Cellular Gene Expression during Hepatitis C Virus Replication as Revealed by Ribosome Profiling

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
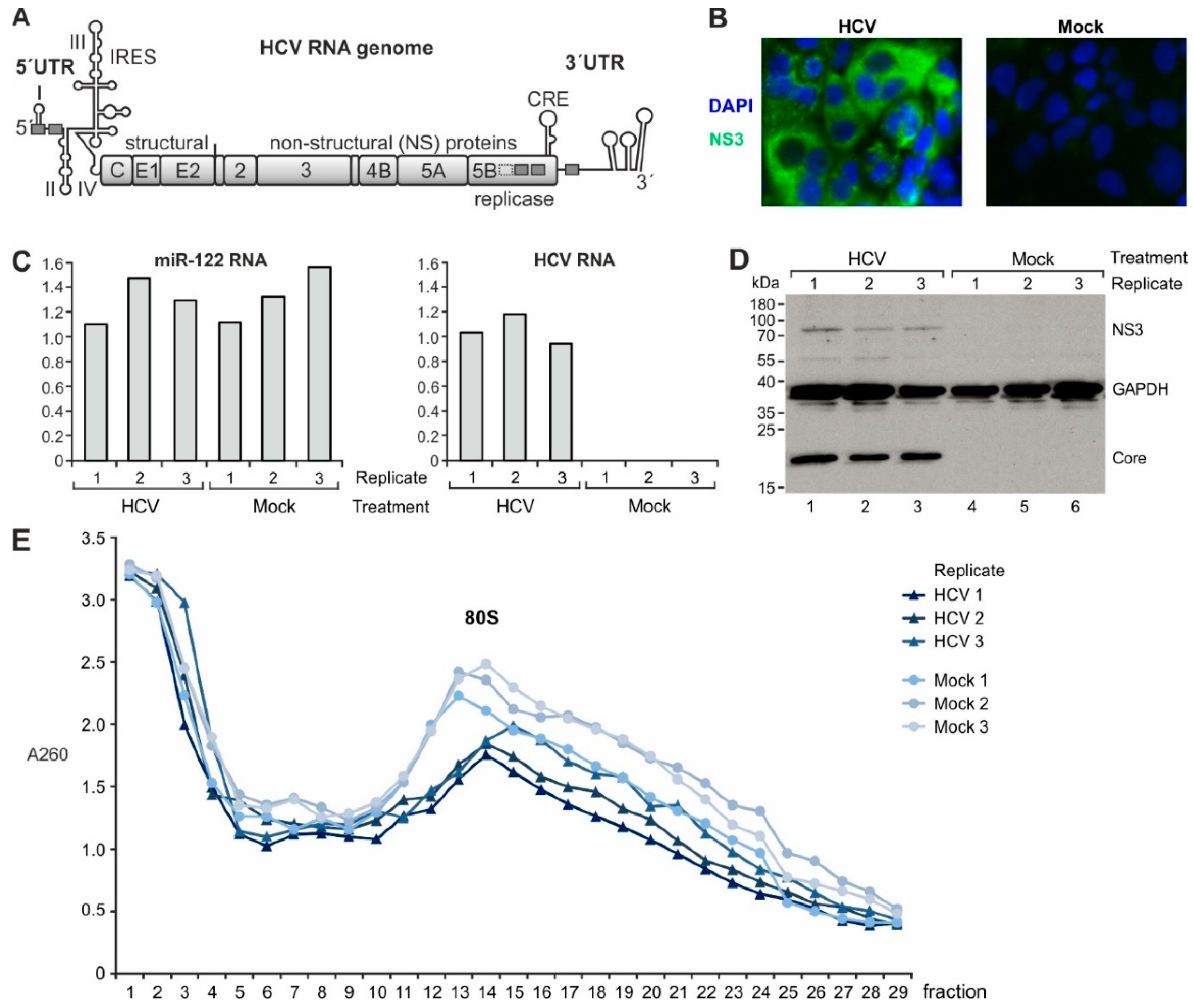
2.1. Gene Expression Analysis in Cells Replicating HCV
2.2. Established Viral Replication Does Not Cause Global Changes in Host Gene Expression
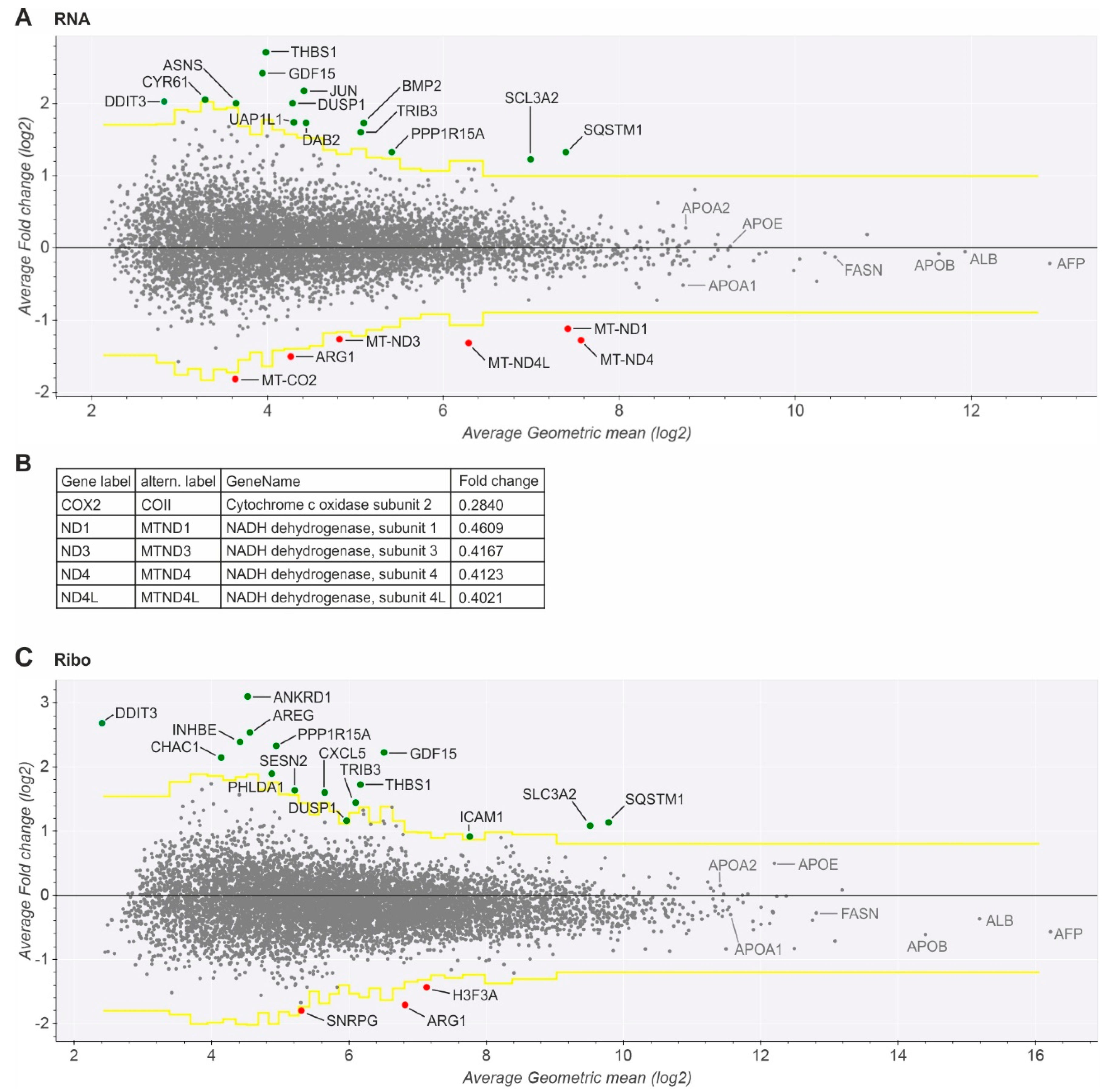
2.3. Key Mitochondrial Respiratory Chain Genes Are Downregulated
2.4. Upregulated Genes Are Related to ER Stress, HCV Replication, and Hepatocarcinogenesis
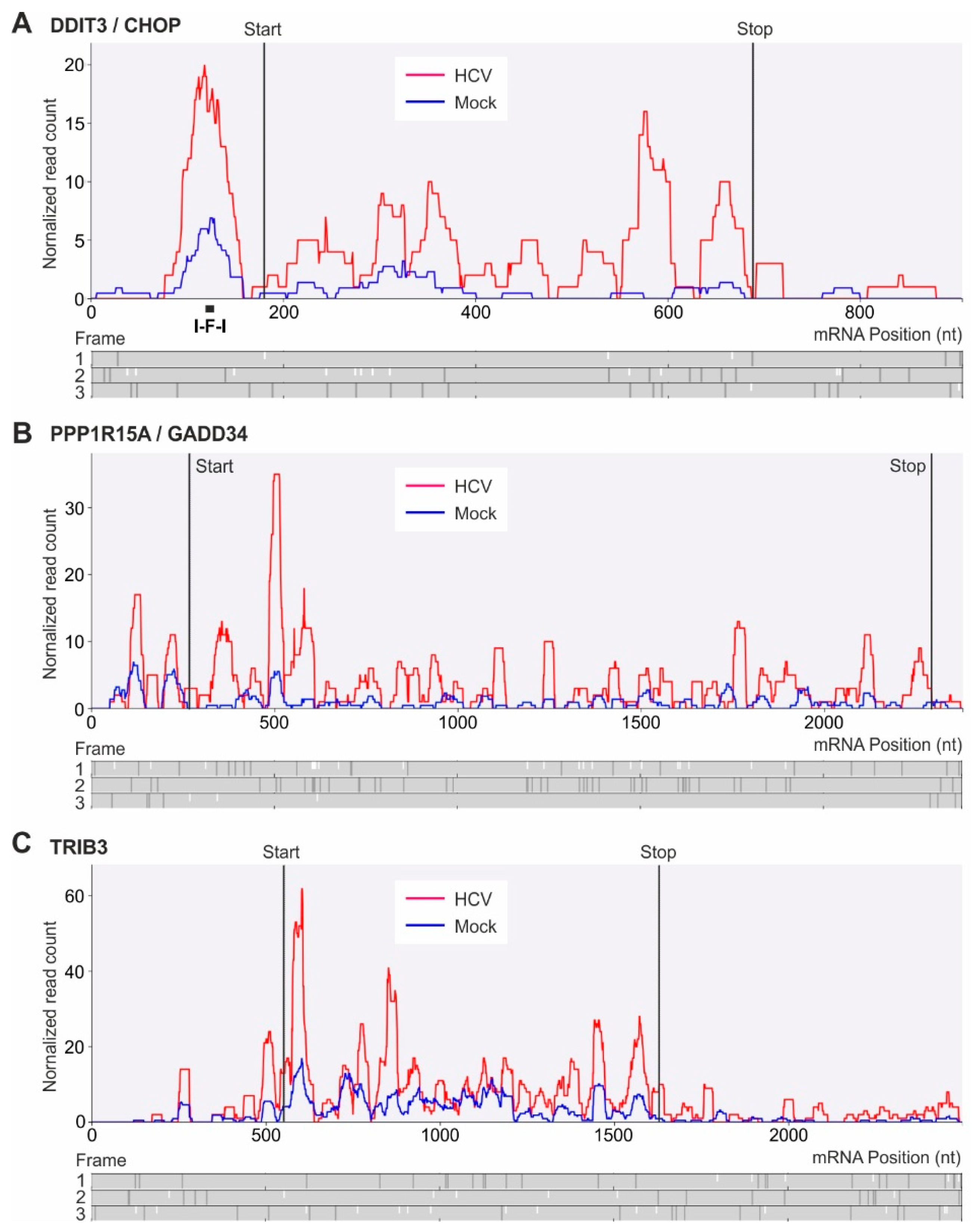
2.5. Translational Upregulation of mRNAs with Upstream Open Reading Frames
3. Conclusions
4. Material and Methods
4.1. Cell Culture
4.2. In Vitro Transcription of HCV-RNA
4.3. miR-122 Preparation
4.4. Transfection
4.5. RT-qPCR
4.6. Western Blot
4.7. Ribosome Profiling and RNA-Seq Library Preparation
4.8. Preprocessing of Reads and Principal Component Analysis
4.9. Trips-Viz Preprocessing and Mapping of Sequencing Data
4.10. Differential Expression Analysis
4.11. Metagene Analysis
4.12. Single Transcript Comparison Plots
4.13. Deposition of Sequences and of Expression Data
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miao, Z.; Xie, Z.; Miao, J.; Ran, J.; Feng, Y.; Xia, X. Regulated Entry of Hepatitis C Virus into Hepatocytes. Viruses 2017, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Yamane, D.; McGivern, D.R.; Masaki, T.; Lemon, S.M. Liver injury and disease pathogenesis in chronic hepatitis C. Curr. Top. Microbiol. Immunol. 2013, 369, 263–288. [Google Scholar] [PubMed]
- WHO. New Hepatitis Data Highlight Need for Urgent Global Response; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Waziry, R.; Hajarizadeh, B.; Grebely, J.; Amin, J.; Law, M.; Danta, M.; George, J.; Dore, G.J. Hepatocellular carcinoma risk following direct-acting antiviral HCV therapy: A systematic review, meta-analyses, and meta-regression. J. Hepatol. 2017, 67, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar] [PubMed]
- Perales, C.; Quer, J.; Gregori, J.; Esteban, J.I.; Domingo, E. Resistance of Hepatitis C Virus to Inhibitors: Complexity and Clinical Implications. Viruses 2015, 7, 5746–5766. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Baumert, T.F.; Bukh, J.; Houghton, M.; Lemon, S.M.; Lindenbach, B.D.; Lohmann, V.; Moradpour, D.; Pietschmann, T.; Rice, C.M.; et al. Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: Considerations for scientists and funding agencies. Virus Res. 2018, 248, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wimmer, E.; Paul, A.V. Cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim. Biophys. Acta 2009, 1789, 495–517. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C virus RNA replication and assembly: Living on the fat of the land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M. Internal translation initiation of picornaviruses and hepatitis C virus. Biochim. Biophys. Acta 2009, 1789, 529–541. [Google Scholar] [CrossRef]
- Strating, J.R.; van Kuppeveld, F.J. Viral rewiring of cellular lipid metabolism to create membranous replication compartments. Curr. Opin. Cell Biol. 2017, 47, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schüttler, C.G.; Fehr, C.; Jünemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V. Hepatitis C Virus RNA Replication. Curr. Top. Microbiol. Immunol. 2013, 369, 167–198. [Google Scholar] [PubMed]
- Fricke, M.; Dünnes, N.; Zayas, M.; Bartenschlager, R.; Niepmann, M.; Marz, M. Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA 2015, 21, 1219–1232. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M.; Shalamova, L.A.; Gerresheim, G.K.; Rossbach, O. Signals Involved in Regulation of Hepatitis C Virus RNA Genome Translation and Replication. Front. Microbiol. 2018, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 2005, 79, 13594–13605. [Google Scholar] [CrossRef] [PubMed]
- Gerold, G.; Bruening, J.; Weigel, B.; Pietschmann, T. Protein Interactions during the Flavivirus and Hepacivirus Life Cycle. Mol. Cell. Proteom. 2017, 16, S75–S91. [Google Scholar] [CrossRef] [PubMed]
- Wrensch, F.; Crouchet, E.; Ligat, G.; Zeisel, M.B.; Keck, Z.Y.; Foung, S.K.H.; Schuster, C.; Baumert, T.F. Hepatitis C Virus (HCV)-Apolipoprotein Interactions and Immune Evasion and Their Impact on HCV Vaccine Design. Front. Immunol. 2018, 9, 1436. [Google Scholar] [CrossRef]
- Ruggieri, A.; Dazert, E.; Metz, P.; Hofmann, S.; Bergeest, J.P.; Mazur, J.; Bankhead, P.; Hiet, M.S.; Kallis, S.; Alvisi, G.; et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 2012, 12, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Binder, M.; Bartenschlager, R. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol. Rev. 2012, 36, 663–683. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61, S14–S25. [Google Scholar] [CrossRef]
