Single-Molecule Imaging and Computational Microscopy Approaches Clarify the Mechanism of the Dimerization and Membrane Interactions of Green Fluorescent Protein


Abstract
1. Introduction
2. Results
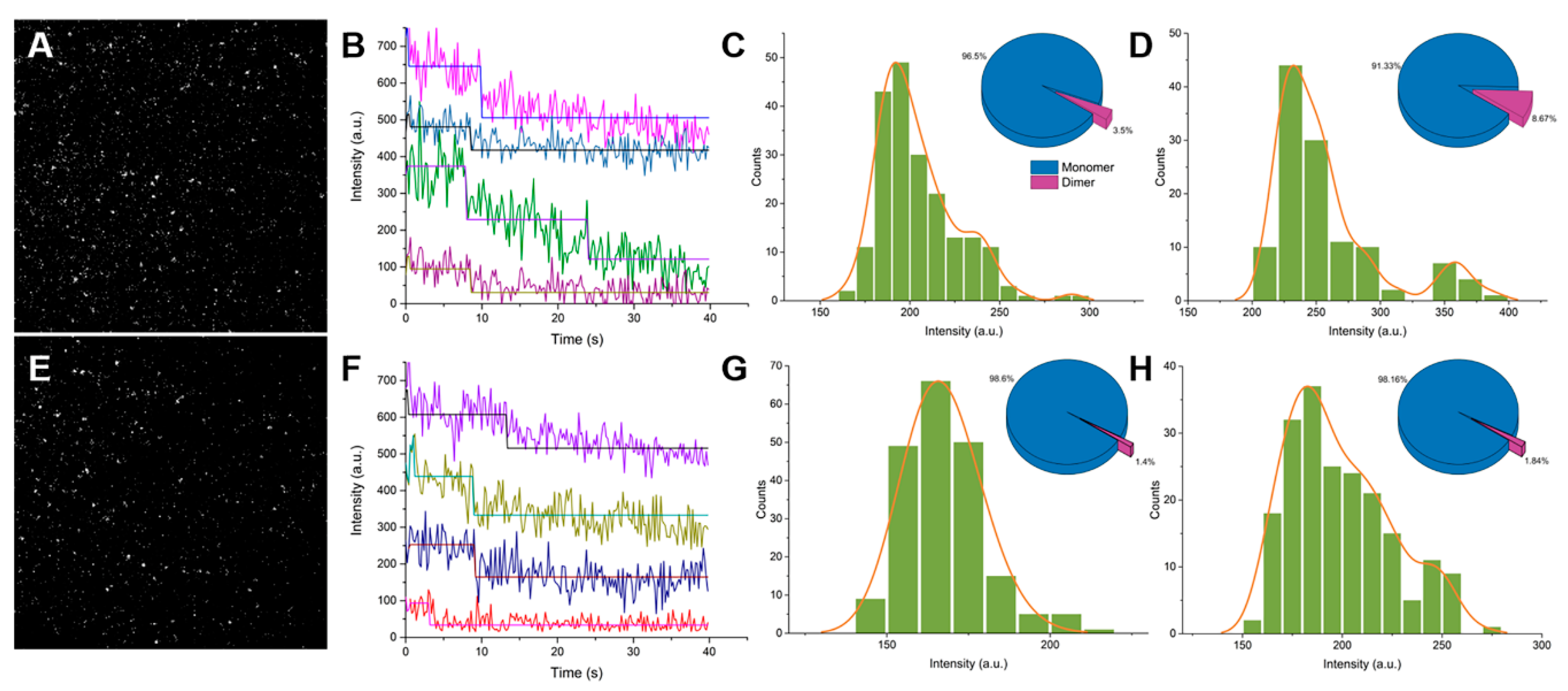
2.1. Single-Molecule Imaging of Wild-Type GFP and GFP Harboring the A206K Mutation (GFP-A206K)
2.2. Interactions between GFP Molecules and Dimer Formation
2.3. N-Myristoylation Targets GFP to the Plasma Membrane
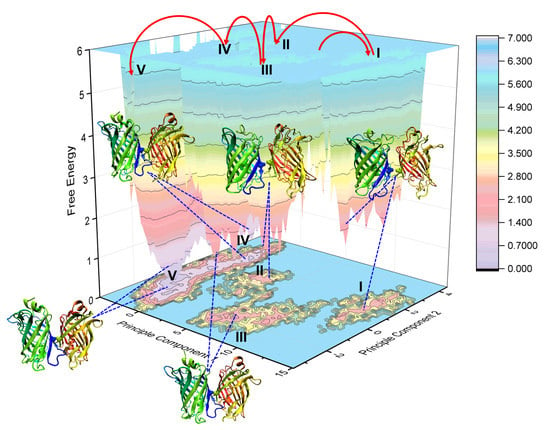
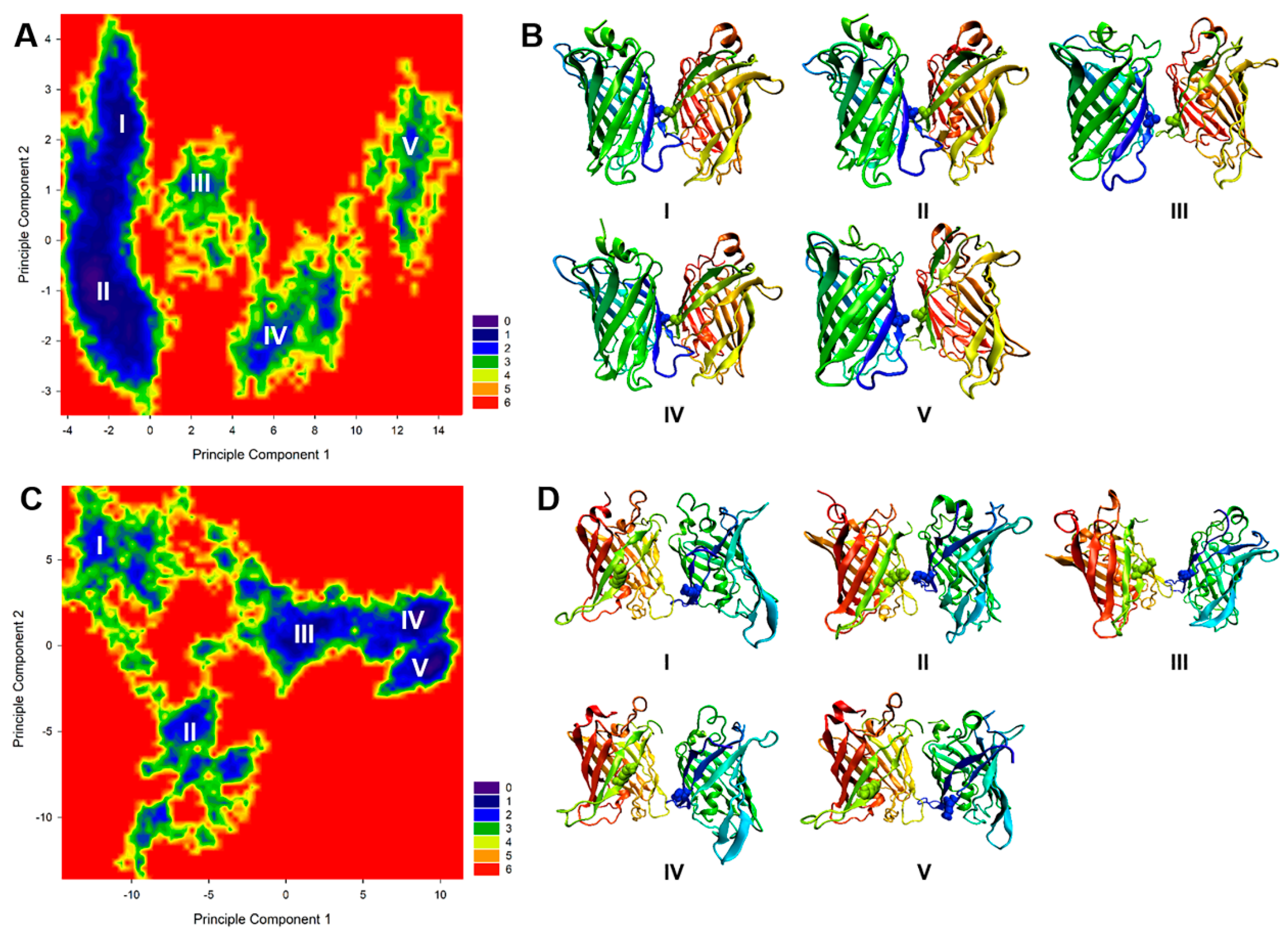
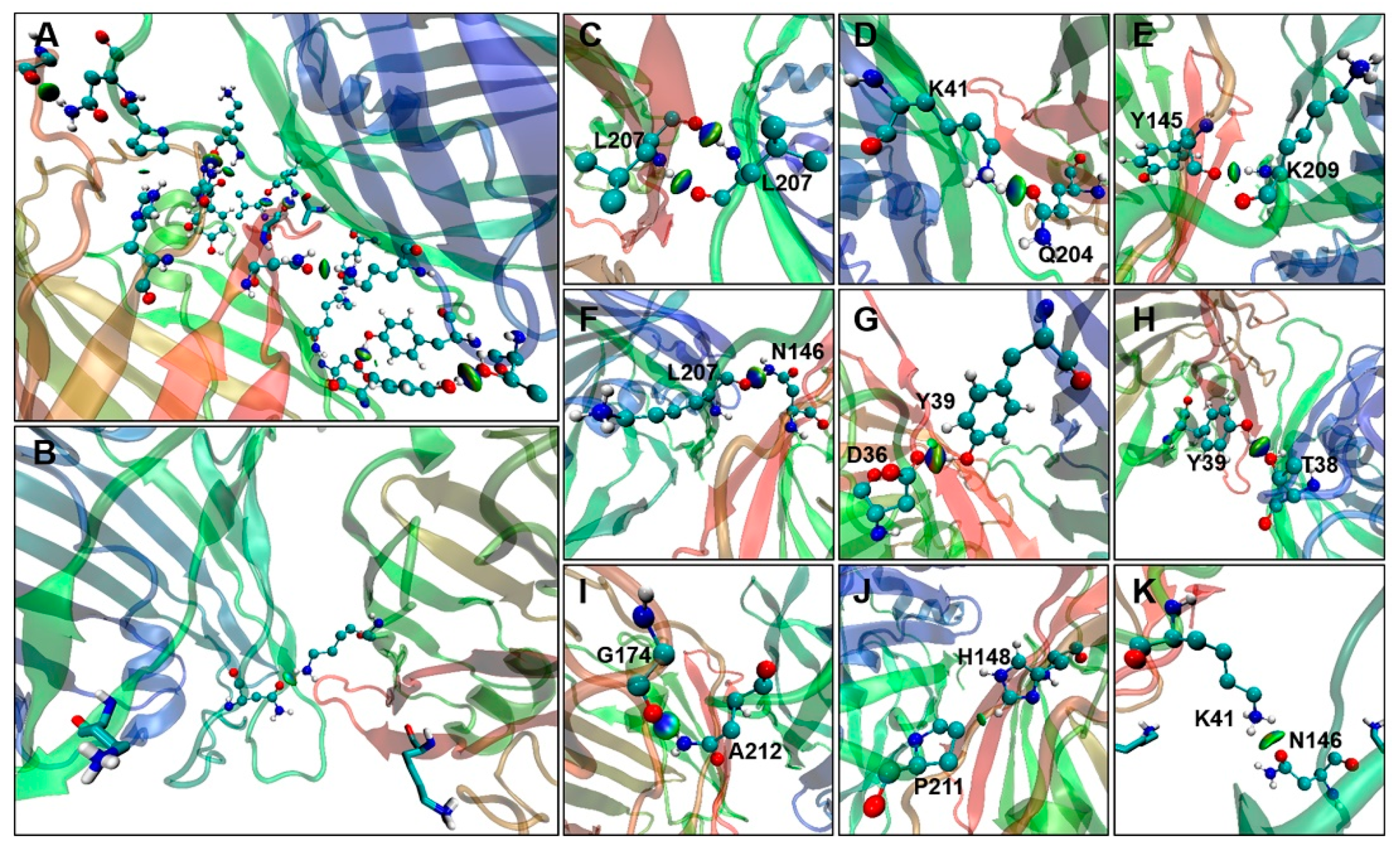
2.4. Binding Free Energy Analysis of Dimerization
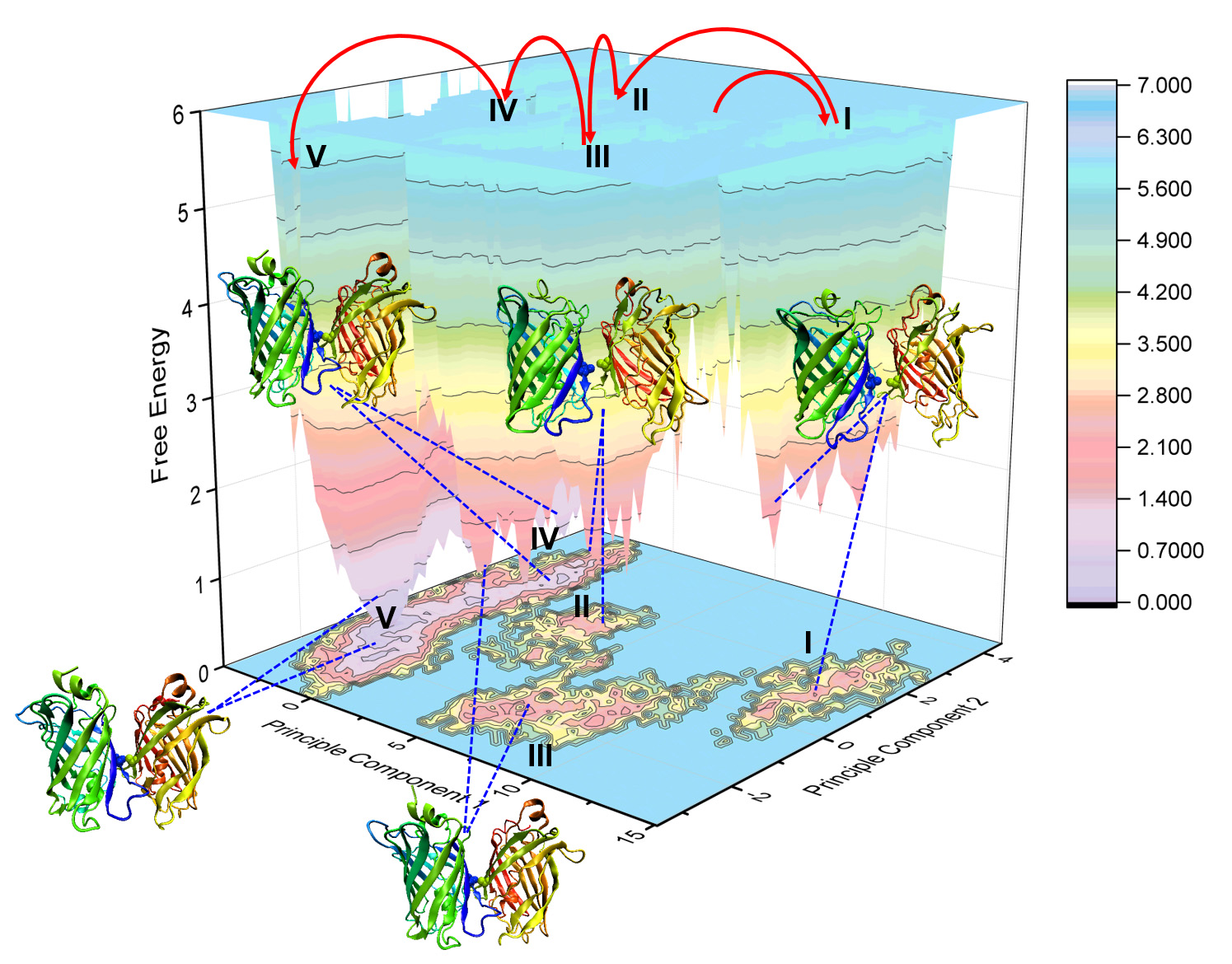
2.5. Binding Free Energy Analysis of Membrane Insertion
3. Discussion
4. Materials and Methods
4.1. Recombinant GFP Expression and Purification
4.2. Single Molecule Imaging and Analysis
4.3. Atomistic Molecular Dynamics Simulation
4.4. Calculation of Absolute Binding Free Energies
4.5. Principal Component Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, D.A.; Violin, J.D.; Newton, A.C.; Tsien, R.Y. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 2002, 296, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant dof yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 2000, 97, 11984–11989. [Google Scholar] [CrossRef]
- Kotera, I.; Iwasaki, T.; Imamura, H.; Noji, H.; Nagai, T. Reversible dimerization of Aequorea victoria fluorescent proteins increases the dynamic range of FRET-based indicators. ACS Chem. Biol. 2010, 5, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Costantini, L.M.; Fossati, M.; Francolini, M.; Snapp, E.L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic 2012, 13, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Segami, S.; Makino, S.; Miyake, A.; Asaoka, M.; Maeshima, M. Dynamics of vacuoles and H+-pyrophosphatase visualized by monomeric green fluorescent protein in Arabidopsis: Artifactual bulbs and native intravacuolar spherical structures. Plant Cell 2014, 26, 3416–3434. [Google Scholar] [CrossRef]
- Trullo, A.; Corti, V.; Arza, E.; Caiolfa, V.R.; Zamai, M. Application limits and data correction in number of molecules and brightness analysis. Microsc. Res. Tech. 2013, 76, 1135–1146. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Deng, X.; Luu, D.T.; Maurel, C.; Lin, J. Single-molecule fluorescence imaging to quantify membrane protein dynamics and oligomerization in living plant cells. Nat. Protoc. 2015, 10, 2054–2063. [Google Scholar] [CrossRef]
- Song, K.; Xue, Y.; Wang, X.; Wan, Y.; Deng, X.; Lin, J. A modified GFP facilitates counting membrane protein subunits by step-wise photobleaching in Arabidopsis. J. Plant Physiol. 2017, 213, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Wong-Ekkabut, J.; Karttunen, M. The good, the bad and the user in soft matter simulations. Biochim. Biophys. Acta 2016, 1858, 2529–2538. [Google Scholar] [CrossRef]
- Senechal, F.; Habrylo, O.; Hocq, L.; Domon, J.M.; Marcelo, P.; Lefebvre, V.; Pelloux, J.; Mercadante, D. Structural and dynamical characterization of the pH-dependence of the pectin methylesterase-pectin methylesterase inhibitor complex. J. Biol. Chem. 2017, 292, 21538–21547. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wan, Y.; Zhu, J.; Yu, Z.; Tian, X.; Han, J.; Zhang, Z.; Han, W. Theoretical study on zearalenol compounds binding with wild type zearalenone hydrolase and V153H mutant. Int. J. Mol. Sci. 2018, 19, 2808. [Google Scholar] [CrossRef]
