Neutrophil Cell Shape Change: Mechanism and Signalling during Cell Spreading and Phagocytosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Biophysics of Cell Surface “Wrinkles”
3. Signalling the Release of Cell Surface Microridges/Wrinkles
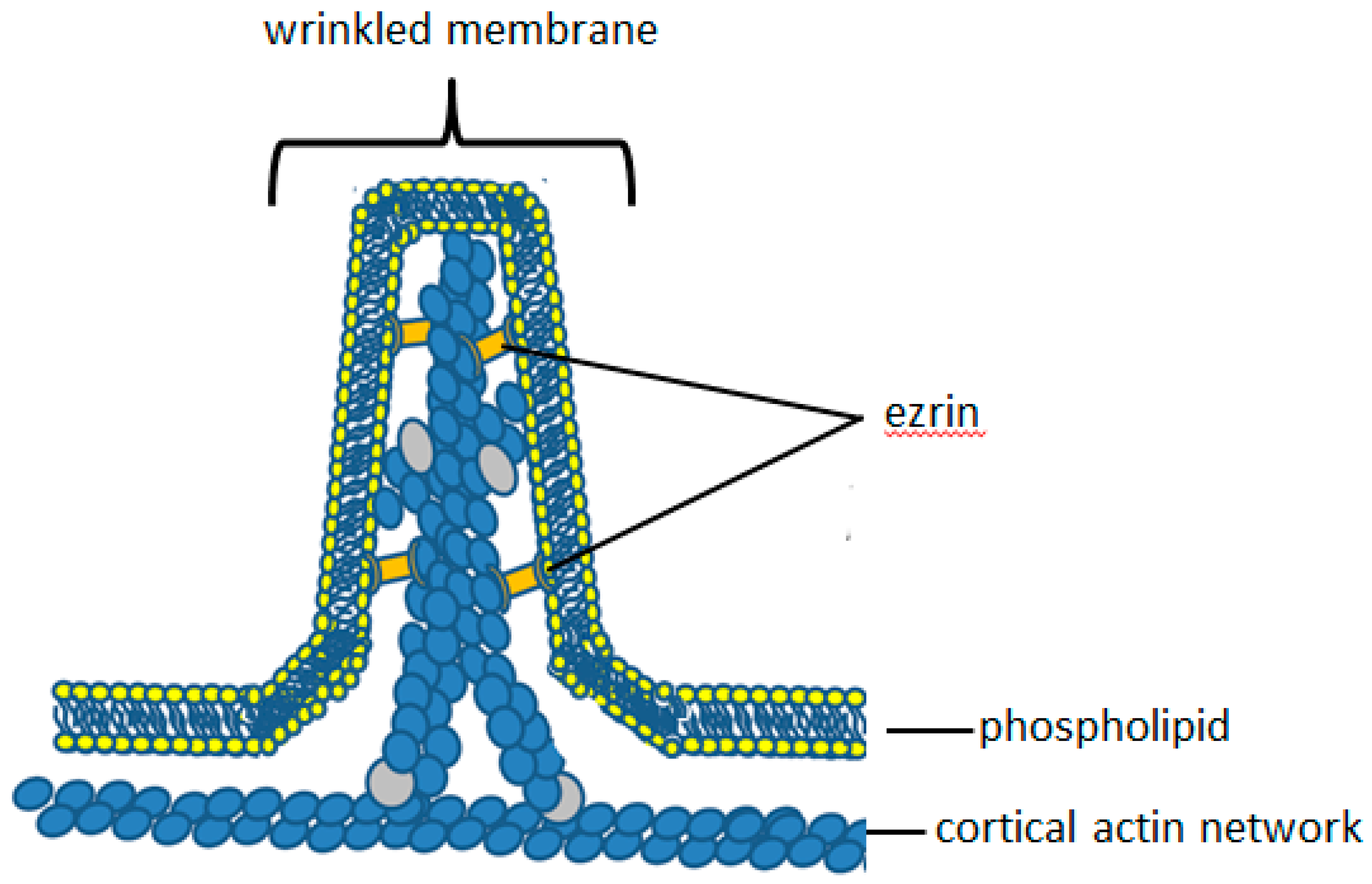
4. Molecular Anatomy of Plasma Membrane Microridges
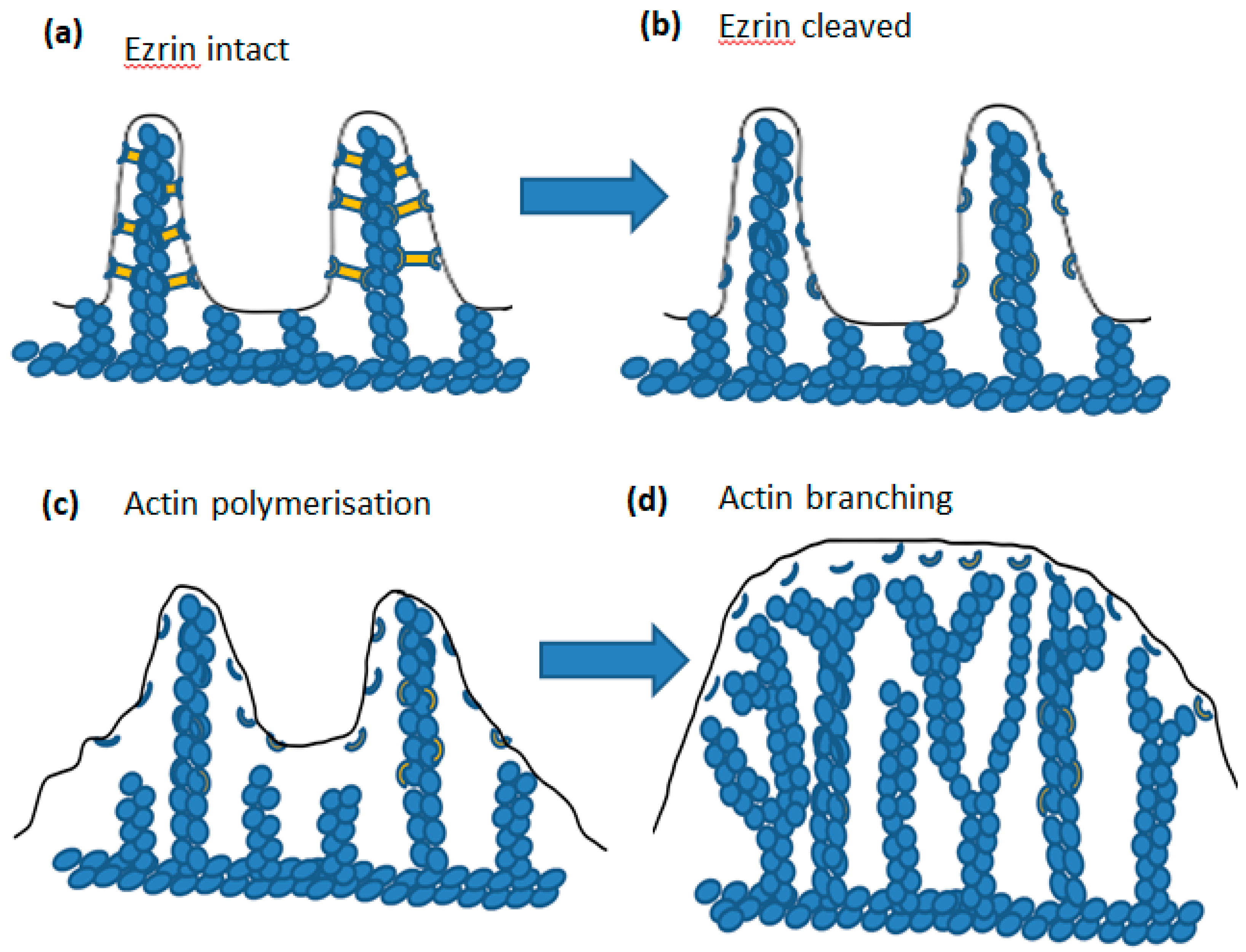
5. Ezrin
6. µ-Calpain
7. µ-Calpain Activation by Ca2+ in Neutrophils
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hallett, M.B.; Dewitt, S. Ironing out the wrinkles of neutrophil phagocytosis. Trends Cell Biol. 2007, 17, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Dewitt, S.; Hallett, M. Leukocyte membrane “expansion”: A central mechanism for leukocyte extravasation. J. Leukoc. Biol. 2017, 81, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Hamill, O.P.; Martinac, B. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 2001, 81, 685–740. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, E.; Duclos, S.; Rondeau, C.; Chevet, E.; Cameron, P.H.; Steele-Mortimer, O.; Paiement, J.; Bergeron, J.J.M.; Desjardins, M. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell 2002, 110, 119–131. [Google Scholar] [CrossRef]
- Bessis, M. Living Blood Cells and Their Ultrastructure; Springer: Berlin, Germany, 1973. [Google Scholar]
- Pettit, E.J.; Davies, E.V.; Hallett, M.B. The microanatomy of calcium stores in human neutrophils: Relationship of structure to function. Histol. Histopathol. 1997, 12, 479–490. [Google Scholar] [PubMed]
- Hallett, M.B.; von Ruhland, C.J.; Dewitt, S. Chemotaxis and the cell surface-area problem. Nat. Rev. Mol. Cell Biol. 2008, 9, 662. [Google Scholar] [CrossRef]
- Hogg, J.C. Neutrophil kinetics and lung injury. Physiol. Rev. 1987, 67, 1249–1295. [Google Scholar] [CrossRef]
- Ting-Beall, H.P.; Needham, D.; Hochmuth, R.M. Volume and osmotic properties of human neutrophils. Blood 1993, 81, 2774–2780. [Google Scholar] [PubMed]
- Evans, E.; Leung, A.; Zhelev, D. Synchrony of cell spreading and contraction force as phagocytes engulf large pathogens. J. Cell Biol. 1993, 122, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Herant, M.; Heinrich, V.; Dembo, M. Mechanics of neutrophil phagocytosis: Behavior of the cortical tension. J. Cell Sci. 2005, 118, 1789–1797. [Google Scholar] [CrossRef]