- Sun, J.; Rajsbaum, R.; Yi, M. Immune and non-immune responses to hepatitis C virus infection. World J. Gastroenterol. 2015, 21, 10739–10748. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Lemon, S.M. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 2011, 30, 1969–1983. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.W.; Jang, J.Y.; Chung, R.T. Hepatitis C virus and hepatocarcinogenesis. Clin. Mol. Hepatol. 2012, 18, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Billie Bian, C.; Hoshida, Y.; Baumert, T.F.; Zeisel, M.B. Chronic hepatitis C virus infection and pathogenesis of hepatocellular carcinoma. Curr. Opin. Virol. 2016, 20, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef]
- Piccoli, C.; Quarato, G.; Ripoli, M.; D’Aprile, A.; Scrima, R.; Cela, O.; Boffoli, D.; Moradpour, D.; Capitanio, N. HCV infection induces mitochondrial bioenergetic unbalance: Causes and effects. Biochim. Biophys. Acta 2009, 1787, 539–546. [Google Scholar] [CrossRef]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta 2015, 1853, 2822–2833. [Google Scholar] [CrossRef]
- Hara, Y.; Yanatori, I.; Ikeda, M.; Kiyokage, E.; Nishina, S.; Tomiyama, Y.; Toida, K.; Kishi, F.; Kato, N.; Imamura, M.; et al. Hepatitis C virus core protein suppresses mitophagy by interacting with parkin in the context of mitochondrial depolarization. Am. J. Pathol. 2014, 184, 3026–3039. [Google Scholar] [CrossRef]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.L.; et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 2006, 103, 7408–7413. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Brar, G.A.; Rouskin, S.; McGeachy, A.M.; Weissman, J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012, 7, 1534–1550. [Google Scholar] [CrossRef]
- Andreev, D.E.; O’Connor, P.B.; Fahey, C.; Kenny, E.M.; Terenin, I.M.; Dmitriev, S.E.; Cormican, P.; Morris, D.W.; Shatsky, I.N.; Baranov, P.V. Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. eLife 2015, 4, e03971. [Google Scholar] [CrossRef]
- Tuller, T.; Carmi, A.; Vestsigian, K.; Navon, S.; Dorfan, Y.; Zaborske, J.; Pan, T.; Dahan, O.; Furman, I.; Pilpel, Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 2010, 141, 344–354. [Google Scholar] [CrossRef]
- Kiniry, S.J.; O’Connor, P.B.F.; Michel, A.M.; Baranov, P.V. Trips-Viz: A transcriptome browser for exploring Ribo-Seq data. Nucleic Acids Res. 2019, 47, D847–D852. [Google Scholar] [CrossRef]
