Hybrid cis-stilbene Molecules: Novel Anticancer Agents



Abstract
1. Introduction
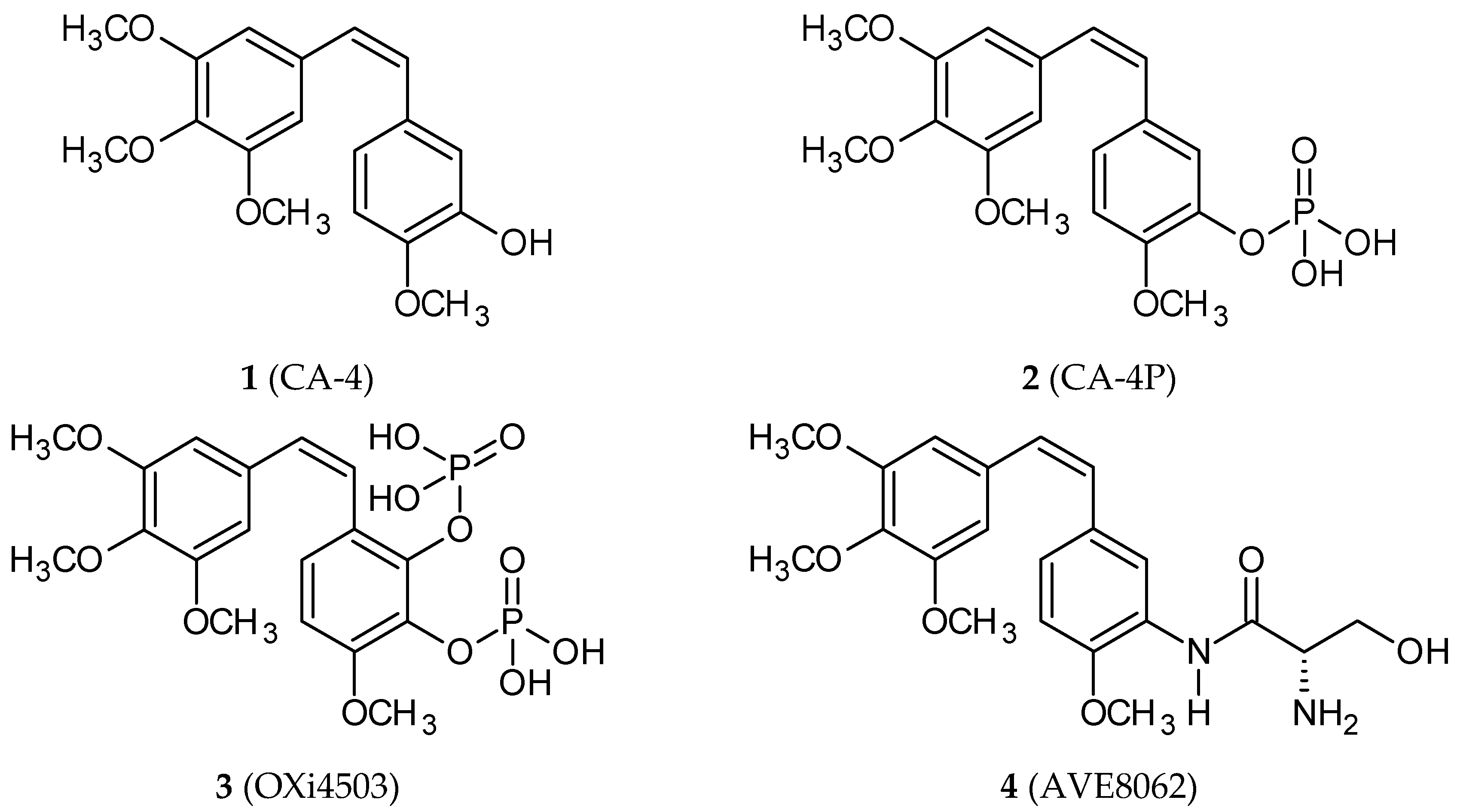
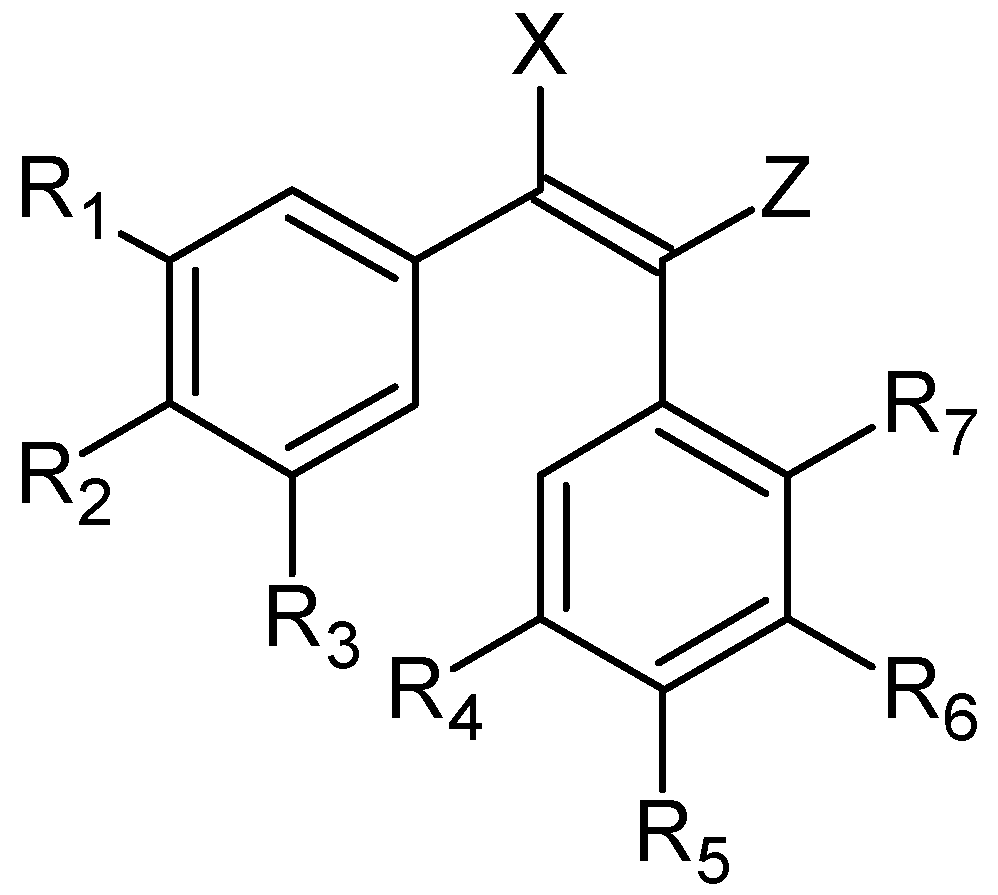
2. Combretastatins: Structure and Activity
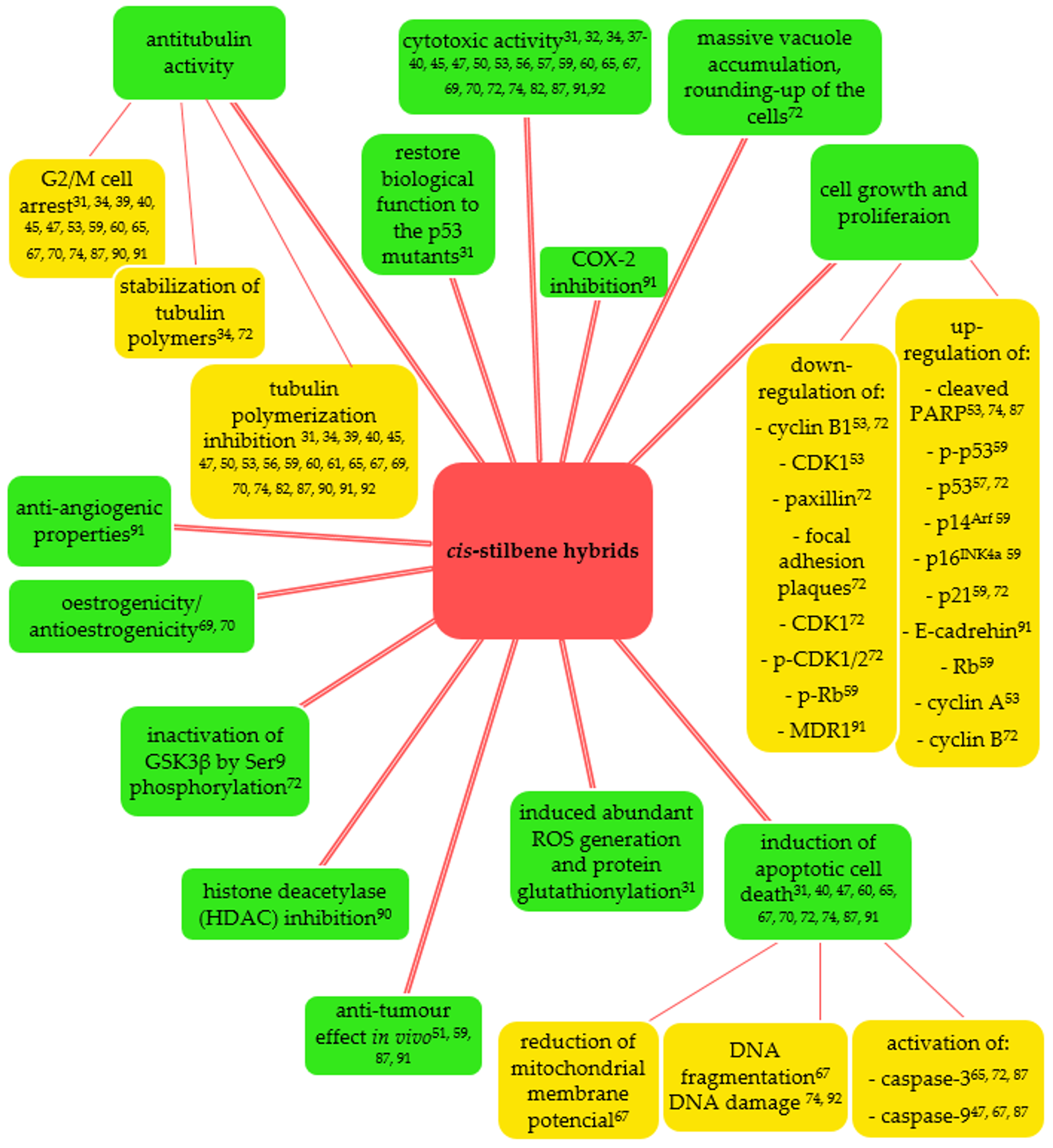
3. cis-Stilbenoid-Related Hybrids and Their Anticancer Activities
3.1. Combretastatin A-4 Based Hybrids
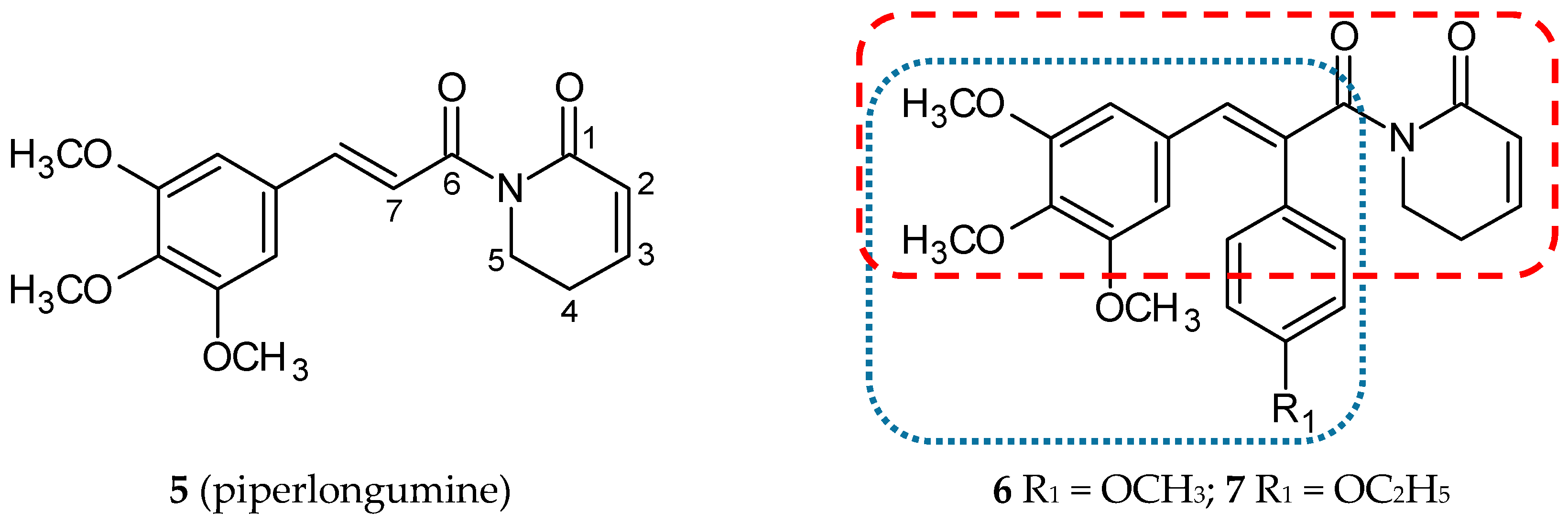
3.1.1. Piperlongumine Hybrids
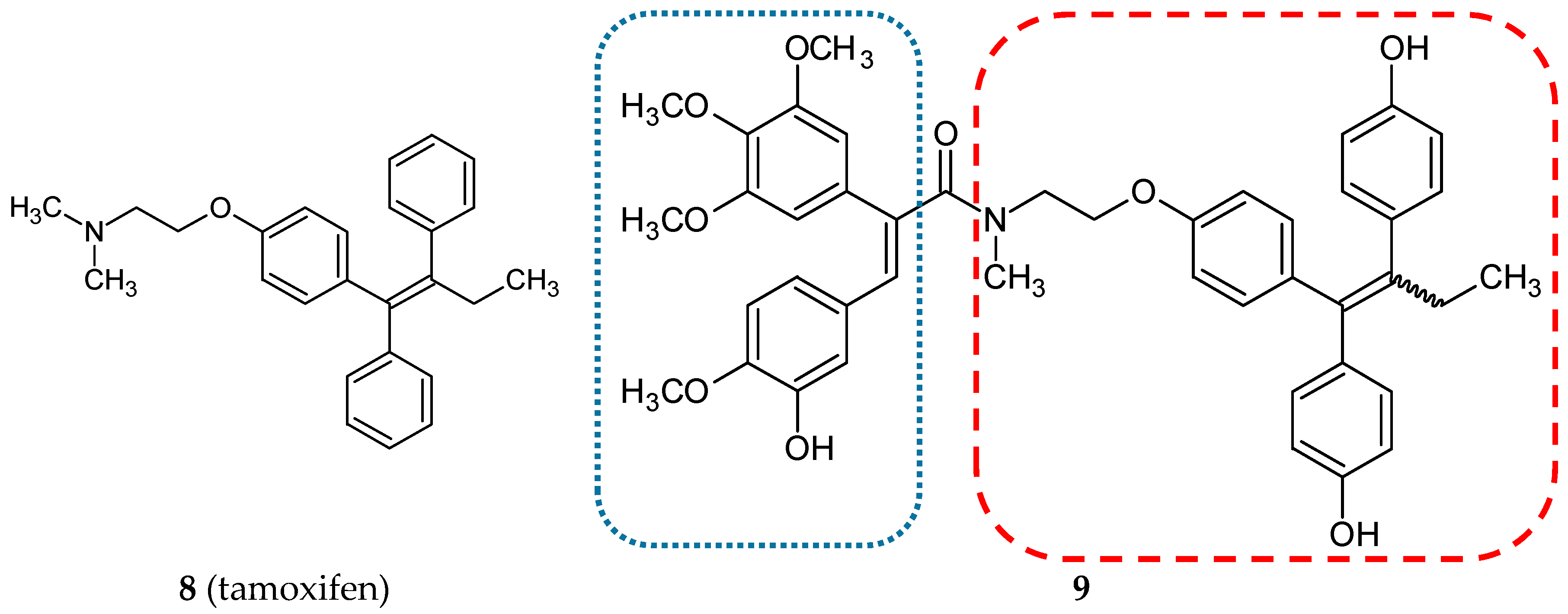
3.1.2. Estrogen Receptor Modulators
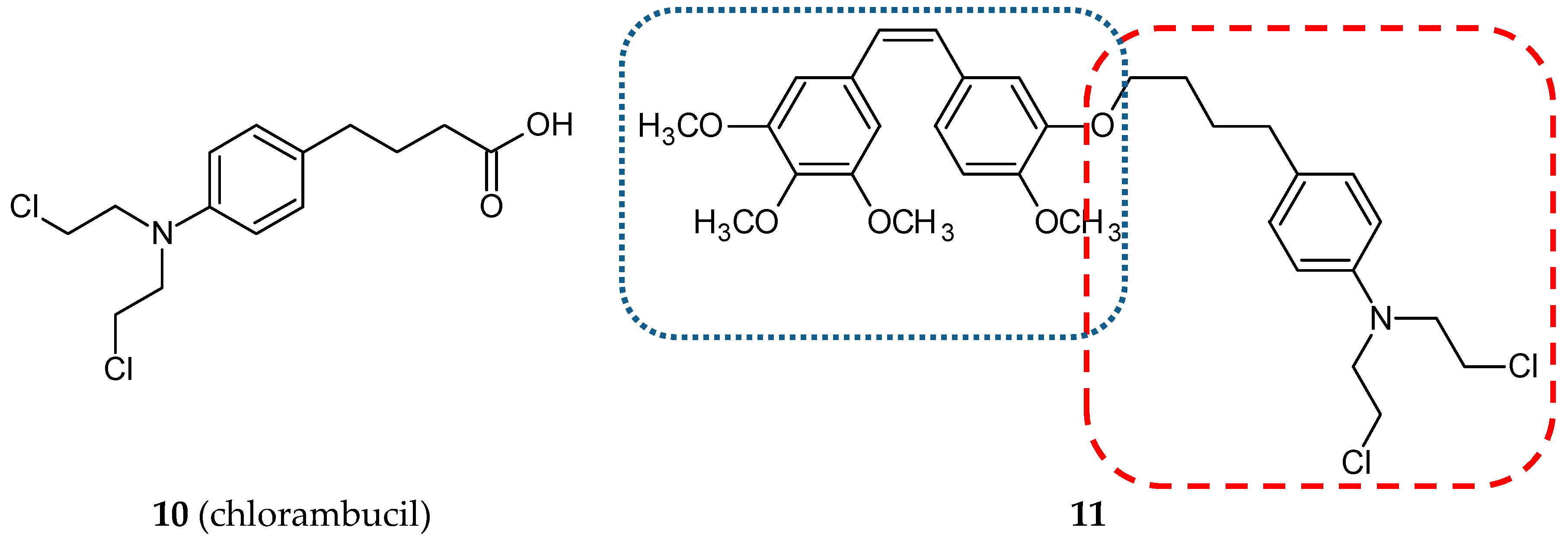
3.1.3. Nitrogen Mustard Hybrids
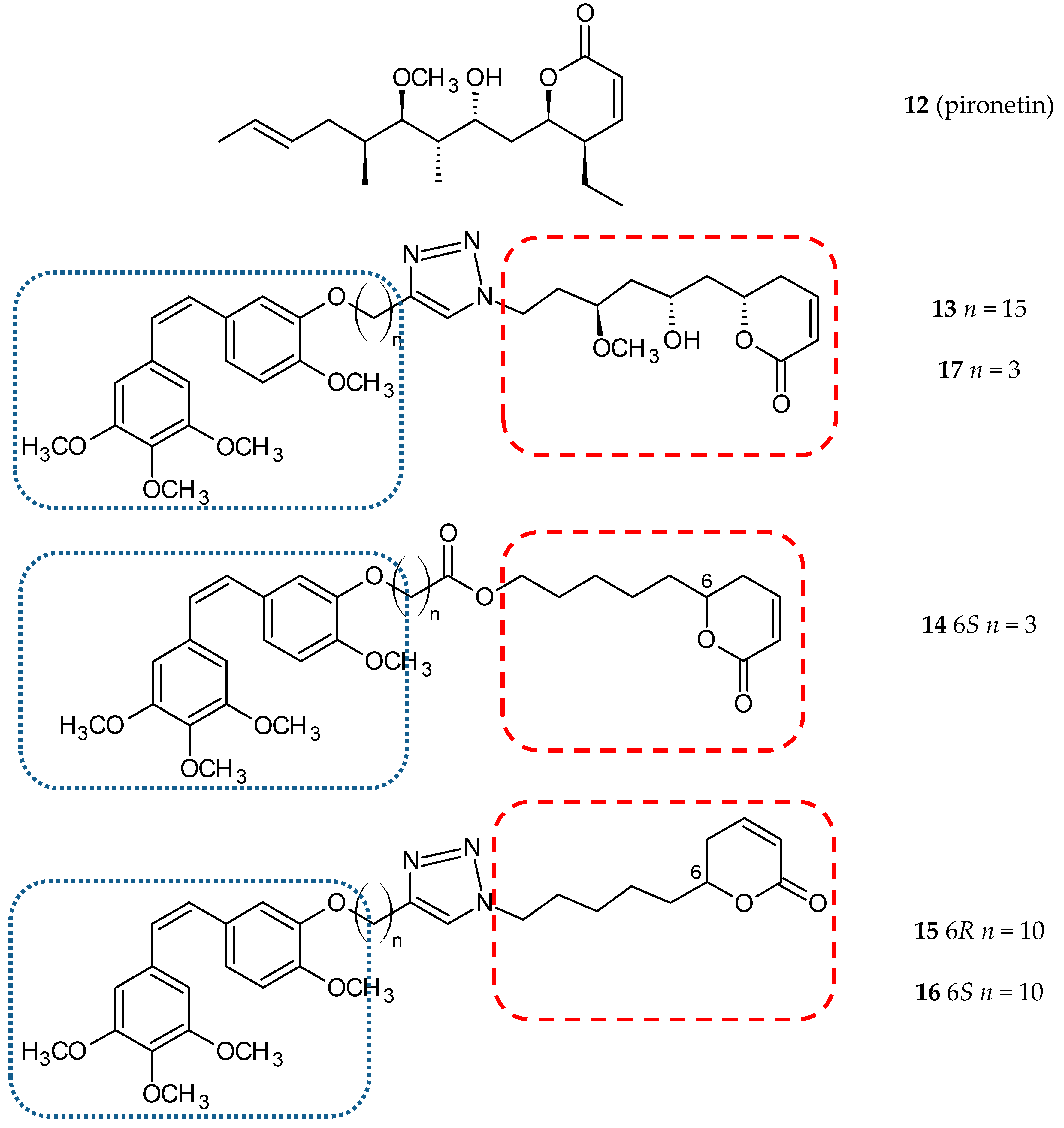
3.1.4. Pironetin Hybrids
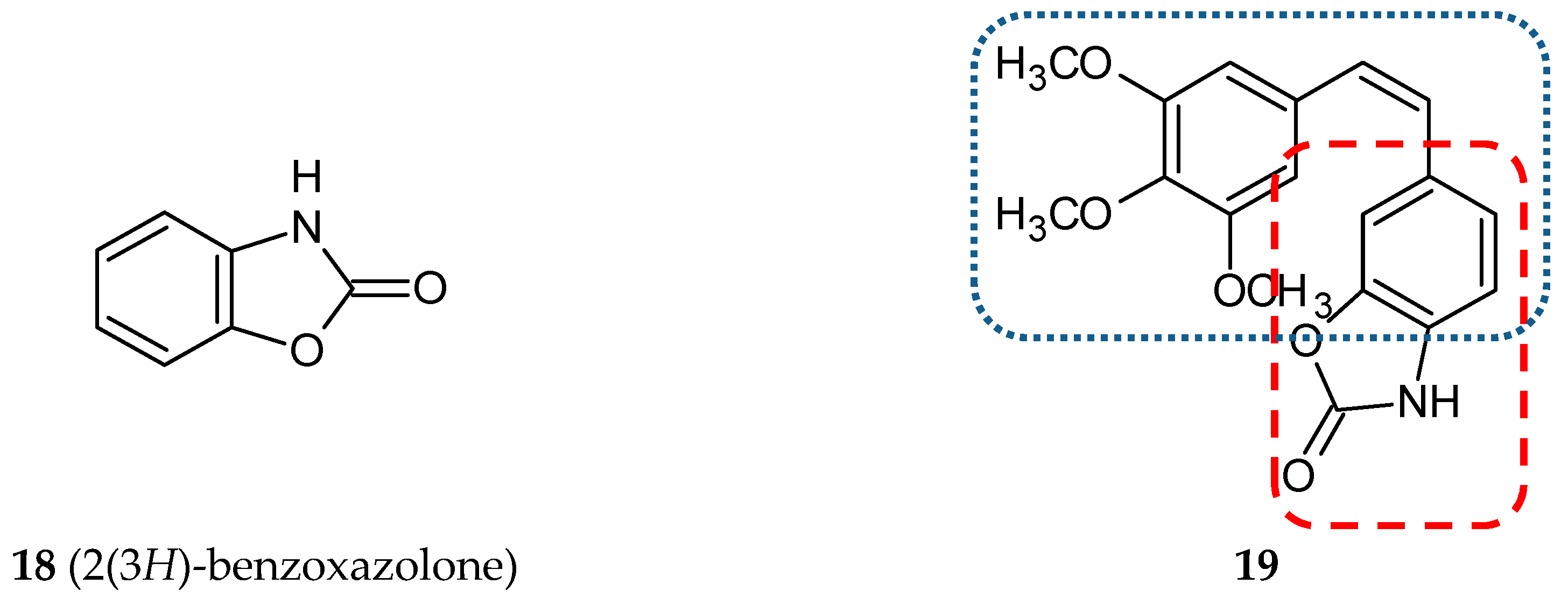
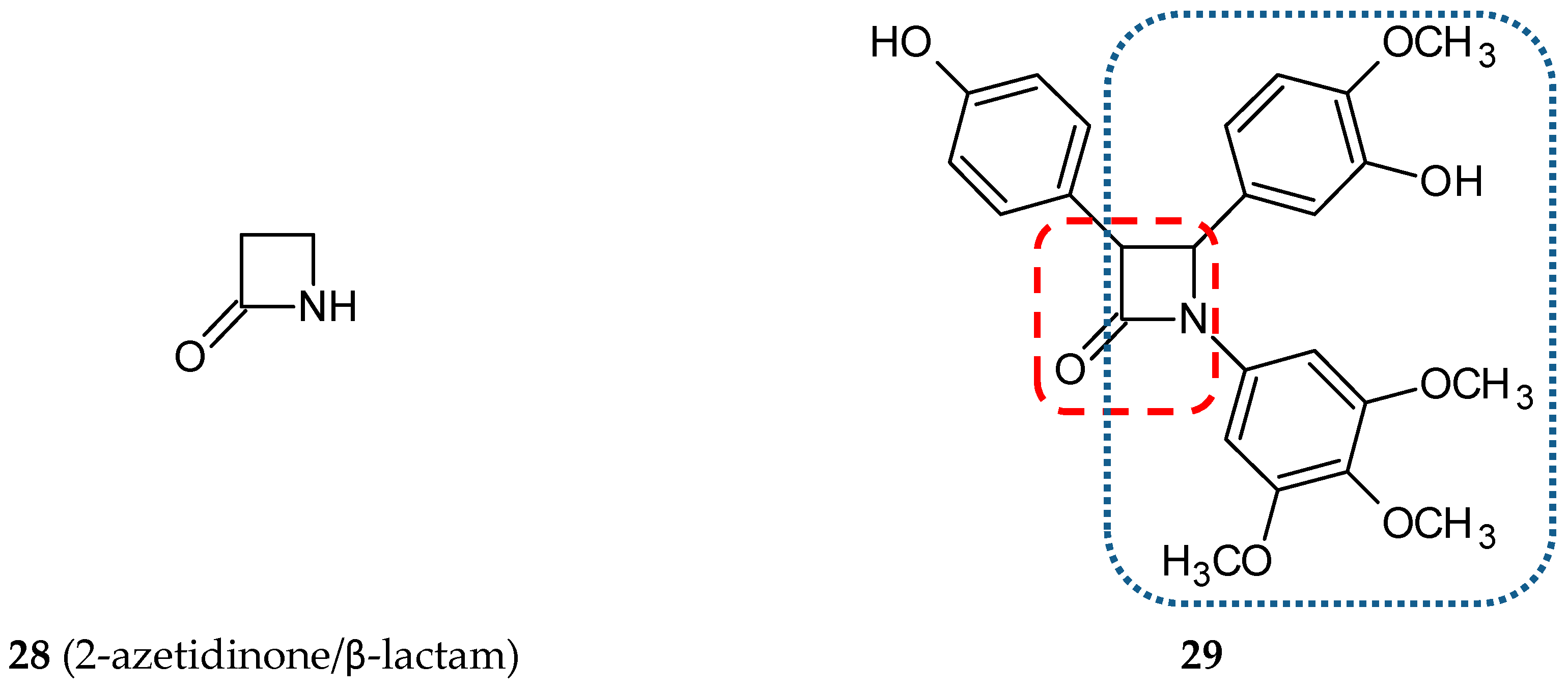
3.1.5. Benzoxazolone Hybrids
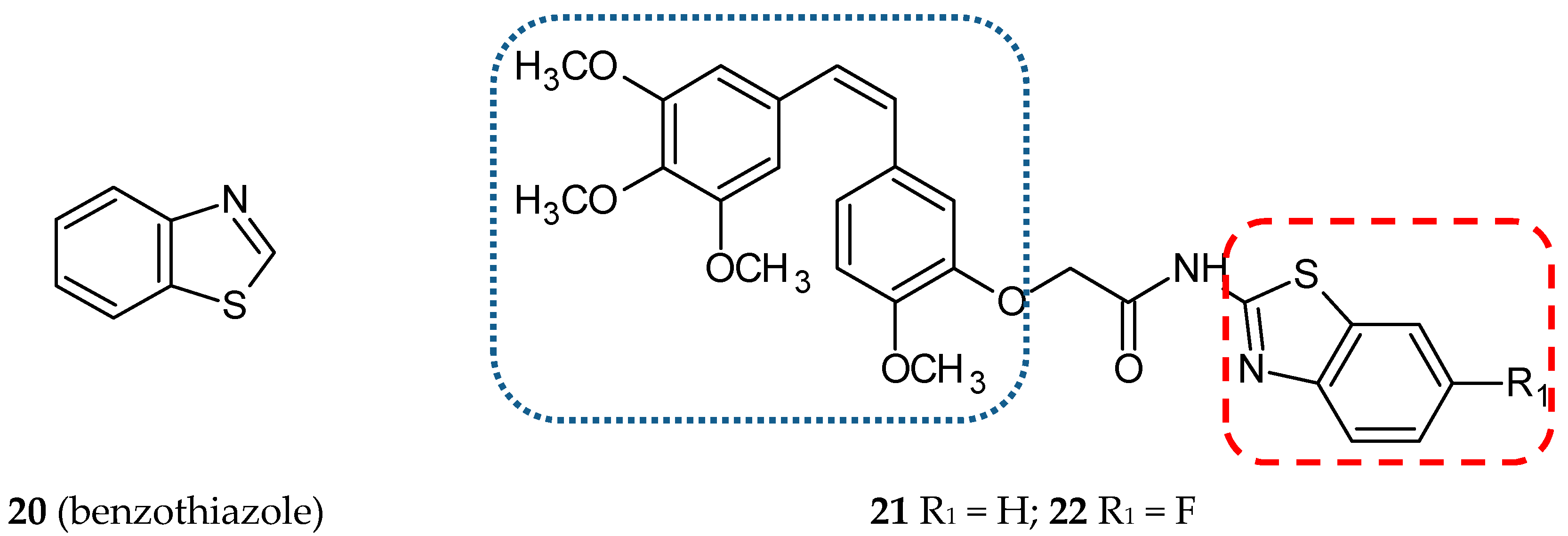
3.1.6. Benzothiazole Hybrids
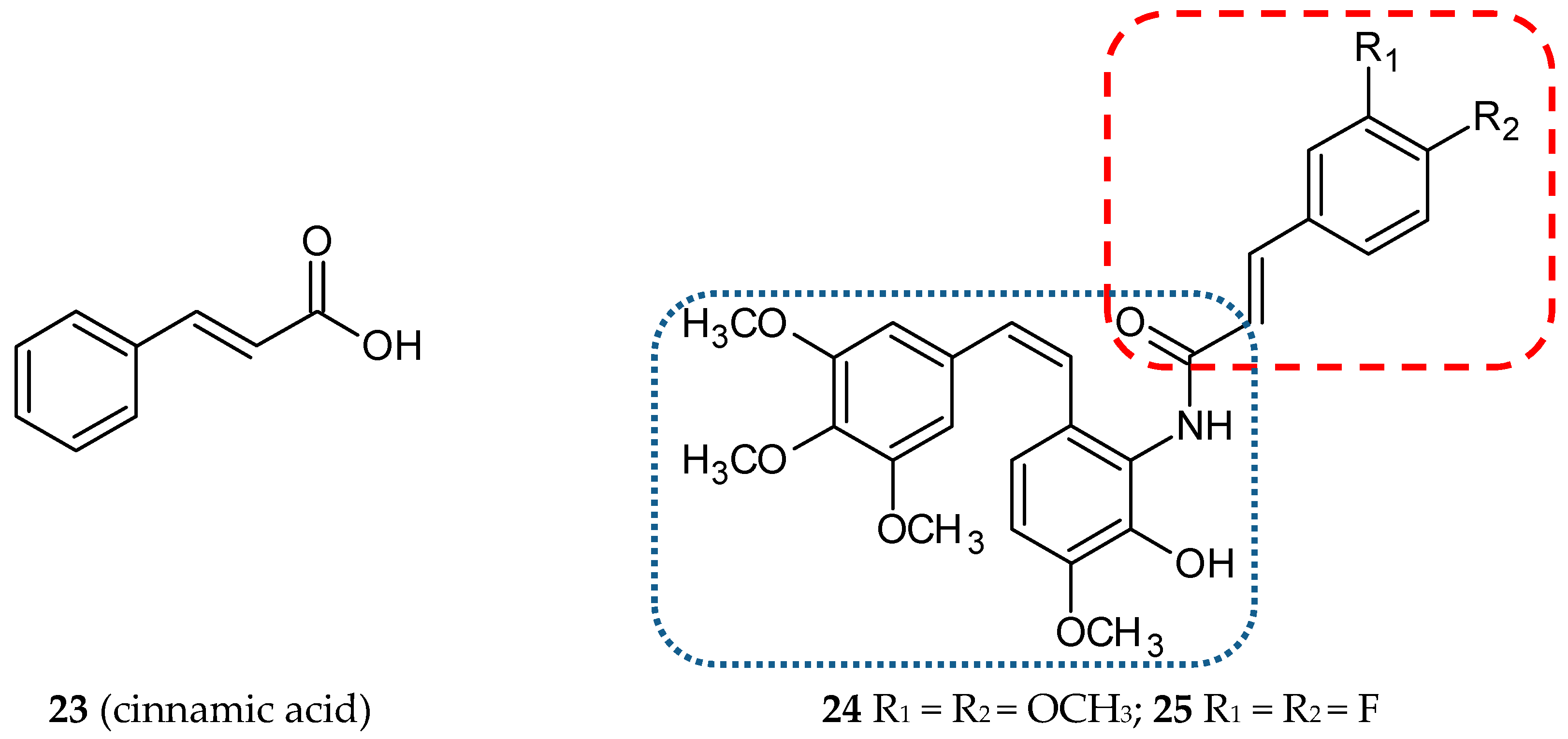
3.1.7. Cinnamic Acid Hybrids
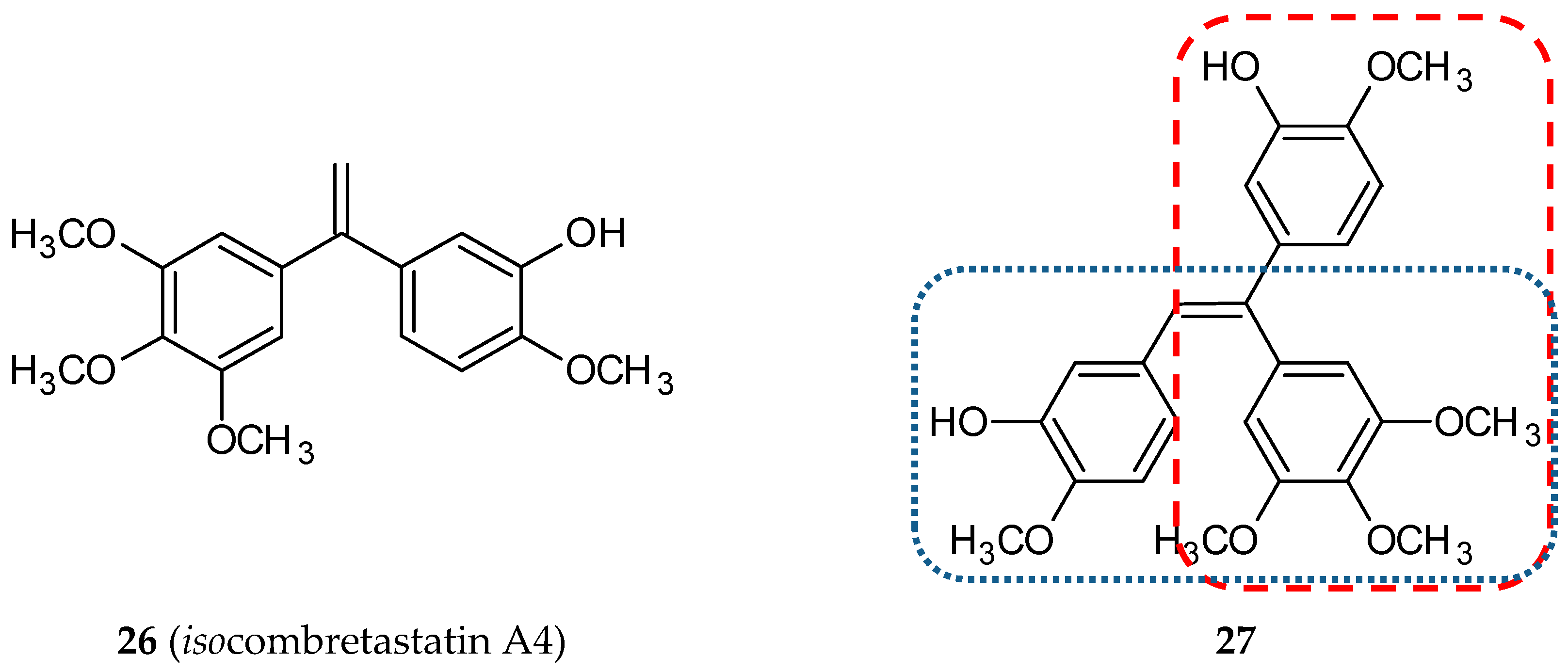
3.1.8. Isocombretastatin Hybrids
3.2. Cis-Restricted Stilbenoid-Based Hybrids
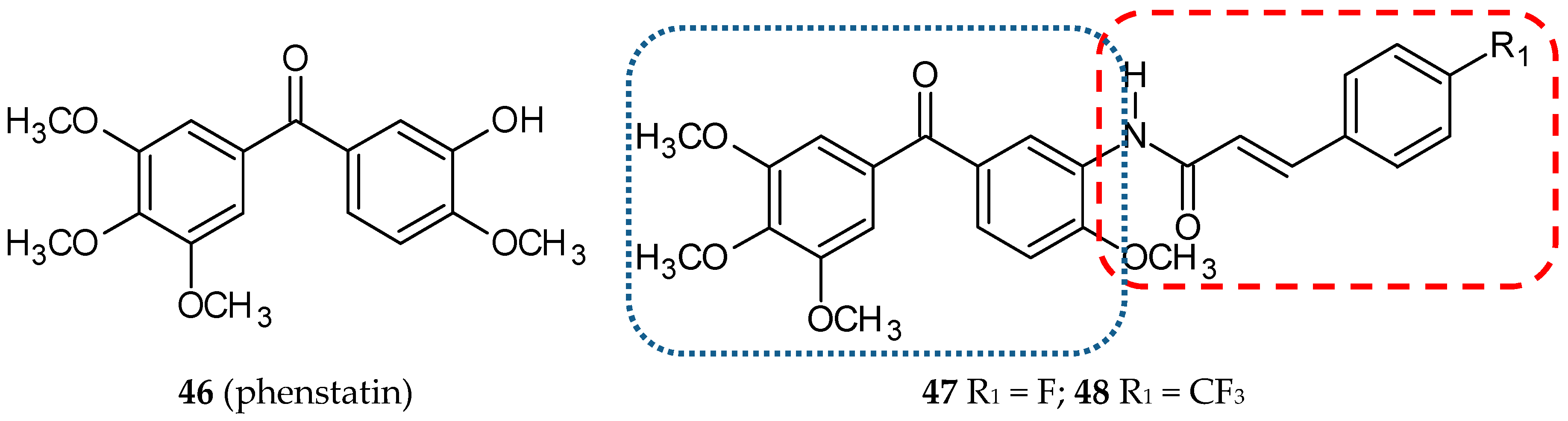
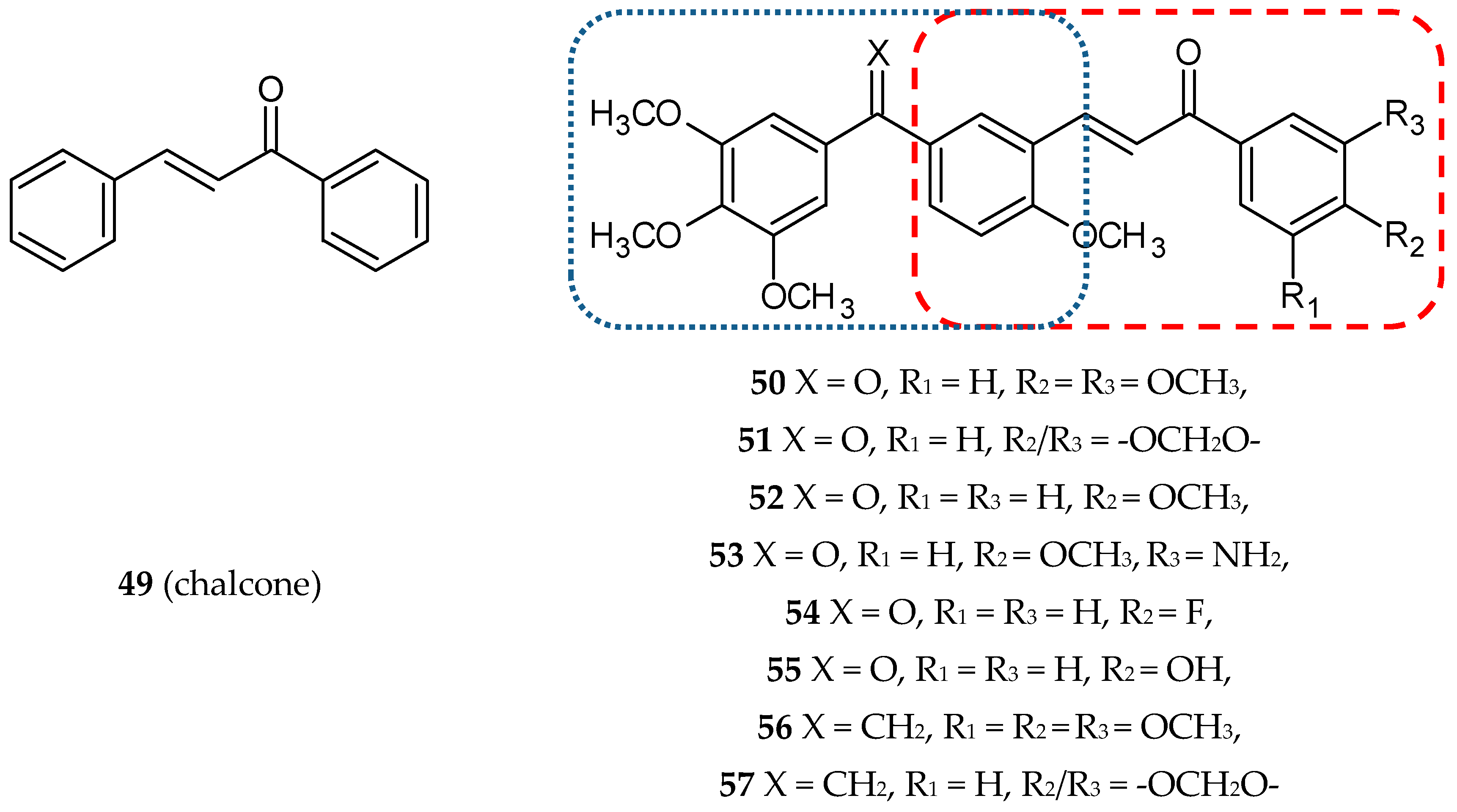
3.3. Phenstatin-Based Hybrids
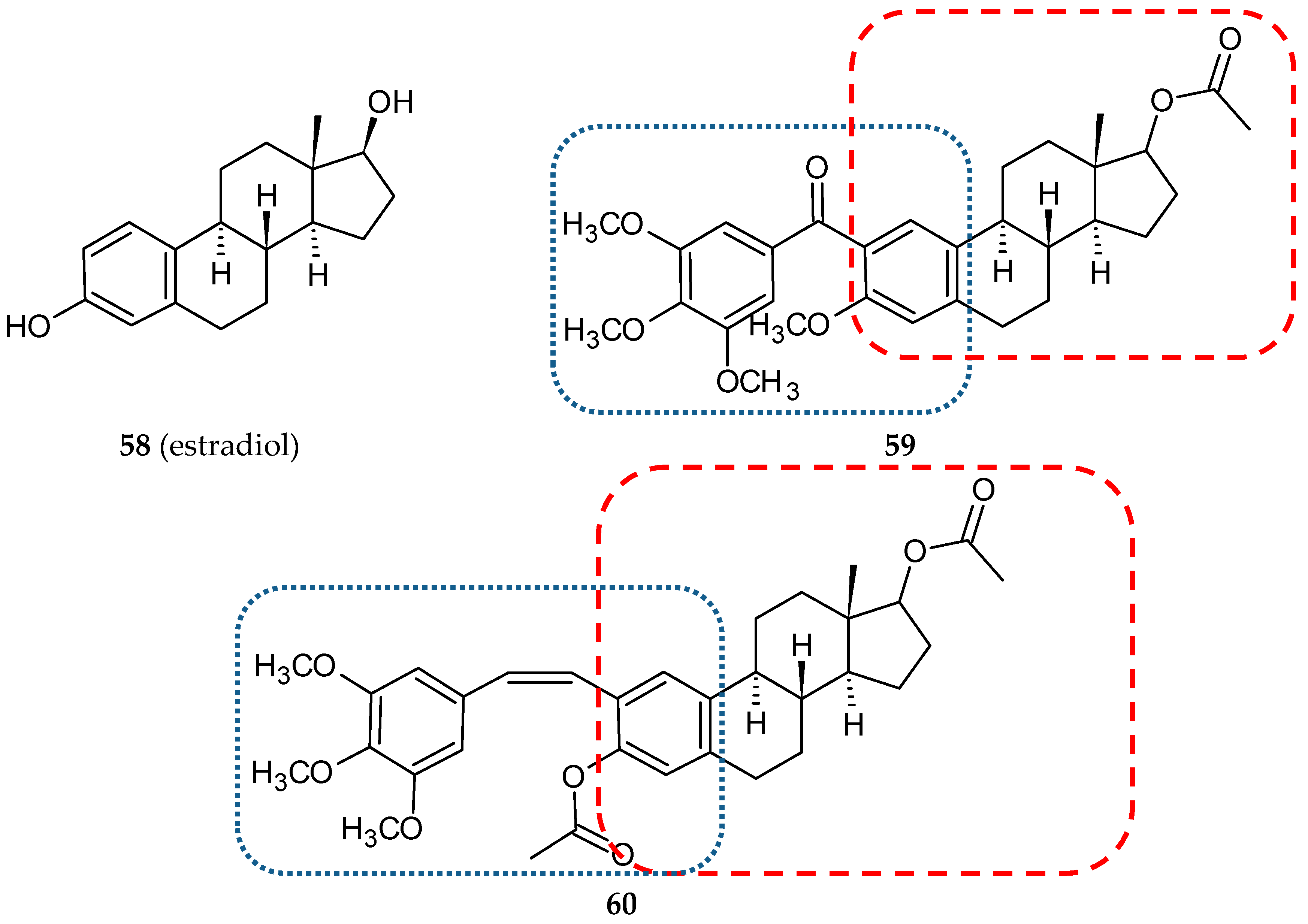
3.4. Steroid-Based Hybrids
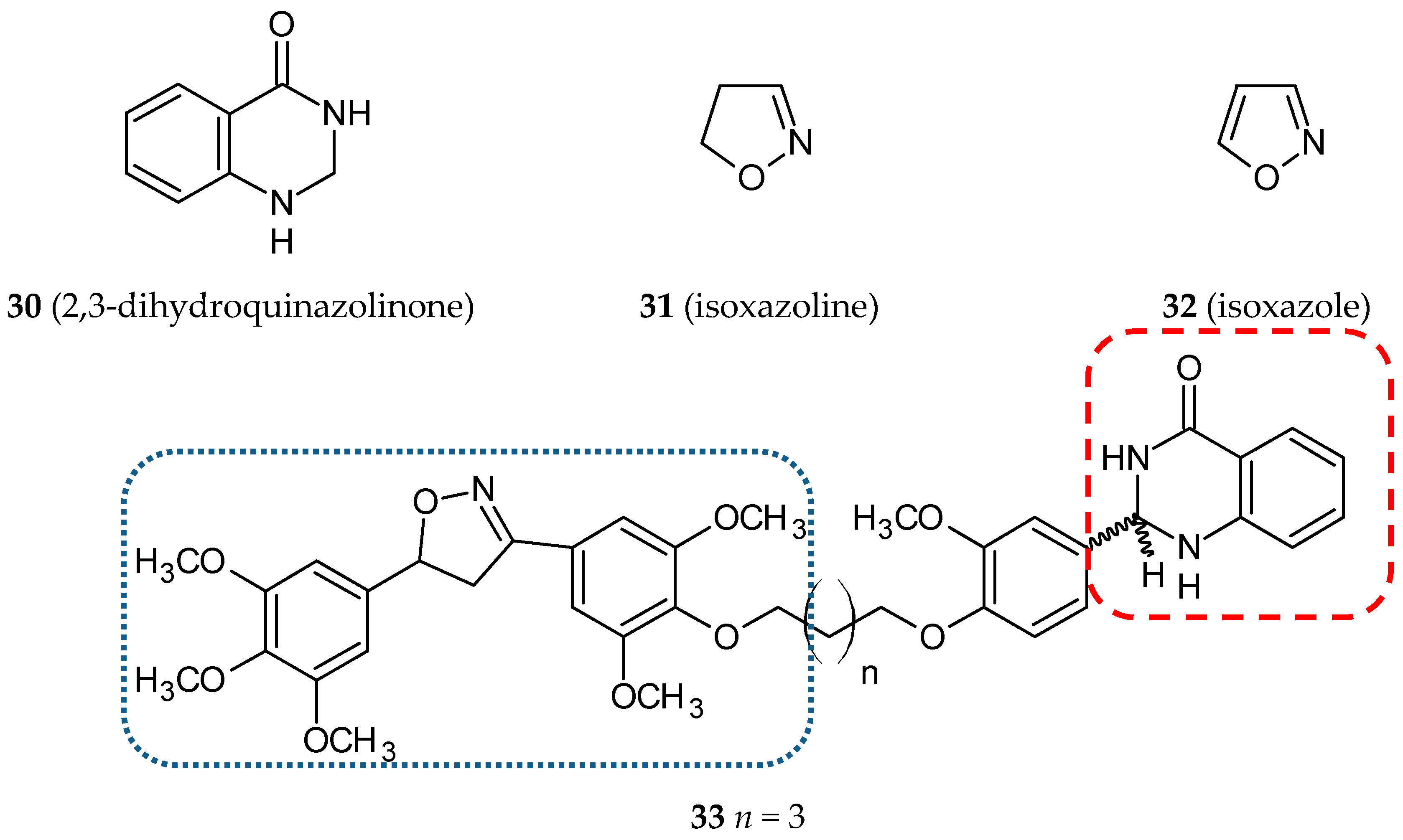
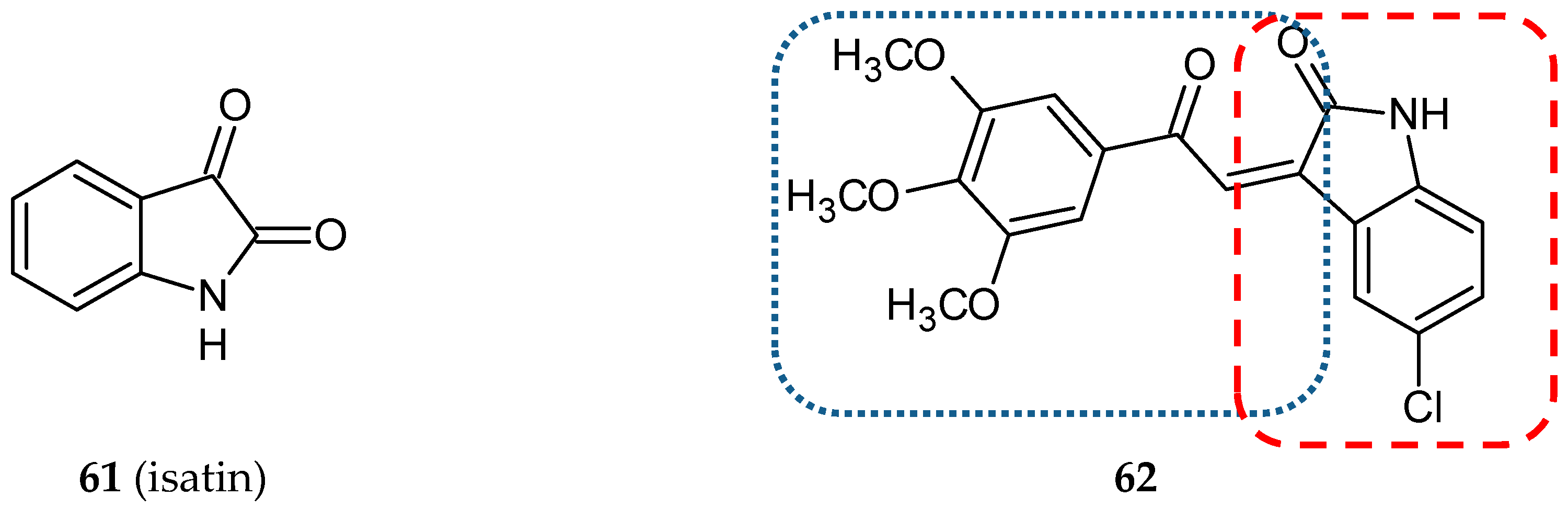
3.5. Other Related Hybrids
4. Further Perspectives
5. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ramos, E.; Egea, J.; de Los Rios, C.; Marco-Contelles, J.; Romero, A. Melatonin as a versatile molecule to design novel multitarget hybrids against neurodegeneration. Future Med. Chem. 2017, 9, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Viegas-Junior, C.; Danuello, A.; da Silva Bolzani, V.; Barreiro, E.J.; Fraga, C.A. Molecular hybridization: A useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent advances (2015-2016) in anticancer hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Kucuksayan, E.; Ozben, T. Hybrid Compounds as Multitarget Directed Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 907–918. [Google Scholar] [CrossRef]
