Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide?



Abstract
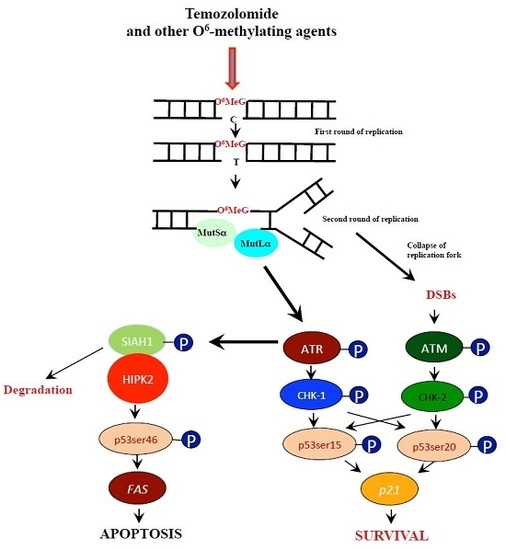
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
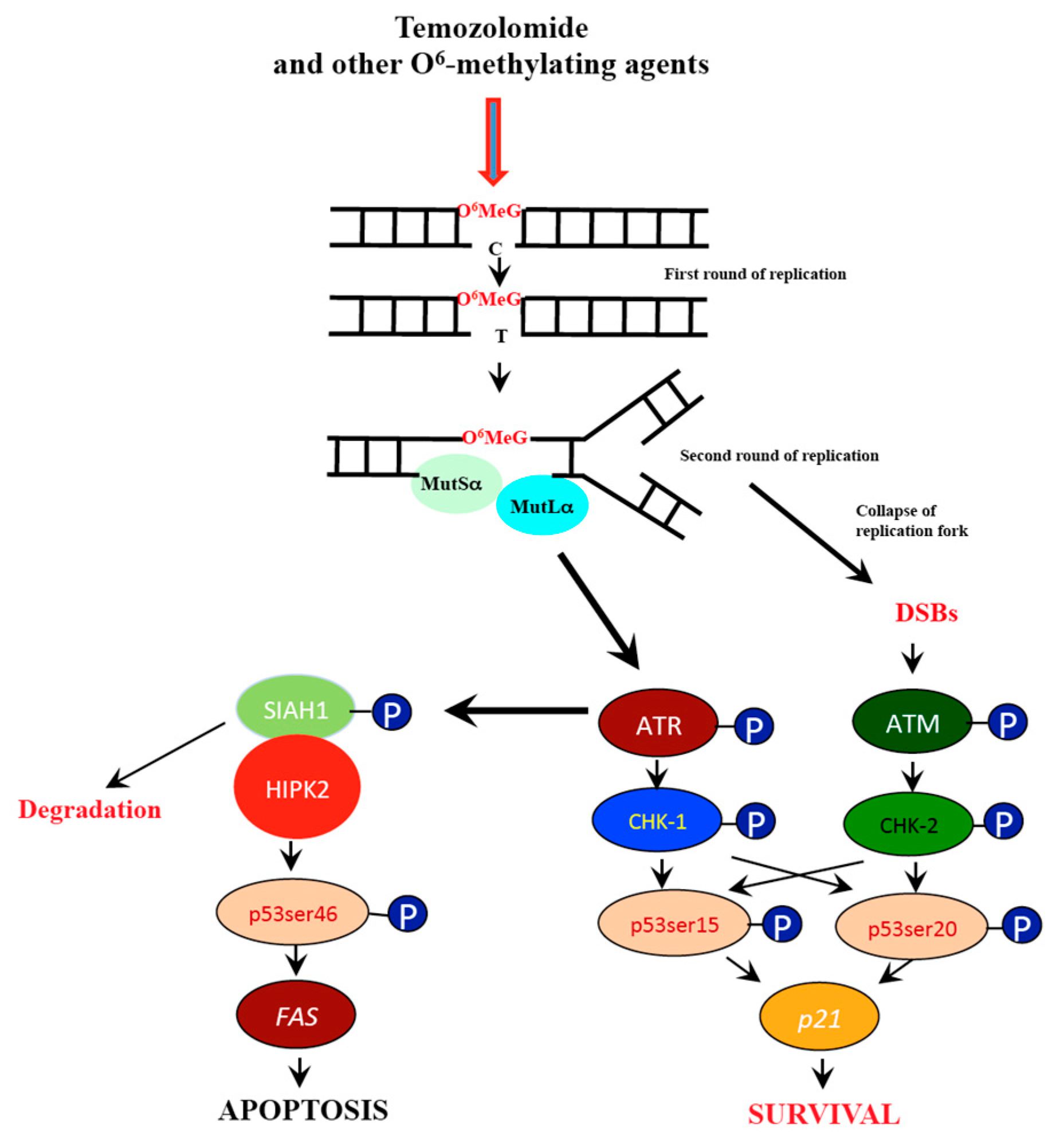
1. Introduction
2. Results
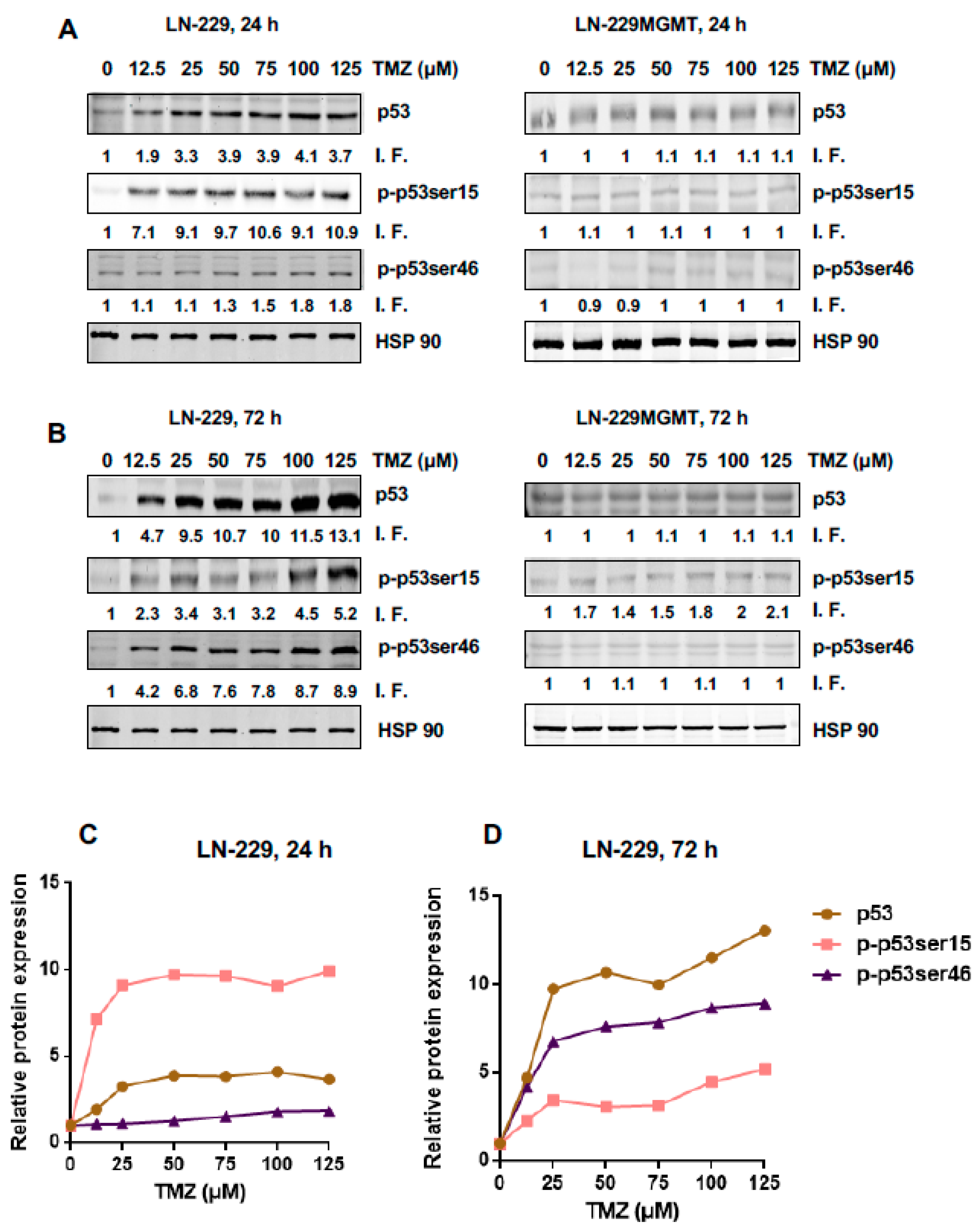
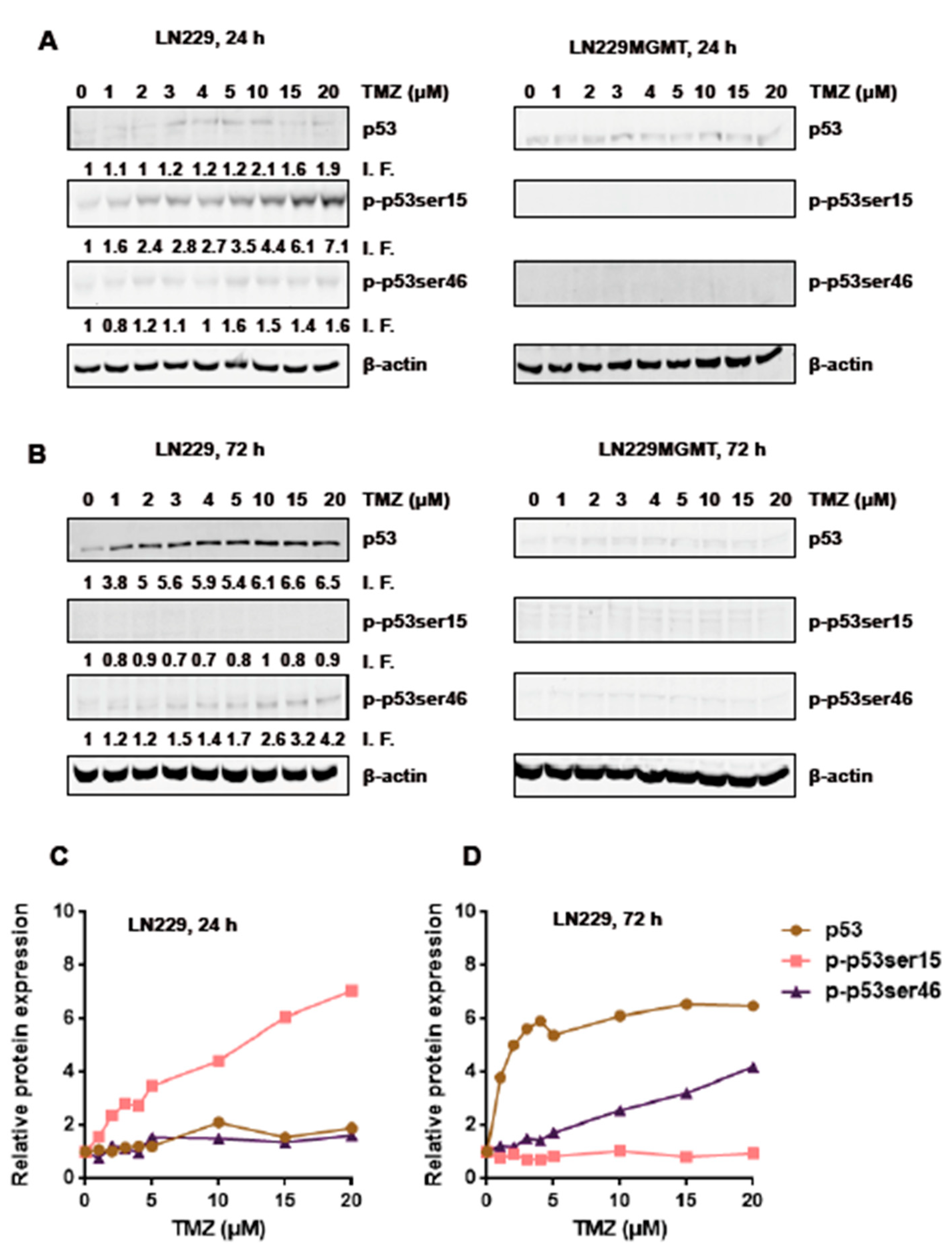
2.1. Dose Response and Time Dependence of p53, p-p53Ser15 and p-53Ser46 in Temozolomide Treated Glioblastoma Cells
2.2. Dose Response and Time-Course of Apoptosis Induction
2.3. Is There a Threshold in p53, p-p53ser15 and p-p53ser46 Induction?
2.4. Is There a Threshold in Apoptosis Induction?
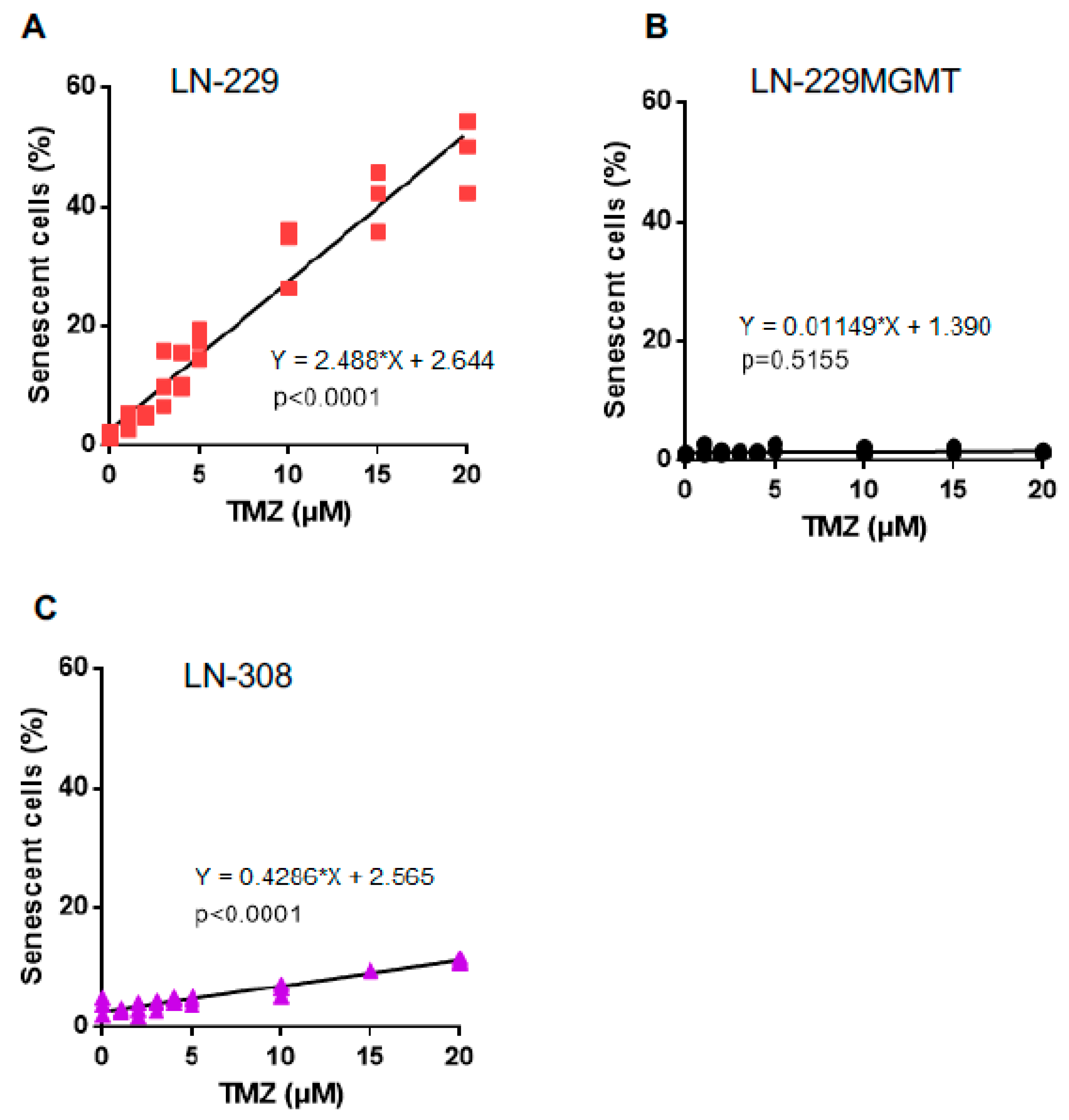
2.5. Dose-Response of TMZ-Induced Senescence and Autophagy
2.6. Dose-Response of TMZ-Induced DNA Double-Strand Breaks
3. Discussion
4. Conclusions and Implications
5. Materials and Methods
5.1. Cell Lines and Culture Conditions
5.2. Cell Seeding and Growth
5.3. Drugs and Drug Treatment
5.4. Colony Survival Assays
5.5. Apoptosis/Necrosis Flow Cytometry
5.6. Whole-Cell Protein Extracts
5.7. Western Blot
5.8. Autophagy Assay
5.9. The γH2AX Foci Assay
5.10. Senescence Measurements with C12FDG Staining
5.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Horvath, M.M.; Wang, X.; Resnick, M.A.; Bell, D.A. Divergent evolution of human p53 binding sites: Cell cycle versus apoptosis. PLoS Genet. 2007, 3, e127. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Liu, F.; Cheng, Z.; Wang, W. Cell fate decision mediated by p53 pulses. Proc. Natl. Acad. Sci. USA 2009, 106, 12245–12250. [Google Scholar] [CrossRef]