- Chen, H.; Egan, J.O.; Chiu, J.F. Regulation and activities of alpha-fetoprotein. Crit. Rev. Eukaryot. Gene Expr. 1997, 7, 11–41. [Google Scholar] [CrossRef]
- Wang, A.B.; Liu, D.P.; Liang, C.C. Regulation of human apolipoprotein B gene expression at multiple levels. Exp. Cell Res. 2003, 290, 1–12. [Google Scholar] [CrossRef]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef]
- Lang, B.F.; Gray, M.W.; Burger, G. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 1999, 33, 351–397. [Google Scholar] [CrossRef]
- Wirth, C.; Brandt, U.; Hunte, C.; Zickermann, V. Structure and function of mitochondrial complex I. Biochim. Biophys. Acta 2016, 1857, 902–914. [Google Scholar] [CrossRef]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef]
- Allen, J.F. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef]
- Fricke, M.; Gerst, R.; Ibrahim, B.; Niepmann, M.; Marz, M. Global importance of RNA secondary structures in protein coding sequences. Bioinformatics 2018, 35, 579–583. [Google Scholar] [CrossRef]
- Herling, A.; Konig, M.; Bulik, S.; Holzhutter, H.G. Enzymatic features of the glucose metabolism in tumor cells. FEBS J. 2011, 278, 2436–2459. [Google Scholar] [CrossRef]
- Bensinger, S.J.; Christofk, H.R. New aspects of the Warburg effect in cancer cell biology. Semin. Cell Dev. Biol. 2012, 23, 352–361. [Google Scholar] [CrossRef]
- Cuezva, J.M.; Krajewska, M.; de Heredia, M.L.; Krajewski, S.; Santamaria, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The bioenergetic signature of cancer: A marker of tumor progression. Cancer Res. 2002, 62, 6674–6681. [Google Scholar]
- Lopez-Rios, F.; Sanchez-Arago, M.; Garcia-Garcia, E.; Ortega, A.D.; Berrendero, J.R.; Pozo-Rodriguez, F.; Lopez-Encuentra, A.; Ballestin, C.; Cuezva, J.M. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res. 2007, 67, 9013–9017. [Google Scholar] [CrossRef]
- Finkielstein, C.V.; Capelluto, D.G. Disabled-2: A modular scaffold protein with multifaceted functions in signaling. Bioessays 2016, 38 (Suppl. 1), S45–S55. [Google Scholar] [CrossRef] [PubMed]
- Than, T.T.; Tran, G.V.; Son, K.; Park, E.M.; Kim, S.; Lim, Y.S.; Hwang, S.B. Ankyrin Repeat Domain 1 is Up-regulated During Hepatitis C Virus Infection and Regulates Hepatitis C Virus Entry. Sci. Rep. 2016, 6, 20819. [Google Scholar] [CrossRef] [PubMed]