- Tangutur, A.D.; Kumar, D.; Krishna, K.V.; Kantevari, S. Microtubule Targeting Agents as Cancer Chemotherapeutics: An Overview of Molecular Hybrids as Stabilizing and Destabilizing Agents. Curr. Top. Med. Chem. 2017, 17, 2523–2537. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Shaik, A.B.; Jain, N.; Kishor, C.; Nagabhushana, A.; Supriya, B.; Bharath Kumar, G.; Chourasiya, S.S.; Suresh, Y.; Mishra, R.K.; et al. Design and synthesis of pyrazole-oxindole conjugates targeting tubulin polymerization as new anticancer agents. Eur. J. Med. Chem. 2015, 92, 501–513. [Google Scholar] [CrossRef]
- Madadi, N.R.; Zong, H.; Ketkar, A.; Zheng, C.; Penthala, N.R.; Janganati, V.; Bommagani, S.; Eoff, R.L.; Guzman, M.L.; Crooks, P.A. Synthesis and evaluation of a series of resveratrol analogues as potent anti-cancer agents that target tubulin. MedChemComm 2015, 6, 788–794. [Google Scholar] [CrossRef]
- Ruan, B.F.; Lu, X.; Tang, J.F.; Wei, Y.; Wang, X.L.; Zhang, Y.B.; Wang, L.S.; Zhu, H.L. Synthesis, biological evaluation, and molecular docking studies of resveratrol derivatives possessing chalcone moiety as potential antitubulin agents. Bioorg. Med. Chem. 2011, 19, 2688–2695. [Google Scholar] [CrossRef]
- Ruan, B.F.; Lu, X.; Li, T.T.; Tang, J.F.; Wei, Y.; Wang, X.L.; Zheng, S.L.; Yao, R.S.; Zhu, H.L. Synthesis, biological evaluation and molecular docking studies of resveratrol derivatives possessing curcumin moiety as potent antitubulin agents. Bioorg. Med. Chem. 2012, 20, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Teiten, M.H.; Dicato, M.; Diederich, M. Hybrid curcumin compounds: A new strategy for cancer treatment. Molecules 2014, 19, 20839–20863. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.; Bansal, Y.; Silakari, O.; Bansal, G. Coumarin hybrids as novel therapeutic agents. Bioorg. Med. Chem. 2014, 22, 3806–3814. [Google Scholar] [CrossRef] [PubMed]