- Herant, M.; Heinrich, V.; Dembo, M. Mechanics of neutrophil phagocytosis: Experiments and quantitative models. J. Cell Sci. 2006, 119, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Herant, M.; Marganski, W.A.; Dembo, M. The mechanics of neutrophils: Synthetic modeling of three experiments. Biophys. J. 2003, 84, 3389–3413. [Google Scholar] [CrossRef]
- Pollard, T. Actin. Current. Opin. Cell Biol. 1990, 2, 33–40. [Google Scholar] [CrossRef]
- Stossel, T. The structure of cortical cytoplasm. Philo. Trans. R. Soc. Lond. Ser. B 1982, 299, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Peskin, C.S.; Odell, G.M.; Oster, G.F. Cellular Motions and Thermal Fluctuations: The Brownian Ratchet. Biophys. J. 1993, 65, 316–324. [Google Scholar] [CrossRef]
- Houk, A.R.; JIlkine, A.; Mejean, C.O.; Boltyanskiy, R.; Dufresne, E.R.; Angenent, S.B.; Altschuler, S.J.; Wu, L.F.; Weiner, O.D. Membrane Tension Maintains Cell Polarity by Confining Signals to the Leading Edge during Neutrophil Migration. Cell 2012, 148, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Francis, E.A.; Heinrich, V. Mechanistic Understanding of Single-Cell Behavior is Essential for Transformative Advances in Biomedicine. Yale J. Biol. Med. 2018, 91, 279–289. [Google Scholar]
- Al-Jumaa, M.A. The Control of the Surface Topography of Neutrophils. Ph.D. Thesis, Cardiff University, Cardiff, UK, 2018. [Google Scholar]
- Al Jumaa, M.A.; Dewitt, S.; Hallett, M.B. Topographical interrogation of the living cell surface reveals its role in rapid cell shape changes during phagocytosis and spreading. Sci. Rep. 2017, 77, 9790. [Google Scholar] [CrossRef]
- Kruskal, B.A.; Shak, S.; Maxfield, F.R. Spreading of human neutrophils is immediately preceded by a large increase in cytoplasmic free calcium. Proc. Natl. Acad. Sci. USA 1986, 83, 2919–2923. [Google Scholar] [CrossRef]
- Jaconi, M.E.; Theler, J.M.; Schlegel, W.; Appel, R.D.; Wright, S.D.; Lew, P.D. Multiple elevations of cytosolic-free Ca2+ in human neutrophils: Initiation by adherence receptors of the integrin family. J. Cell Biol. 1991, 112, 1249–1257. [Google Scholar] [CrossRef]
- NgSikorski, J.; Andersson, R.; Patarroyo, M.; Andersson, T. Calcium signalling capacity of the CD11b/CD18 integrin on human neutrophils. Exp. Cell Res. 1991, 195, 504–508. [Google Scholar] [CrossRef]
- Petersen, M.; Williams, J.D.; Hallett, M.B. Cross-linking of CD11b or CD18 signals release of localised Ca2+ from intracellular stores. Immunology 1993, 60, 157–159. [Google Scholar]
- Hellberg, C.; Molony, L.; Zheng, L.M.; Andersson, T. Ca2+ signalling mechanisms of the beta(2) integrin on neutrophils: Involvement of phospholipase C gamma 2 and Ins(1,4,5)P-3. Biochem. J. 1996, 317, 403–409. [Google Scholar] [CrossRef]