- Gjymishka, A.; Su, N.; Kilberg, M.S. Transcriptional induction of the human asparagine synthetase gene during the unfolded protein response does not require the ATF6 and IRE1/XBP1 arms of the pathway. Biochem. J. 2009, 417, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Ochiai, K.; Kondo, S.; Tsumagari, K.; Murakami, T.; Cavener, D.R.; Imaizumi, K. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J. Biol. Chem. 2011, 286, 4809–4818. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Corre, J.; Hebraud, B.; Bourin, P. Concise review: Growth differentiation factor 15 in pathology: A clinical role? Stem Cells Transl. Med. 2013, 2, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Brüning, A.; Matsingou, C.; Brem, G.J.; Rahmeh, M.; Mylonas, I. Inhibin beta E is upregulated by drug-induced endoplasmic reticulum stress as a transcriptional target gene of ATF4. Toxicol. Appl. Pharmacol. 2012, 264, 300–304. [Google Scholar] [CrossRef]
- Chabicovsky, M.; Herkner, K.; Rossmanith, W. Overexpression of activin beta(C) or activin beta(E) in the mouse liver inhibits regenerative deoxyribonucleic acid synthesis of hepatic cells. Endocrinology 2003, 144, 3497–3504. [Google Scholar] [CrossRef]
- Rhee, S.G.; Bae, S.H. The antioxidant function of sestrins is mediated by promotion of autophagic degradation of Keap1 and Nrf2 activation and by inhibition of mTORC1. Free Radic. Biol. Med. 2015, 88, 205–211. [Google Scholar] [CrossRef]
- Liu, C.; Li, X.; Li, C.; Zhang, Z.; Gao, X.; Jia, Z.; Chen, H.; Jia, Q.; Zhao, X.; Liu, J.; et al. SLC3A2 is a novel endoplasmic reticulum stress-related signaling protein that regulates the unfolded protein response and apoptosis. PLoS ONE 2018, 13, e0208993. [Google Scholar] [CrossRef]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Tan, T.H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.A. Pleckstrin homology-like domain, family A, member 1 (PHLDA1) and cancer. Biomed. Rep. 2016, 4, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A. The split protein phosphatase system. Biochem. J. 2018, 475, 3707–3723. [Google Scholar] [CrossRef]
- Ord, T.; Ord, D.; Koivomagi, M.; Juhkam, K. Human TRB3 is upregulated in stressed cells by the induction of translationally efficient mRNA containing a truncated 5′-UTR. Gene 2009, 444, 24–32. [Google Scholar] [CrossRef]