- Pawelczyk, A.; Olender, D.; Sowa-Kasprzak, K.; Zaprutko, L. Hybrid Compounds Strategy in the Synthesis of Oleanolic Acid Skeleton-NSAID Derivatives. Molecules 2016, 21, 420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tortorella, M.D.; Liao, J.; Qin, X.; Chen, T.; Luo, J.; Guan, J.; Talley, J.J.; Tu, Z. Synthesis and Evaluation of Novel Erlotinib-NSAID Conjugates as More Comprehensive Anticancer Agents. ACS Med. Chem. Lett. 2015, 6, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Cragg, G.M.; Herald, D.L.; Schmidt, J.M.; Lohavanijaya, P. Isolation and structure of combretastatin. Can. J. Chem. 1982, 60, 1374–1376. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, S.; Genazzani, A.A. Medicinal chemistry of combretastatin A4: Present and future directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef]
- Siemann, D.W.; Chaplin, D.J.; Walicke, P.A. A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P). Expert Opin. Investig. Drugs 2009, 18, 189–197. [Google Scholar] [CrossRef]
- Mikstacka, R.; Stefanski, T.; Rozanski, J. Tubulin-interactive stilbene derivatives as anticancer agents. Cell Mol. Biol. Lett. 2013, 18, 368–397. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003, 8, 413–450. [Google Scholar] [CrossRef]
- Singh, P.; Rathinasamy, K.; Mohan, R.; Panda, D. Microtubule assembly dynamics: An attractive target for anticancer drugs. IUBMB Life 2008, 60, 368–375. [Google Scholar] [CrossRef]
- Nagle, A.A.; Gan, F.F.; Jones, G.; So, C.L.; Wells, G.; Chew, E.H. Induction of tumor cell death through targeting tubulin and evoking dysregulation of cell cycle regulatory proteins by multifunctional cinnamaldehydes. PLoS ONE 2012, 7, e50125. [Google Scholar] [CrossRef]
- Field, J.J.; Diaz, J.F.; Miller, J.H. The binding sites of microtubule-stabilizing agents. Chem. Biol. 2013, 20, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed]
- Stefanski, T.; Mikstacka, R.; Kurczab, R.; Dutkiewicz, Z.; Kucinska, M.; Murias, M.; Zielinska-Przyjemska, M.; Cichocki, M.; Teubert, A.; Kaczmarek, M.; et al. Design, synthesis, and biological evaluation of novel combretastatin A-4 thio derivatives as microtubule targeting agents. Eur. J. Med. Chem. 2018, 144, 797–816. [Google Scholar] [CrossRef]