- Pettit, E.J.; Hallett, M.B. Release of ‘caged’ cytosolic Ca2+ triggers rapid spreading of human neutrophils adherent via integrin engagement. J. Cell Sci. 1998, 111, 2209–2215. [Google Scholar] [PubMed]
- Dewitt, S.; Francis, R.J.; Hallett, M.B. Ca2+ and calpain control membrane expansion during the rapid cell spreading of neutrophils. J. Cell Sci. 2013, 126, 4627–4635. [Google Scholar] [CrossRef] [PubMed]
- Dewitt, S.; Hallett, M.B. Cytosolic free Ca2+ changes and calpain activation are required for beta integrin-accelerated phagocytosis by human neutrophils. J. Cell Biol. 2002, 159, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef]
- Adams, S.E.; Parr, C.; Miller, D.J.; Allemann, R.K.; Hallett, M.B. Potent inhibition of Ca2+-dependent activation of calpain-1 by novel mercaptoacrylates. MedChemCommun 2012, 3, 566–570. [Google Scholar] [CrossRef]
- Adams, S.E.; Robinson, E.J.; Miller, D.J.; Hallett, M.B.; Allemann, R.K. Conformationally restricted calpain inhibitors. Chem. Sci. 2015, 6, 6865–6871. [Google Scholar] [CrossRef]
- Adams, S.E.; Rizkallah, P.J.; Miller, D.J.; Hallett, M.B.; Allemann, R. K The structural basis of differential inhibition of human calpain by indole and phenyl alpha-mercaptoacrylic acids. J. Struct. Biol. 2014, 187, 236–241. [Google Scholar] [CrossRef]
- Stewart, M.P.; McDowell, A.; Hogg, N. LFA-1-mediated adhesion is regulated by cytoskeletal restraint and by a Ca2+-dependent protease, calpain. J. Cell Biol. 1998, 140, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Leitinger, B.; McDowall, A.; Stanley, P.; Hogg, N. The regulation of integrin function by Ca2+. Biochim. Biophys. Acta 2000, 1498, 91–98. [Google Scholar] [CrossRef]
- Heinrich, V. Controlled One-on-One encounters between immune cells and microbes reveal mechanisms of phagocytosis. Biophys. J. 2015, 109, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Francis, E.A.; Heinrich, V. Extension of chemotactic pseudopods by nonadherent human neutrophils does not require or cause calcium bursts. Sci. Signal. 2018, 11, eaal4289. [Google Scholar] [CrossRef] [PubMed]