- Zhang, X.P.; Liu, F.; Wang, W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Kaina, B. Transcriptional regulation of human DNA repair genes following genotoxic stress: Trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 2013, 41, 8403–8420. [Google Scholar] [CrossRef]
- Villano, J.L.; Seery, T.E.; Bressler, L.R. Temozolomide in malignant gliomas: Current use and future targets. Cancer Chemother. Pharmacol. 2009, 64, 647–655. [Google Scholar] [CrossRef]
- Beranek, D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 1990, 231, 11–30. [Google Scholar] [CrossRef]
- Kaina, B.; Fritz, G.; Mitra, S.; Coquerelle, T. Transfection and expression of human O6-methylguanine-DNA methyltransferase (MGMT) cDNA in chinese hamster cells: The role of MGMT in protection against the genotoxic effects of alkylating agents. Carcinogenesis 1991, 12, 1857–1867. [Google Scholar]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst) 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Quiros, S.; Roos, W.P.; Kaina, B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle 2010, 9, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell. Mol. Life Sci. 2016, 73, 2829–2850. [Google Scholar] [CrossRef]
- He, Y.; Roos, W.P.; Wu, Q.; Hofmann, T.G.; Kaina, B. The Siah1-Hipk2-p53ser46 damage response pathway is involved in temozolomide-induced glioblastoma cell death. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; Van Meir, E.G. Frequent co-alterations of tp53, p16/cdkn2a, p14arf, pten tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Aasland, D.; Reich, T.R.; Tomicic, M.T.; Switzeny, O.J.; Kaina, B.; Christmann, M. Repair gene O(6)-methylguanine-DNA methyltransferase is controlled by SP1 and up-regulated by glucocorticoids, but not by temozolomide and radiation. J. Neurochem. 2018, 144, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Batista, L.F.Z.; Naumann, S.; Wick, W.; Weller, M.; Menck, C.F.M.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef]