- Kamal, A.; Shaik, A.B.; Rao, B.B.; Khan, I.; Bharath Kumar, G.; Jain, N. Design and synthesis of pyrazole/isoxazole linked arylcinnamides as tubulin polymerization inhibitors and potential antiproliferative agents. Org. Biomol. Chem. 2015, 13, 10162–10178. [Google Scholar] [CrossRef] [PubMed]
- Madadi, N.R.; Penthala, N.R.; Howk, K.; Ketkar, A.; Eoff, R.L.; Borrelli, M.J.; Crooks, P.A. Synthesis and biological evaluation of novel 4,5-disubstituted 2H-1,2,3-triazoles as cis-constrained analogues of combretastatin A-4. Eur. J. Med. Chem. 2015, 103, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef]
- Jin, H.O.; Lee, Y.H.; Park, J.A.; Lee, H.N.; Kim, J.H.; Kim, J.Y.; Kim, B.; Hong, S.E.; Kim, H.A.; Kim, E.K.; et al. Piperlongumine induces cell death through ROS-mediated CHOP activation and potentiates TRAIL-induced cell death in breast cancer cells. J. Cancer Res. Clin. Oncol. 2014, 140, 2039–2046. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef]
- Punganuru, S.R.; Madala, H.R.; Venugopal, S.N.; Samala, R.; Mikelis, C.; Srivenugopal, K.S. Design and synthesis of a C7-aryl piperlongumine derivative with potent antimicrotubule and mutant p53-reactivating properties. Eur. J. Med. Chem. 2016, 107, 233–244. [Google Scholar] [CrossRef]
- Keely, N.O.; Carr, M.; Yassin, B.; Ana, G.; Lloyd, D.G.; Zisterer, D.; Meegan, M.J. Design, Synthesis and Biochemical Evaluation of Novel Selective Estrogen Receptor Ligand Conjugates Incorporating an Endoxifen-Combretastatin Hybrid Scaffold. Biomedicines 2016, 4, 15. [Google Scholar] [CrossRef]
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433. [Google Scholar] [CrossRef]
- Coggiola, B.; Pagliai, F.; Allegrone, G.; Genazzani, A.A.; Tron, G.C. Synthesis and biological activity of mustard derivatives of combretastatins. Bioorg. Med. Chem. Lett. 2005, 15, 3551–3554. [Google Scholar] [CrossRef]
