 1989, 21, 625–637. [Google Scholar] [CrossRef]
- Hearse, D.J.; Kusama, Y.; Bernier, M. Rapid electrophysiological changes leading to arrhythmias in the aerobic rat heart. Photosensitization studies with rose bengal-derived reactive oxygen intermediates. Circ. Res. 1989, 65, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A.; Przyklenk, K.; Whittaker, P. Deleterious effects of oxygen radicals in ischemia/reperfusion. Resolved and unresolved issues. Circulation 1989, 80, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.S.; Coltart, D.J.; Hearse, D.J. Ischemia and reperfusion-induced arrhythmias in the rat. Effects of xanthine oxidase inhibition with allopurinol. Circ. Res. 1984, 55, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Bernier, M.; Hearse, D.J.; Manning, A.S. Reperfusion-induced arrhythmias and oxygen-derived free radicals. Studies with “anti-free radical” interventions and a free radical-generating system in the isolated perfused rat heart. Circ. Res. 1986, 58, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Gelvan, D.; Saltman, P.; Powell, S.R. Cardiac reperfusion damage prevented by a nitroxide free radical. Proc. Natl. Acad. Sci. USA 1991, 88, 4680–4684. [Google Scholar] [CrossRef] [PubMed]
- Murohara, Y.; Yui, Y.; Hattori, R.; Kawai, C. Effects of superoxide dismutase on reperfusion arrhythmias and left ventricular function in patients undergoing thrombolysis for anterior wall acute myocardial infarction. Am. J. Cardiol. 1991, 67, 765–767. [Google Scholar] [CrossRef]
- Haines, D.D.; Bak, I.; Ferdinandy, P.; Mahmoud, F.F.; Al-Harbi, S.A.; Blasig, I.E.; Tosaki, A.J. Cardioprotective effects of the calcineurin inhibitor FK506 and the PAF receptor antagonist and free radical scavenger, EGb 761, in isolated ischemic/reperfused rat hearts. Cardiovasc. Pharmacol. 2000, 35, 37–44. [Google Scholar] [CrossRef]
- Tosaki, A.; Droy-Lefaix, M.T.; Pali, T.; Das, D.K. Effects of SOD, catalase, and a novel antiarrhythmic drug, EGB 761, on reperfusion-induced arrhythmias in isolated rat hearts. Free Radic. Biol. Med. 1993, 14, 361–370. [Google Scholar] [CrossRef]
- Yap, S.-C.; Sakhi, R.; Theuns, D.A.M.J.; Yasar, Y.E.; Bhagwandien, R.E.; Diletti, R.; Zijlstra, F.; Szili-Torok, T. Increased risk of ventricular arrhythmias in survivors of out-of-hospital cardiac arrest with chronic total coronary occlusion. Heart Rhythm 2018, 15, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Tse, G.; Gong, M.; Wong, C.W.; Chan, C.; Georgopoulos, S.; Chan, Y.S.; Yan, B.P.; Li, G.; Whittaker, P.; Ciobanu, A.; et al. International Health Informatics Study (IHIS) Network. Ann. Noninvasive Electrocardiol. 2018, 23, e12495. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A.; Shook, T.; Przyklenk, K.; Davis, V.G.; Junio, L.; Matthews, R.V.; Burstein, S.; Gibson, M.; Poole, W.K.; Cannon, C.P. Previous angina alters in-hospital outcome in TIMI 4. A clinical correlate to preconditioning? Circulation 1995, 91, 37–45. [Google Scholar] [CrossRef]
- Deutsch, E.; Berger, M.; Kussmaul, W.G.; Hirshfeld, J.W.; Herrmann, H.C.; Laskey, W.K. Adaptation to ischemia during percutaneous transluminal coronary angioplasty. Clinical, hemodynamic, and metabolic features. Circulation 1990, 82, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Tosaki, A.; Szekeres, L.; Hearse, D.J. Diltiazem and the reduction of reperfusion-induced arrhythmias in the rat: Protection is secondary to modification of ischemic injury and heart rate. J. Mol. Cell. Cardiol. 1987, 19, 441–451. [Google Scholar] [CrossRef]
- Tosaki, A.; Balint, S.; Szekeres, L. Pacing and reperfusion induced arrhythmias: Protection by slow heart rate in the rat heart. Cardiovasc. Res. 1988, 22, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Das, D.K.; Tosaki, A.J. Pacing-induced ventricular fibrillation leading to oxygen free radical formation in aerobically perfused rat hearts. Mol. Cell. Cardiol. 1993, 25, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Horstkotte, D.; Fassbender, D.; Piper, C. Congenital heart disease and acquired valvular lesions in pregnancy. Herz 2003, 28, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K.; Jeremy, R.W.; Schaper, J.; Becker, L.C. Progression of myocardial necrosis during reperfusion of ischemic myocardium. Circulation 1998, 97, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Redfors, B.; Shao, Y.; Omerovic, E. Myocardial infarct size and area at risk assessment in mice. Exp. Clin. Cardiol. 2012, 17, 268–272. [Google Scholar]