- Nikolova, T.; Marini, F.; Kaina, B. Genotoxicity testing: Comparison of the γH2AX focus assay with the alkaline and neutral comet assays. Mutat. Res. Gen. Tox. En. 2017, 822, 10–18. [Google Scholar] [CrossRef]
- Khoury, I.; Zalko, D.; Audebert, M. Validation of the high-throughput genotoxicity assay screening using γH2AX in-cell western assay on HepG2 cells. Environ. Mol. Mutagen. 2013, 54, 737–746. [Google Scholar] [CrossRef]
- Wiewrodt, D.; Nagel, G.; Dreimuller, N.; Hundsberger, T.; Perneczky, A.; Kaina, B. MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int. J. Cancer 2008, 122, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Hamilton, S.R.; Burger, P.C.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999, 59, 793–797. [Google Scholar]
- Christmann, M.; Nagel, G.; Horn, S.; Krahn, U.; Wiewrodt, D.; Sommer, C.; Kaina, B. MGMT activity, promoter methylation and immunohistochemistry of pretreatment and recurrent malignant gliomas: A comparative study on astrocytoma and glioblastoma. Int. J. Cancer 2010, 127, 2106–2118. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase iii study: 5-year analysis of the eortc-ncic trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Chekhonin, V.P. Recent advances in oncolytic virotherapy and immunotherapy for glioblastoma: A glimmer of hope in the search for an effective therapy? Cancers 2018, 10, 492. [Google Scholar] [CrossRef]