- Vilanova, C.; Torijano-Gutierrez, S.; Diaz-Oltra, S.; Murga, J.; Falomir, E.; Carda, M.; Alberto Marco, J. Design and synthesis of pironetin analogue/combretastatin A-4 hybrids containing a 1,2,3-triazole ring and evaluation of their cytotoxic activity. Eur. J. Med. Chem. 2014, 87, 125–130. [Google Scholar] [CrossRef]
- Kamal, A.; Mallareddy, A.; Janaki Ramaiah, M.; Pushpavalli, S.N.; Suresh, P.; Kishor, C.; Murty, J.N.; Rao, N.S.; Ghosh, S.; Addlagatta, A.; et al. Synthesis and biological evaluation of combretastatin-amidobenzothiazole conjugates as potential anticancer agents. Eur. J. Med. Chem. 2012, 56, 166–178. [Google Scholar] [CrossRef]
- Kamal, A.; Bajee, S.; Lakshma Nayak, V.; Venkata Subba Rao, A.; Nagaraju, B.; Ratna Reddy, C.; Jeevak Sopanrao, K.; Alarifi, A. Synthesis and biological evaluation of arylcinnamide linked combretastatin-A4 hybrids as tubulin polymerization inhibitors and apoptosis inducing agents. Bioorg. Med. Chem. Lett. 2016, 26, 2957–2964. [Google Scholar] [CrossRef]
- Greene, L.M.; Wang, S.; O’Boyle, N.M.; Bright, S.A.; Reid, J.E.; Kelly, P.; Meegan, M.J.; Zisterer, D.M. Combretazet-3 a novel synthetic cis-stable combretastatin A-4-azetidinone hybrid with enhanced stability and therapeutic efficacy in colon cancer. Oncol. Rep. 2013, 29, 2451–2458. [Google Scholar] [CrossRef]
- Kamal, A.; Kumar, G.B.; Vishnuvardhan, M.V.; Shaik, A.B.; Reddy, V.S.; Mahesh, R.; Sayeeda, I.B.; Kapure, J.S. Synthesis of phenstatin/isocombretastatin-chalcone conjugates as potent tubulin polymerization inhibitors and mitochondrial apoptotic inducers. Org. Biomol. Chem. 2015, 13, 3963–3981. [Google Scholar] [CrossRef]
- Parihar, S.; Gupta, A.; Chaturvedi, A.K.; Agarwal, J.; Luqman, S.; Changkija, B.; Manohar, M.; Chanda, D.; Chanotiya, C.S.; Shanker, K.; et al. Gallic acid based steroidal phenstatin analogues for selective targeting of breast cancer cells through inhibiting tubulin polymerization. Steroids 2012, 77, 878–886. [Google Scholar] [CrossRef]