- Leiderman, P.; Huppert, D.; Agmon, N. Transition in the temperature-dependence of GFP fluorescence: From proton wires to proton exit. Biophys. J. 2006, 90, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, R.; Wu, Y.; Song, M.; Li, J.; Yang, Q.; Chen, X.; Bao, J.; Zhao, Q. L1198F mutation resensitizes crizotinib to ALK by altering the conformation of inhibitor and ATP binding sites. Int. J. Mol. Sci. 2017, 18, 482. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery, C.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Li, X.; Luu, D.T.; Maurel, C.; Lin, J. Probing plasma membrane dynamics at the single-molecule level. Trends Plant Sci. 2013, 18, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xing, J.; Qiu, Z.; He, Q.; Lin, J. Quantification of membrane protein dynamics and interactions in plant cells by fluorescence correlation spectroscopy. Mol. Plant 2016, 9, 1229–1239. [Google Scholar] [CrossRef]
- Cui, Y.; Yu, M.; Yao, X.; Xing, J.; Lin, J.; Li, X. Single-particle tracking for the quantification of membrane protein dynamics in living plant cells. Mol. Plant 2018, 11, 1315–1327. [Google Scholar] [CrossRef]
- McGuire, H.; Aurousseau, M.R.; Bowie, D.; Blunck, R. Automating single subunit counting of membrane proteins in mammalian cells. J. Biol. Chem. 2012, 287, 35912–35921. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Yang, Y.; Li, R.; He, Q.; Fang, X.; Luu, D.T.; Maurel, C.; Lin, J. Single-molecule analysis of PIP2;1 dynamics and partitioning reveals multiple modes of Arabidopsis plasma membrane aquaporin regulation. Plant Cell 2011, 23, 3780–3897. [Google Scholar] [CrossRef]