- Ishak, R.; Hallett, M.B. Defective rapid cell shape and transendothelial migration by calpain-1 null neutrophils. Biochem. Biophys. Res. Commun. 2018, 506, 1065–1070. [Google Scholar] [CrossRef]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell. Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Saotome, I.; Curto, M.; McClatchey, A.I. Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev. Cell 2004, 6, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, S.; Tsukita, S.; Tsukita, S. Direct involvement of ezrin/radixin/moesin (ERM)-binding membrane proteins in the organization of microvilli in collaboration with activated ERM proteins. J. Cell Biol. 1999, 145, 1497–1509. [Google Scholar] [CrossRef]
- Berryman, M.; Gary, R.; Bretscher, A. Ezrin oligomers are major cytoskeletal components of placental microvilli: A proposal for their involvement in cortical morphogenesis. J. Cell Biol. 1995, 131, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, A. Purification of an 80,000-dalton protein that is a component of the isolated microvillus cytoskeleton, and its localization in nonmuscle cells. J. Cell Biol. 1983, 97, 425–432. [Google Scholar] [CrossRef]
- Ivetic, A.; Ridley, A.J. Ezrin/radixin/moesin proteins and Rho GTPase signalling in leucocytes. Immunology 2004, 112, 165–176. [Google Scholar] [CrossRef]
- Shiue, H.; Musch, M.W.; Wang, Y.M.; Chang, E.B.; Turner, J.R. Akt2 phosphorylates ezrin to trigger NHE3 translocation and activation. J. Biol. Chem. 2005, 280, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Reinheckel, T.; North, J.A.; Li, R.; Bescos, P.B.; Shringarpure, R.; Davies, K.J.A. Ezrin turnover and cell shape changes catalyzed by proteasome in oxidatively stressed cells. FASEB J. 2002, 16, 1602–1610. [Google Scholar] [CrossRef]
- Gary, R.; Bretscher, A. Ezrin self-association involves binding of an N-terminal domain to a normally masked C-terminal domain that includes the F-actin binding site. Mol. Biol. Cell 1995, 6, 1061–1075. [Google Scholar] [CrossRef]
- Bretscher, A.; Reczek, D.; Berryman, M. Ezrin: A protein requiring conformational activation to link microfilaments to the plasma membrane in the assembly of cell surface structures. J. Cell Sci. 1997, 110, 3011–3018. [Google Scholar] [PubMed]
- Ohtani, K.; Sakamoto, H.; Rutherford, T.; Chen, Z.; Kikuchi, A.; Yamamoto, T.; Satoh, K.; Naftolin, F. Ezrin, a membrane-cytoskeletal linking protein, is highly expressed in atypical endometrial hyperplasia and uterine endometrioid adenocarcinoma. Cancer Lett. 2002, 179, 79–86. [Google Scholar] [CrossRef]