- Wick, W.; Steinbach, J.P.; Kuker, W.M.; Dichgans, J.; Bamberg, M.; Weller, M. One week on/one week off: A novel active regimen of temozolomide for recurrent glioblastoma. Neurology 2004, 62, 2113–2115. [Google Scholar] [CrossRef]
- Strik, H.M.; Marosi, C.; Kaina, B.; Neyns, B. Temozolomide dosing regimens for glioma patients. Curr. Neurol. Neurosci. Rep. 2012, 12, 286–293. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Winkler, F. Regimen of procarbazine, lomustine, and vincristine versus temozolomide for gliomas. Cancer 2018, 124, 2674–2676. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Steinbach, J.P.; Wick, W. Temozolomide: A milestone in the pharmacotherapy of brain tumors. Future Oncol. 2005, 1, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, T. Temozolomide for malignant brain tumours. Lancet 2000, 355, 1115–1116. [Google Scholar] [CrossRef]
- Danson, S.J.; Middleton, M.R. Temozolomide: A novel oral alkylating agent. Expert Rev. Anticancer Ther. 2001, 1, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar] [PubMed]
- Marzolini, C.; Decosterd, L.A.; Shen, F.; Gander, M.; Leyvraz, S.; Bauer, J.; Buclin, T.; Biollaz, J.; Lejeune, F. Pharmacokinetics of temozolomide in association with fotemustine in malignant melanoma and malignant glioma patients: Comparison of oral, intravenous, and hepatic intra-arterial administration. Cancer Chemother. Pharmacol. 1998, 42, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Newlands, E.; Stevens, M.; Wedge, S.; Wheelhouse, R.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef]