- Leisle, L.; Chadda, R.; Lueck, J.D.; Infield, D.T.; Galpin, J.D.; Krishnamani, V.; Robertson, J.L.; Ahern, C.A. Cellular encoding of Cy dyes for single-molecule imaging. Elife 2016. [Google Scholar] [CrossRef] [PubMed]
- Stansfeld, P.J.; Sansom, M.S.P. Molecular simulation approaches to membrane proteins. Structure 2011, 19, 1562–1572. [Google Scholar] [CrossRef] [PubMed]
- Psachoulia, E.; Marshall, D.P.; Sansom, M.S.P. Molecular dynamics simulations of the dimerization of transmembrane α-helices. Acc. Chem. Res. 2010, 43, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Hummer, G.; Wikstrom, M. Molecular simulation and modeling of complex I. Biochim. Biophys. Acta 2016, 1857, 915–921. [Google Scholar] [CrossRef]
- Resh, M.D. Covalent lipid modifications of proteins. Curr. Biol. 2013, 23, R431–R435. [Google Scholar] [CrossRef]
- Martin, D.D.; Beauchamp, E.; Berthiaume, L.G. Post-translational myristoylation: Fat matters in cellular life and death. Biochimie 2011, 93, 18–31. [Google Scholar] [CrossRef]
- Sorek, N.; Bloch, D.; Yalovsky, S. Protein lipid modifications in signaling and subcellular targeting. Curr. Opin. Plant Biol. 2009, 12, 714–720. [Google Scholar] [CrossRef]
- Martinez, A.; Traverso, J.A.; Valot, B.; Ferro, M.; Espagne, C.; Ephritikhine, G.; Zivy, M.; Giglione, C.; Meinnel, T. Extent of N-terminal modifications in cytosolic proteins from eukaryotes. Proteomics 2008, 8, 2809–2831. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Khartabil, H.; Boisson, J.C.; Contreras-Garcia, J.; Piquemal, J.P.; Henon, E. The Independent Gradient Model: A new approach for probing strong and weak interactions in molecules from wave function calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Garcia, J.; Yang, W.; Johnson, E.R. Analysis of hydrogen-bond interaction potentials from the electron density: Integration of noncovalent interaction regions. J. Phys. Chem. A 2011, 115, 12983–12990. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.C.; Contreras-Garcia, J.; Henon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Meirovitch, H. Recent developments in methodologies for calculating the entropy and free energy of biological systems by computer simulation. Curr. Opin. Struct. Biol. 2007, 17, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Parenti, M.D.; Rastelli, G. Advances and applications of binding affinity prediction methods in drug discovery. Biotechnol. Adv. 2012, 30, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Swanson, J.M.J.; Henchman, R.H.; McCammon, J.A. Revisiting free energy calculations: A theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys. J. 2004, 86, 67–74. [Google Scholar] [CrossRef]
- Liu, F.F.; Dong, X.Y.; He, L.; Middelberg, A.P.; Sun, Y. Molecular insight into conformational transition of amyloid beta-peptide 42 inhibited by (-)-epigallocatechin-3-gallate probed by molecular simulations. J. Phys. Chem. B 2011, 115, 11879–11887. [Google Scholar] [CrossRef]
- Petukh, M.; Li, M.; Alexov, E. Predicting binding free energy change caused by point mutations with knowledge-modified MM/PBSA method. PLoS Comput. Biol. 2015, 11, e1004276. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, S.W.; Cho, A.E. Molecular insights into the adsorption mechanism of human beta-defensin-3 on bacterial membranes. Langmuir 2016, 32, 1782–1790. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GFP-A206 | GFP-K206 | Myristoylated-GFP–POPC | |