- Gautreau, A.; Louvard, D.; Arpin, M. Morphogenic effects of ezrin require a phosphorylation-induced transition from oligomers to monomers at the plasma membrane. J. Cell Biol. 2000, 150, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Fievet, B.T.; Gautreau, A.; Roy, C.; Del Maestro, L.; Mangeat, P.; Louvard, D.; Arpin, M. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J. Cell Biol. 2004, 164, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Viswanatha, R.; Wayt, J.; Ohouo, P.Y.; Smolka, M.B.; Bretscher, A. Interactome analysis reveals ezrin can adopt multiple conformational states. J. Biol. Chem. 2013, 288, 35437–35451. [Google Scholar] [CrossRef]
- Gould, K.L.; Bretscher, A.; Esch, F.S.; Hunter, T. cDNA cloning and sequencing of the protein-tyrosine kinase substrate, ezrin, reveals homology to band 4.1. EMBO J. 1989, 8, 4133–4142. [Google Scholar] [CrossRef]
- Correas, I.; Leto, T.L.; Speicher, D.W.; Marchesi, V.T. Identification of the functional site of erythrocyte protein 4.1 involved in spectrin-actin associations. J. Biol. Chem. 1986, 261, 3310–3315. [Google Scholar]
- Algrain, M.; Turunen, O.; Vaheri, A.; Louvard, D.; Arpin, M. Ezrin contains cytoskeleton and membrane binding domains accounting for its proposed role as a membrane-cytoskeletal linker. J. Cell Biol. 1993, 120, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Ivetic, A.; Deka, J.; Ridley, A.; Ager, A. The cytoplasmic tail of L-selectin interacts with members of the Ezrin-Radixin-Moesin (ERM) family of proteins: Cell activation-dependent binding of Moesin but not Ezrin. J. Biol. Chem. 2002, 277, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Erlansenm, S.L.; Hasslen, S.R.; Nelson, R.D. Detection and spatial distribution of beta-2 integrin (MAC-1) and L-selectin (LECAM-1) adherence receptors on human neutrophils by high resolution field emission SEM. J. Histochem. Cytochem. 1993, 41, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Kansas, G.F.; Ley, K.; Munro, J.M.; Tedder, T.F. Regukation of leukocyte rolling and adhesion to high endothelial venules through the cytoplasmic domain of L-selectin. J. Exp. Med. 1993, 177, 833–838. [Google Scholar] [CrossRef]
- Dwir, O.; Kansas, G.S.; Alon, R. Cytoplasmic anchorage of L-selectin controls leukocyte capture and rolling by increasing the mechanical stability of the selectin tether. J. Cell Biol. 2001, 155, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Barret, C.; Roy, C.; Montcourrier, P.; Mangeat, P.; Niggli, V. Mutagenesis of the phosphatidylinositol 4,5-bisphosphate (PIP2) binding site in the NH2-terminal domain of ezrin correlates with its altered cellular distribution. J. Cell Biol. 2002, 151, 1067–1080. [Google Scholar] [CrossRef]