- Eich, M.; Roos, W.P.; Nikolova, T.; Kaina, B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol. Cancer Ther. 2013, 12, 2529–2540. [Google Scholar] [CrossRef] [PubMed]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at ser 46 and mediates apoptosis. Nat. Cell Biol. 2002, 4, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.G.; Moller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Droge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 2002, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Sombroek, D.; Dauth, I.; Moehlenbrink, J.; Scheuermann, K.; Crone, J.; Hofmann, T.G. Control of hipk2 stability by ubiquitin ligase Siah-1 and checkpoint kinases ATM and ATR. Nat. Cell Biol. 2008, 10, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Calzado, M.A.; de la Vega, L.; Munoz, E.; Schmitz, M.L. Autoregulatory control of the p53 response by Siah-1-mediated Hipk2 degradation. Biol. Chem. 2009, 390, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Wilder, S.; Bannasch, D.; Israeli, D.; Lehlbach, K.; Li-Weber, M.; Friedman, S.L.; Galle, P.R.; Stremmel, W.; Oren, M.; et al. p53 activates the CD95 (Apo-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 1998, 188, 2033–2045. [Google Scholar] [CrossRef]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Boisseau, C.; Kitzinger, R.; Berac, C.; Allmann, S.; Sommer, T.; Aasland, D.; Kaina, B.; Tomicic, M.T. Adaptive upregulation of DNA repair genes following benzo(a)pyrene diol epoxide protects against cell death at the expense of mutations. Nucleic Acids Res. 2016, 44, 10727–10743. [Google Scholar] [CrossRef]
- Thomas, A.D.; Fahrer, J.; Johnson, G.E.; Kaina, B. Theoretical considerations for thresholds in chemical carcinogenesis. Mutat. Res. Rev. Mutat. Res. 2015, 765, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Ochs, K.; Grosch, S.; Fritz, G.; Lips, J.; Tomicic, M.; Dunkern, T.; Christmann, M. BER, MGMT, and MMR in defense against alkylation-induced genotoxicity and apoptosis. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 41–54. [Google Scholar] [PubMed]