- Lamaa, D.; Lin, H.P.; Zig, L.; Bauvais, C.; Bollot, G.; Bignon, J.; Levaique, H.; Pamlard, O.; Dubois, J.; Ouaissi, M.; et al. Design and Synthesis of Tubulin and Histone Deacetylase Inhibitor Based on iso-Combretastatin A-4. J. Med. Chem. 2018, 61, 6574–6591. [Google Scholar] [CrossRef]
- Punganuru, S.R.; Madala, H.R.; Mikelis, C.M.; Dixit, A.; Arutla, V.; Srivenugopal, K.S. Conception, synthesis, and characterization of a rofecoxib-combretastatin hybrid drug with potent cyclooxygenase-2 (COX-2) inhibiting and microtubule disrupting activities in colon cancer cell culture and xenograft models. Oncotarget 2018, 9, 26109–26129. [Google Scholar] [CrossRef]
- Marco, J.A.; Garcia-Pla, J.; Carda, M.; Murga, J.; Falomir, E.; Trigili, C.; Notararigo, S.; Diaz, J.F.; Barasoain, I. Design and synthesis of pironetin analogues with simplified structure and study of their interactions with microtubules. Eur. J. Med. Chem. 2011, 46, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Setter, J.; Waight, A.B.; Bargsten, K.; Murga, J.; Diaz, J.F.; Steinmetz, M.O. Pironetin Binds Covalently to alphaCys316 and Perturbs a Major Loop and Helix of alpha-Tubulin to Inhibit Microtubule Formation. J. Mol. Biol. 2016, 428, 2981–2988. [Google Scholar] [CrossRef] [PubMed]
- Torijano-Gutiérrez, S.; Vilanova, C.; Díaz-Oltra, S.; Murga, J.; Falomir, E.; Carda, M.; Alberto Marco, J. Design and Synthesis of Pironetin Analogue/Combretastatin A-4 Hybrids and Evaluation of Their Cytotoxic Activity. Eur. J. Org. Chem. 2014, 2014, 2284–2296. [Google Scholar] [CrossRef]
- Torijano-Gutierrez, S.; Vilanova, C.; Diaz-Oltra, S.; Murga, J.; Falomir, E.; Carda, M.; Redondo-Horcajo, M.; Diaz, J.F.; Barasoain, I.; Marco, J.A. The Mechanism of the Interactions of Pironetin Analog/Combretastatin A-4 Hybrids with Tubulin. Arch. Pharm. 2015, 348, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Gerova, M.S.; Stateva, S.R.; Radonova, E.M.; Kalenderska, R.B.; Rusew, R.I.; Nikolova, R.P.; Chanev, C.D.; Shivachev, B.L.; Apostolova, M.D.; Petrov, O.I. Combretastatin A-4 analogues with benzoxazolone scaffold: Synthesis, structure and biological activity. Eur. J. Med. Chem. 2016, 120, 121–133. [Google Scholar] [CrossRef]