- Gautreau, A.; Louvard, D.; Arpin, M. ERM proteins and NF2 tumor suppressor: The Yin and Yang of cortical actin organization and cell growth signaling. Curr. Opin Cell Biol. 2002, 14, 104–109. [Google Scholar] [CrossRef]
- Roberts, R.E. The μ-Calpain-Ezrin Axis: A Potential Target for Therapy in Inflammatory Disease. Ph.D. Thesis, Cardiff University, Cardiff, UK, 2017. [Google Scholar]
- Roberts, R.E.; Elumalai, G.L.; Hallett, M.B. Phagocytosis and motility in human neutrophils is competent but compromised by pharmacological inhibition of ezrin phosphorylation. Curr. Mol. Pharmacol. 2018, 11, 305–315. [Google Scholar] [CrossRef]
- Imajoh, S.; Aoki, K.; Ohno, S.; Emori, Y.; Kawasaki, H.; Sugihara, H.; Suzuki, K. Molecular cloning of the cDNA for the large subunit of the high-Ca2+-requiring form of human Ca2+-activated neutral protease. Biochemistry 1988, 27, 8122–8128. [Google Scholar] [CrossRef]
- Blanchard, H.; Grochulski, P.; Li, Y.; Arthur, J.S.; Davies, P.L.; Elce, J.S.; Cygler, M. Structure of a calpain Ca2+-binding domain reveals a novel EF-hand and Ca2+-induced conformational changes. Nat. Struct. Biol. 1997, 4, 532–538. [Google Scholar] [CrossRef]
- Ohno, S.; Emori, Y.; Suzuki, K. Nucleotide sequence of a cDNA coding for the small subunit of human calcium-dependent protease. Nucleic Acids Res. 1986, 14, 5559. [Google Scholar] [PubMed]
- Tompa, P.; Emori, Y.; Sorimachi, H.; Suzuki, K.; Friedrich, P. Domain III of calpain is a Ca2+-regulated phospholipid-binding domain. Biochem. Biophys. Res. Commun. 2001, 280, 1333–1339. [Google Scholar] [CrossRef]
- Shcherbina, A.; Bretscher, A.; Kenney, D.M.; Remold-O’Donnell, E. Moesin, the major ERM protein of lymphocytes and platelets, differs from ezrin in its insensitivity to calpain. FEBS Lett. 1999, 443, 31–36. [Google Scholar] [CrossRef]
- Shuster, C.B.; Herman, I.M. Indirect association of ezrin with F-actin: Isoform specificity and calcium sensitivity. J. Cell Biol. 1995, 128, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Thibodeau, A.; Forte, J.G. Ezrin-calpain I interactions in gastric parietal cells. Am. J. Physiol. 1993, 265, C36–C46. [Google Scholar] [CrossRef] [PubMed]
- Hallett, M.B.; Campbell, A.K. Measurement of changes in cytoplasmic free Ca2+ in fused cell hybrids. Nature 1982, 295, 155–158. [Google Scholar] [CrossRef]
- Dewitt, S.; Tian, W.; Hallett, M.B. Localised PtdIns(3,4,5)P-3 or PtdIns(3,4)P-2 at the phagocytic cup is required for both phagosome closure and Ca2+ signalling in HL60 neutrophils. J. Cell Sci. 2008, 119, 443–451. [Google Scholar] [CrossRef]