|---|---|---|---|
| ΔEelec | −252.8 ± 25.6 | −73.4 ± 72.5 | −286.4 ± 28.9 |
| ΔEvdW | −344.6 ± 17.5 | −89.3 ± 12.4 | −238.9 ± 35.3 |
| ΔGpolar | 736.2 ± 74.8 | 217.9 ± 103.2 | 341.5 ± 126.8 |
| ΔGnonpolar | −251.3 ± 49.4 | −18.1 ± 2.6 | −57.8 ± 6.8 |
| ΔGbinding | −112.5 ± 43.7 | 37.1 ± 15.4 | −241.6 ± 90.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Song, K.; Li, Y.; Tang, L.; Deng, X. Single-Molecule Imaging and Computational Microscopy Approaches Clarify the Mechanism of the Dimerization and Membrane Interactions of Green Fluorescent Protein. Int. J. Mol. Sci. 2019, 20, 1410. https://doi.org/10.3390/ijms20061410
Wang X, Song K, Li Y, Tang L, Deng X. Single-Molecule Imaging and Computational Microscopy Approaches Clarify the Mechanism of the Dimerization and Membrane Interactions of Green Fluorescent Protein. International Journal of Molecular Sciences. 2019; 20(6):1410. https://doi.org/10.3390/ijms20061410
Chicago/Turabian StyleWang, Xiaohua, Kai Song, Yang Li, Ling Tang, and Xin Deng. 2019. "Single-Molecule Imaging and Computational Microscopy Approaches Clarify the Mechanism of the Dimerization and Membrane Interactions of Green Fluorescent Protein" International Journal of Molecular Sciences 20, no. 6: 1410. https://doi.org/10.3390/ijms20061410
APA StyleWang, X., Song, K., Li, Y., Tang, L., & Deng, X. (2019). Single-Molecule Imaging and Computational Microscopy Approaches Clarify the Mechanism of the Dimerization and Membrane Interactions of Green Fluorescent Protein. International Journal of Molecular Sciences, 20(6), 1410. https://doi.org/10.3390/ijms20061410