- Fritz, G.; Tano, K.; Mitra, S.; Kaina, B. Inducibility of the DNA repair gene encoding O6-methylguanine-DNA methyltransferase in mammalian cells by DNA-damaging treatments. Mol. Cell. Biol. 1991, 11, 4660–4668. [Google Scholar] [CrossRef] [PubMed]
- Rafferty, J.A.; Clarke, A.R.; Sellappan, D.; Koref, M.S.; Frayling, I.M.; Margison, G.P. Induction of murine O6-alkylguanine-DNA-alkyltransferase in response to ionising radiation is p53 gene dose dependent. Oncogene 1996, 12, 693–697. [Google Scholar] [PubMed]
- Bayer, N.; Schober, D.; Prchla, E.; Murphy, R.F.; Blaas, D.; Fuchs, R. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: Implications for viral uncoating and infection. J. Virol. 1998, 72, 9645–9655. [Google Scholar] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Kaina, B. Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide? Int. J. Mol. Sci. 2019, 20, 1562. https://doi.org/10.3390/ijms20071562
He Y, Kaina B. Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide? International Journal of Molecular Sciences. 2019; 20(7):1562. https://doi.org/10.3390/ijms20071562
Chicago/Turabian StyleHe, Yang, and Bernd Kaina. 2019. "Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide?" International Journal of Molecular Sciences 20, no. 7: 1562. https://doi.org/10.3390/ijms20071562
APA StyleHe, Y., & Kaina, B. (2019). Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide? International Journal of Molecular Sciences, 20(7), 1562. https://doi.org/10.3390/ijms20071562