- Tran, S.C.; Pham, T.M.; Nguyen, L.N.; Park, E.M.; Lim, Y.S.; Hwang, S.B. Nonstructural 3 Protein of Hepatitis C Virus Modulates the Tribbles Homolog 3/Akt Signaling Pathway for Persistent Viral Infection. J. Virol. 2016, 90, 7231–7247. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Zhao, H.; Wang, J.; Zhang, Q. CXCL5 secreted from adipose tissue-derived stem cells promotes cancer cell proliferation. Oncol. Lett. 2018, 15, 1403–1410. [Google Scholar]
- Menendez, J.A.; Vellon, L.; Espinoza, I.; Lupu, R. The metastasis inducer CCN1 (CYR61) activates the fatty acid synthase (FASN)-driven lipogenic phenotype in breast cancer cells. Oncoscience 2016, 3, 242–257. [Google Scholar]
- Meng, Q.; Xia, Y. c-Jun, at the crossroad of the signaling network. Protein Cell 2011, 2, 889–898. [Google Scholar] [CrossRef]
- Huang, T.; Sun, L.; Yuan, X.; Qiu, H. Thrombospondin-1 is a multifaceted player in tumor progression. Oncotarget 2017, 8, 84546–84558. [Google Scholar] [CrossRef]
- Pei, R.; Chen, H.; Lu, L.; Zhu, W.; Beckebaum, S.; Cicinnati, V.; Lu, M.; Chen, X. Hepatitis C virus infection induces the expression of amphiregulin, a factor related to the activation of cellular survival pathways and required for efficient viral assembly. J. Gen. Virol. 2011, 92, 2237–2248. [Google Scholar] [CrossRef]
- Lai, C.Y.; Liu, H.; Tin, K.X.; Huang, Y.; Yeh, K.H.; Peng, H.W.; Chen, H.D.; He, J.Y.; Chiang, Y.J.; Liu, C.S.; et al. Identification of UAP1L1 as a critical factor for protein O-GlcNAcylation and cell proliferation in human hepatoma cells. Oncogene 2019, 38, 317–331. [Google Scholar] [CrossRef]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef]
- Palam, L.R.; Baird, T.D.; Wek, R.C. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem. 2011, 286, 10939–10949. [Google Scholar] [CrossRef]
- Young, S.K.; Palam, L.R.; Wu, C.; Sachs, M.S.; Wek, R.C. Ribosome Elongation Stall Directs Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 6546–6558. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Cevallos, R.C.; Jan, E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J. Biol. Chem. 2009, 284, 6661–6673. [Google Scholar] [CrossRef]
- Young, S.K.; Willy, J.A.; Wu, C.; Sachs, M.S.; Wek, R.C. Ribosome Reinitiation Directs Gene-specific Translation and Regulates the Integrated Stress Response. J. Biol. Chem. 2015, 290, 28257–28271. [Google Scholar] [CrossRef]
- Young, S.K.; Wek, R.C. Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef]