- Poupaert, J.; Carato, P.; Colacino, E.; Yous, S. 2(3H)-benzoxazolone and bioisosters as “privileged scaffold” in the design of pharmacological probes. Curr. Med. Chem. 2005, 12, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Erol, D.D.; Aytemir, M.D.; Yuluğ, N. Synthesis and antimicrobial activity of thiazolinomethyl-2(3H)-benzoxazolone derivatives (I). Eur. J. Med. Chem. 1995, 30, 521–524. [Google Scholar] [CrossRef]
- Ivanova, Y.; Momekov, G.; Petrov, O.; Karaivanova, M.; Kalcheva, V. Cytotoxic Mannich bases of 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones. Eur. J. Med. Chem. 2007, 42, 1382–1387. [Google Scholar] [CrossRef]
- Lad, N.P.; Manohar, Y.; Mascarenhas, M.; Pandit, Y.B.; Kulkarni, M.R.; Sharma, R.; Salkar, K.; Suthar, A.; Pandit, S.S. Methylsulfonyl benzothiazoles (MSBT) derivatives: Search for new potential antimicrobial and anticancer agents. Bioorg. Med. Chem. Lett. 2017, 27, 1319–1324. [Google Scholar] [CrossRef]
- Novak, I.; Klasinc, L.; McGlynn, S.P. Photoelectron spectra and biological activity of cinnamic acid derivatives revisited. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 189, 129–132. [Google Scholar] [CrossRef]
- Khelifi, I.; Naret, T.; Renko, D.; Hamze, A.; Bernadat, G.; Bignon, J.; Lenoir, C.; Dubois, J.; Brion, J.D.; Provot, O.; et al. Design, synthesis and anticancer properties of IsoCombretaQuinolines as potent tubulin assembly inhibitors. Eur. J. Med. Chem. 2017, 127, 1025–1034. [Google Scholar] [CrossRef]
- Messaoudi, S.; Treguier, B.; Hamze, A.; Provot, O.; Peyrat, J.F.; De Losada, J.R.; Liu, J.M.; Bignon, J.; Wdzieczak-Bakala, J.; Thoret, S.; et al. Isocombretastatins a versus combretastatins a: The forgotten isoCA-4 isomer as a highly promising cytotoxic and antitubulin agent. J. Med. Chem. 2009, 52, 4538–4542. [Google Scholar] [CrossRef]