- Pettit, E.J.; Hallett, M.B. Early Ca2+ signalling events in neutrophils detected by rapid confocal laser scanning. Biochem. J. 1995, 310, 445–448. [Google Scholar] [CrossRef]
- Lewis, K.J. Control of Neutrophil Infiltration into Inflamed Tissue. the Role of μ-Calpain in Neutrophil Shape Change. Ph.D. Thesis, Cardiff University, Cardiff, UK, 2011. [Google Scholar]
- Gil-Parrado, S.; Popp, O.; Knoch, T.A.; Zahler, S.; Bestvater, F.; Felgentrager, M.; Holloschi, A.; Fernandez-Montalvan, A.; Auerswald, E.A.; Fritz, H.; et al. Subcellular localization and in vivo subunit interactions of ubiquitous mu-calpain. J. Biol. Chem. 2003, 278, 16336–16346. [Google Scholar] [CrossRef]
- Davies, E.V.; Hallett, M.B. Near membrane Ca2+ changes resulting from store release in neutrophils: Detection by FFP-18. Cell Calcium 1996, 19, 355–362. [Google Scholar] [CrossRef]
- Davies, E.V.; Hallett, M.B. High micromolar Ca2+ beneath the plasma membrane in stimulated neutrophils. Biochem. Biophys. Res. Commun. 1998, 248, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Omann, G.M.; Axelrod, D. Membrane-proximal calcium transients in stimulated neutrophils detected by total internal reflection fluorescence. Biophys. J. 1996, 71, 2885–2891. [Google Scholar] [CrossRef]
- Brasen, J.C.; Olsen, L.F.; Hallett, M.B. Cell surface topology creates high Ca2+ signalling microdomains. Cell Calcium 2010, 47, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Saido, T.C.; Suzuki, H.; Yamazaki, H.; Tanoue, K.; Suzuki, K. In situ capture of mu-calpain activation in platelets. J. Biol. Chem. 1993, 268, 7422–7426. [Google Scholar]
- Campbell, J.S.; Hallett, M.B. Active calpain in phagocytically competent human neutrophils: Electroinjection of fluorogenic calpain substrate. Biochem. Biophys. Res. Commun. 2015, 457, 341–346. [Google Scholar] [CrossRef]
- Lewis, K.J.; Masterman, B.; Laffafian, I.; Dewitt, S.; Campbell, J.S.; Hallett, M.B. Minimal impact electro-injection of cells undergoing dynamic shape change reveals calpain activation. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1182–1187. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; McDonald, M.C.; Mazzon, E.; Siriwardena, D.; Serraino, I.; Dugo, L.; Britti, D.; Mazzullo, G.; Caputi, A.P.; Thiemermann, C. Calpain inhibitor I reduces the development of acute and chronic inflammation. Am. J. Pathol. 2000, 157, 2065–2079. [Google Scholar] [CrossRef]
- Ikeda, Y.; Young, L.H.; Lefer, A.M. Attenuation of neutrophil-mediated myocardial ischemia-reperfusion injury by a calpain inhibitor. Am. J. Physiol. 2002, 282, H1421–H1426. [Google Scholar] [CrossRef]