- Gerresheim, G.K.; Dünnes, N.; Nieder-Röhrmann, A.; Shalamova, L.A.; Fricke, M.; Hofacker, I.; Höner Zu Siederdissen, C.; Marz, M.; Niepmann, M. microRNA-122 target sites in the hepatitis C virus RNA NS5B coding region and 3′ untranslated region: Function in replication and influence of RNA secondary structure. Cell. Mol. Life Sci. 2017, 74, 747–760. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Michel, A.M.; Mullan, J.P.; Velayudhan, V.; O’Connor, P.B.; Donohue, C.A.; Baranov, P.V. RiboGalaxy: A browser based platform for the alignment, analysis and visualization of ribosome profiling data. RNA Biol. 2016, 13, 316–319. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Rodriguez-Rivas, J.; Di Domenico, T.; Vázquez, J.; Valencia, A.; Tress, M.L. APPRIS 2017: principal isoforms for multiple gene sets. Nucleic Acids Res. 2018, 46, D213–D217. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerresheim, G.K.; Bathke, J.; Michel, A.M.; Andreev, D.E.; Shalamova, L.A.; Rossbach, O.; Hu, P.; Glebe, D.; Fricke, M.; Marz, M.; et al. Cellular Gene Expression during Hepatitis C Virus Replication as Revealed by Ribosome Profiling. Int. J. Mol. Sci. 2019, 20, 1321. https://doi.org/10.3390/ijms20061321
Gerresheim GK, Bathke J, Michel AM, Andreev DE, Shalamova LA, Rossbach O, Hu P, Glebe D, Fricke M, Marz M, et al. Cellular Gene Expression during Hepatitis C Virus Replication as Revealed by Ribosome Profiling. International Journal of Molecular Sciences. 2019; 20(6):1321. https://doi.org/10.3390/ijms20061321
Chicago/Turabian StyleGerresheim, Gesche K., Jochen Bathke, Audrey M. Michel, Dmitri E. Andreev, Lyudmila A. Shalamova, Oliver Rossbach, Pan Hu, Dieter Glebe, Markus Fricke, Manja Marz, and et al. 2019. "Cellular Gene Expression during Hepatitis C Virus Replication as Revealed by Ribosome Profiling" International Journal of Molecular Sciences 20, no. 6: 1321. https://doi.org/10.3390/ijms20061321
APA StyleGerresheim, G. K., Bathke, J., Michel, A. M., Andreev, D. E., Shalamova, L. A., Rossbach, O., Hu, P., Glebe, D., Fricke, M., Marz, M., Goesmann, A., Kiniry, S. J., Baranov, P. V., Shatsky, I. N., & Niepmann, M. (2019). Cellular Gene Expression during Hepatitis C Virus Replication as Revealed by Ribosome Profiling. International Journal of Molecular Sciences, 20(6), 1321. https://doi.org/10.3390/ijms20061321