- Mukherjee, S.; Ray, D.; Lekli, I.; Bak, I.; Tosaki, A.; Das, D.K. Effects of Longevinex (modified resveratrol) on cardioprotection and its mechanisms of action. Can. J. Physiol. Pharmacol. 2010, 88, 1017–1025. [Google Scholar] [CrossRef]
- Lekli, I.; Ray, D.; Mukherjee, S.; Gurusamy, N.; Ahsan, M.K.; Juhasz, B.; Bak, I.; Tosaki, A.; Gherghiceanu, M.; Popescu, L.M.; et al. Co-ordinated autophagy with resveratrol and γ-tocotrienol confers synergetic cardioprotection. J. Cell. Mol. Med. 2010, 14, 2506–2518. [Google Scholar] [CrossRef]
- Goodsell, D.S. The molecular perspective: Caspases. Stem Cells 2000, 18, 457–458. [Google Scholar] [CrossRef]
- Varghese, J.; Khandre, N.S.; Sarin, A. Caspase-3 activation is an early event and initiates apoptotic damage in a human leukemia cell line. Apoptosis 2003, 8, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Gervais, F.G.; Xu, D.; Robertson, G.S.; Vaillancourt, J.P.; Zhu, Y.; Huang, J.; LeBlanc, A.; Smith, D.; Rigby, M.; Shearman, M.S.; et al. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 1999, 97, 395–406. [Google Scholar] [CrossRef]
- Czompa, A.; Gyongyosi, A.; Czegledi, A.; Csepanyi, E.; Bak, I.; Haines, D.D.; Tosaki, A.; Lekli, I.J. Cardioprotection afforded by sour cherry seed Kernel: The role of heme oxygenase-1. Cardiovasc. Pharmacol. 2014, 64, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Csepanyi, E.; Czompa, A.; Haines, D.; Lekli, I.; Bakondi, E.; Balla, G.; Tosaki, A.; Bak, I. Cardiovascular effects of low versus high-dose beta-carotene in a rat model. Pharmacol. Res. 2015, 100, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Chiang, G.G.; Abraham, R.T. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 2005, 280, 25485–25490. [Google Scholar] [CrossRef]
- Haines, D.D.; Juhasz, B.; Tosaki, A.J. Management of multicellular senescence and oxidative stress. Cell. Mol. Med. 2013, 17, 936–957. [Google Scholar] [CrossRef]
- Lekli, I.; Haines, D.D.; Balla, G.; Tosaki, A.J. Autophagy: An adaptive physiological countermeasure to cellular senescence and ischaemia/reperfusion-associated cardiac arrhythmias. Cell. Mol. Med. 2017, 21, 1058–1072. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; Czompa, A.; Reboul, C.; Csepanyi, E.; Czegledi, A.; Bak, I.; Balla, G.; Balla, J.; Tosaki, A.; Lekli, I. The Cellular Autophagy Markers Beclin-1 and LC3B-II are Increased During Reperfusion in Fibrillated Mouse Hearts. Curr. Pharm. Des. 2013, 19, 6912–6918. [Google Scholar] [CrossRef]
- Rusten, T.E.; Stenmark, H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Klionsky, D.J. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Ghosh, R.; Gilda, J.E.; Gomes, A.V. The necessity of and strategies for improving confidence in the accuracy of western blots. Expert Rev. Proteom. 2014, 11, 549–560. [Google Scholar] [CrossRef]
- Mozos, I.; Caraba, A. Electrocardiographic Predictors of Cardiovascular Mortality. Dis. Markers 2015, 2015, 727401. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czegledi, A.; Tosaki, A.; Gyongyosi, A.; Zilinyi, R.; Tosaki, A.; Lekli, I. Electrically-Induced Ventricular Fibrillation Alters Cardiovascular Function and Expression of Apoptotic and Autophagic Proteins in Rat Hearts. Int. J. Mol. Sci. 2019, 20, 1628. https://doi.org/10.3390/ijms20071628
Czegledi A, Tosaki A, Gyongyosi A, Zilinyi R, Tosaki A, Lekli I. Electrically-Induced Ventricular Fibrillation Alters Cardiovascular Function and Expression of Apoptotic and Autophagic Proteins in Rat Hearts. International Journal of Molecular Sciences. 2019; 20(7):1628. https://doi.org/10.3390/ijms20071628
Chicago/Turabian StyleCzegledi, Andras, Agnes Tosaki, Alexandra Gyongyosi, Rita Zilinyi, Arpad Tosaki, and Istvan Lekli. 2019. "Electrically-Induced Ventricular Fibrillation Alters Cardiovascular Function and Expression of Apoptotic and Autophagic Proteins in Rat Hearts" International Journal of Molecular Sciences 20, no. 7: 1628. https://doi.org/10.3390/ijms20071628
APA StyleCzegledi, A., Tosaki, A., Gyongyosi, A., Zilinyi, R., Tosaki, A., & Lekli, I. (2019). Electrically-Induced Ventricular Fibrillation Alters Cardiovascular Function and Expression of Apoptotic and Autophagic Proteins in Rat Hearts. International Journal of Molecular Sciences, 20(7), 1628. https://doi.org/10.3390/ijms20071628