- Marzocco, S.; Di Paola, R.; Autore, G.; Mazzon, E.; Pinto, A.; Caputi, A.P.; Thiemermann, C.; Cuzzocrea, S. Calpain inhibitor I reduces ischemia-reperfusion injury in the rat. Shock 2004, 21, 38–44. [Google Scholar] [CrossRef]
- Tissier, S.; Lancel, S.; Marechal, X.; Mordon, S.; Depontieu, F.; Scherpereel, A.; Chopin, C.; Neviere, R. Calpain inhibitors improve myocardial dysfunction and inflammation induced by endotoxin in rats. Shock 2004, 21, 352–357. [Google Scholar] [CrossRef]
- Yoshifuji, H.; Umehara, H.; Maruyama, H.; Itoh, M.; Tanaka, M.; Kawabata, D.; Fujii, T.; Mimori, T. Amelioration of experimental arthritis by a calpain-inhibitory compound: Regulation of cytokine production by E-64-d in vivo and in vitro. Int. Immunol. 2005, 1, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.K.; Todorovic, Z.; Sivarajah, A.; Mota-Filipe, H.; Brown, P.A.J.; Stewart, K.N.; Mazzon, E.; Cuzzocrea, S.; Thiemermann, C. Inhibitors of calpain activation (PD150606 and E-64) and renal ischemia-reperfusion injury. Biochem. Pharmacol. 2005, 69, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Donkar, I.O. Calpain inhibitors: A survey of compounds reported in the patent and scientific literature. Exp. Opin. Ther. Pat. 2011, 21, 601–636. [Google Scholar] [CrossRef]
- Miller, D.J.; Adams, S.E.; Hallett, M.B.; Allemann, R.K. Calpain-1 inhibitors for selective treatment of rheumatoid arthritis: What is the future? Future Med. Chem. 2013, 5, 2057–2074. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, R.E.; Hallett, M.B. Neutrophil Cell Shape Change: Mechanism and Signalling during Cell Spreading and Phagocytosis. Int. J. Mol. Sci. 2019, 20, 1383. https://doi.org/10.3390/ijms20061383
Roberts RE, Hallett MB. Neutrophil Cell Shape Change: Mechanism and Signalling during Cell Spreading and Phagocytosis. International Journal of Molecular Sciences. 2019; 20(6):1383. https://doi.org/10.3390/ijms20061383
Chicago/Turabian StyleRoberts, Rhiannon E., and Maurice B. Hallett. 2019. "Neutrophil Cell Shape Change: Mechanism and Signalling during Cell Spreading and Phagocytosis" International Journal of Molecular Sciences 20, no. 6: 1383. https://doi.org/10.3390/ijms20061383
APA StyleRoberts, R. E., & Hallett, M. B. (2019). Neutrophil Cell Shape Change: Mechanism and Signalling during Cell Spreading and Phagocytosis. International Journal of Molecular Sciences, 20(6), 1383. https://doi.org/10.3390/ijms20061383