- Rasolofonjatovo, E.; Provot, O.; Hamze, A.; Bignon, J.; Thoret, S.; Brion, J.D.; Alami, M. Regioselective hydrostannation of diarylalkynes directed by a labile ortho bromine atom: An easy access to stereodefined triarylolefins, hybrids of combretastatin A-4 and isocombretastatin A-4. Eur. J. Med. Chem. 2010, 45, 3617–3626. [Google Scholar] [CrossRef]
- He, D.; Wang, M.; Zhao, S.; Shu, Y.; Zeng, H.; Xiao, C.; Lu, C.; Liu, Y. Pharmaceutical prospects of naturally occurring quinazolinone and its derivatives. Fitoterapia 2017, 119, 136–149. [Google Scholar] [CrossRef]
- Kamal, A.; Bharathi, E.V.; Reddy, J.S.; Ramaiah, M.J.; Dastagiri, D.; Reddy, M.K.; Viswanath, A.; Reddy, T.L.; Shaik, T.B.; Pushpavalli, S.N.; et al. Synthesis and biological evaluation of 3,5-diaryl isoxazoline/isoxazole linked 2,3-dihydroquinazolinone hybrids as anticancer agents. Eur. J. Med. Chem. 2011, 46, 691–703. [Google Scholar] [CrossRef]
- Chittchang, M.; Gleeson, M.P.; Ploypradith, P.; Ruchirawat, S. Assessing the drug-likeness of lamellarins, a marine-derived natural product class with diverse oncological activities. Eur. J. Med. Chem. 2010, 45, 2165–2172. [Google Scholar] [CrossRef]
- Malla Reddy, S.; Srinivasulu, M.; Satyanarayana, N.; Kondapi, A.K.; Venkateswarlu, Y. New potent cytotoxic lamellarin alkaloids from Indian ascidian Didemnum obscurum. Tetrahedron 2005, 61, 9242–9247. [Google Scholar] [CrossRef]
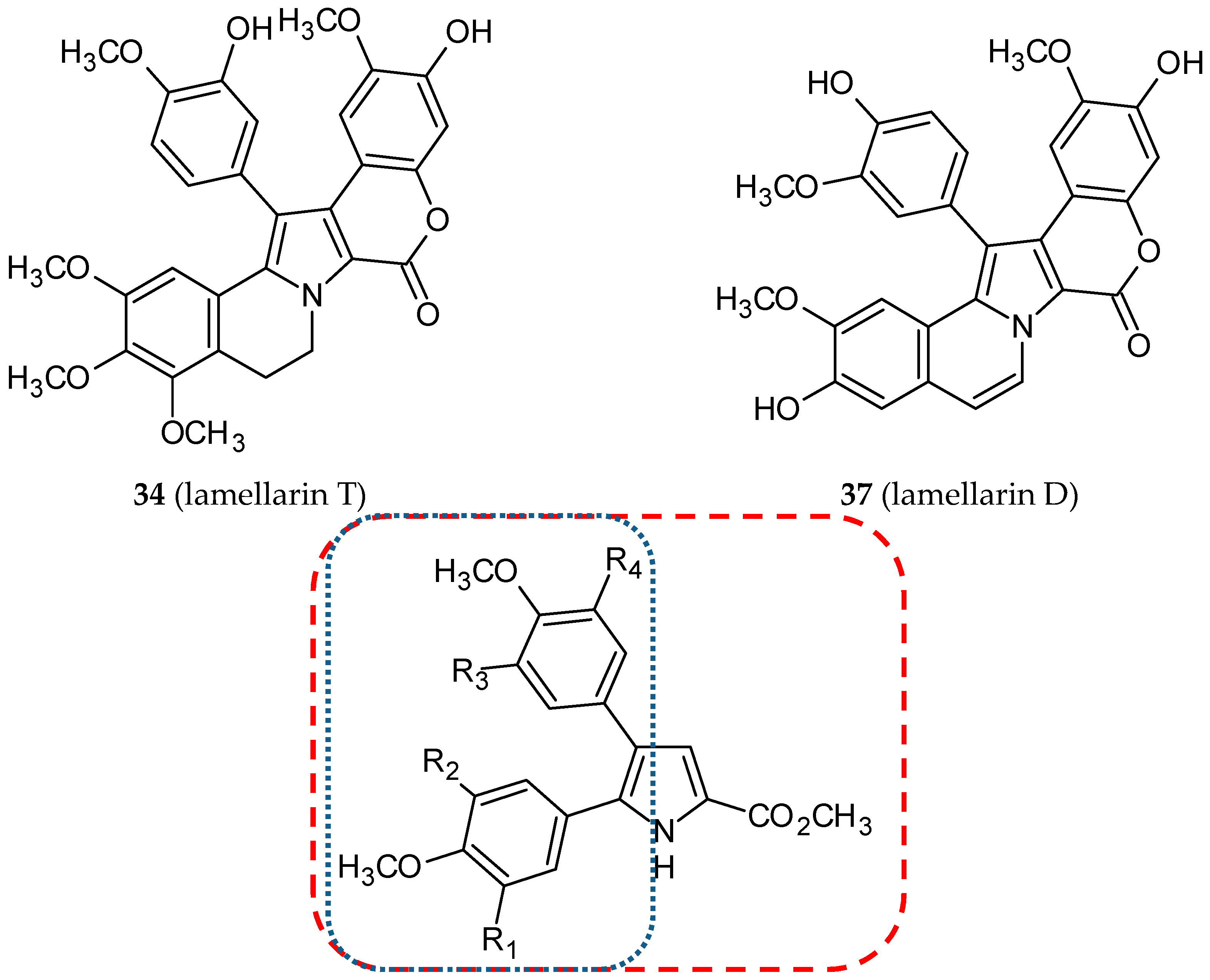
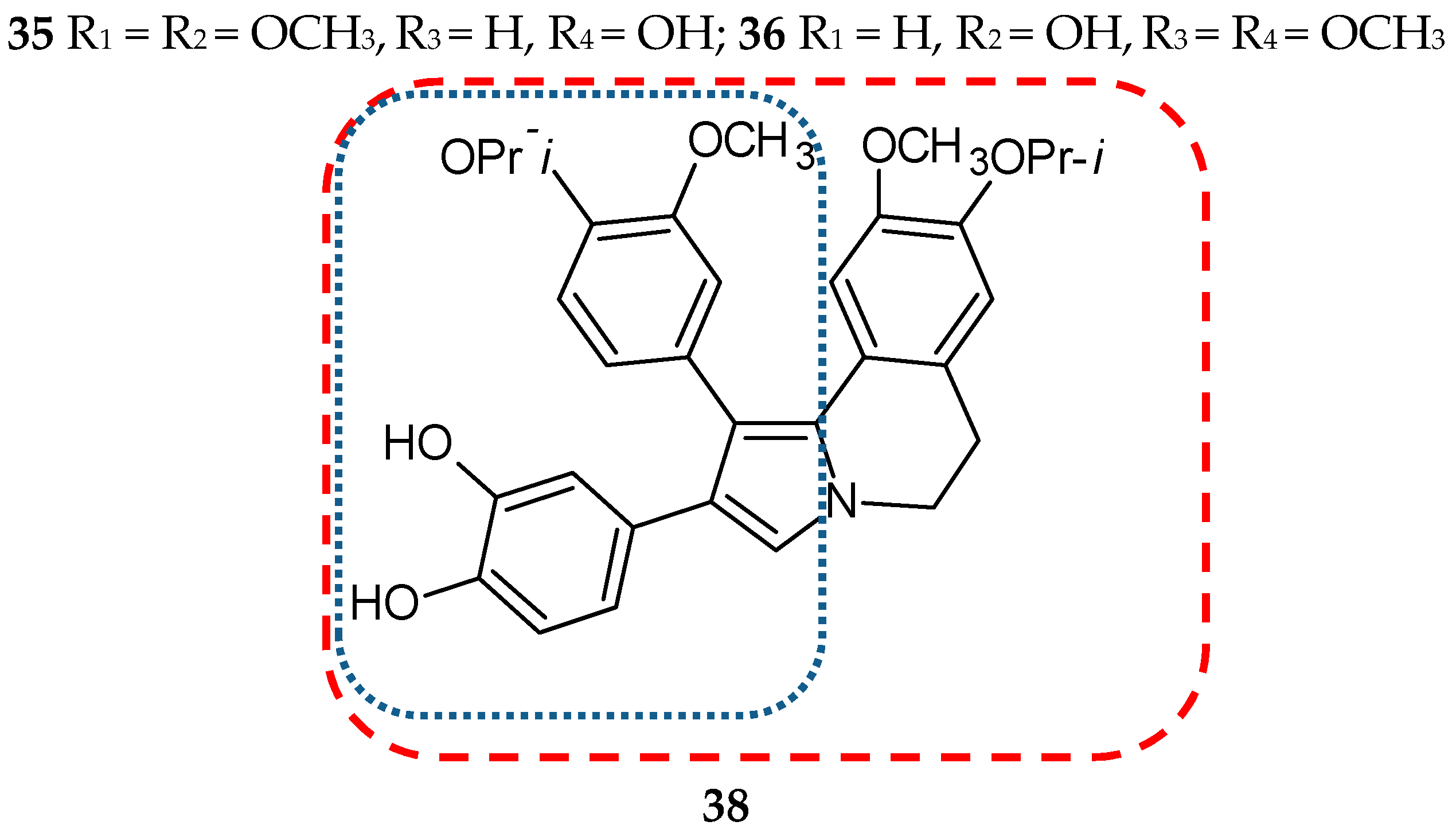
- Banwell, M.G.; Hamel, E.; Hockless, D.C.; Verdier-Pinard, P.; Willis, A.C.; Wong, D.J. 4,5-Diaryl-1H-pyrrole-2-carboxylates as combretastatin A-4/lamellarin T hybrids: Synthesis and evaluation as anti-mitotic and cytotoxic agents. Bioorg. Med. Chem. 2006, 14, 4627–4638. [Google Scholar] [CrossRef]
- Shen, L.; Yang, X.; Yang, B.; He, Q.; Hu, Y. Novel hybrids from lamellarin D and combretastatin A 4 as cytotoxic agents. Eur. J. Med. Chem. 2010, 45, 11–18. [Google Scholar] [CrossRef]
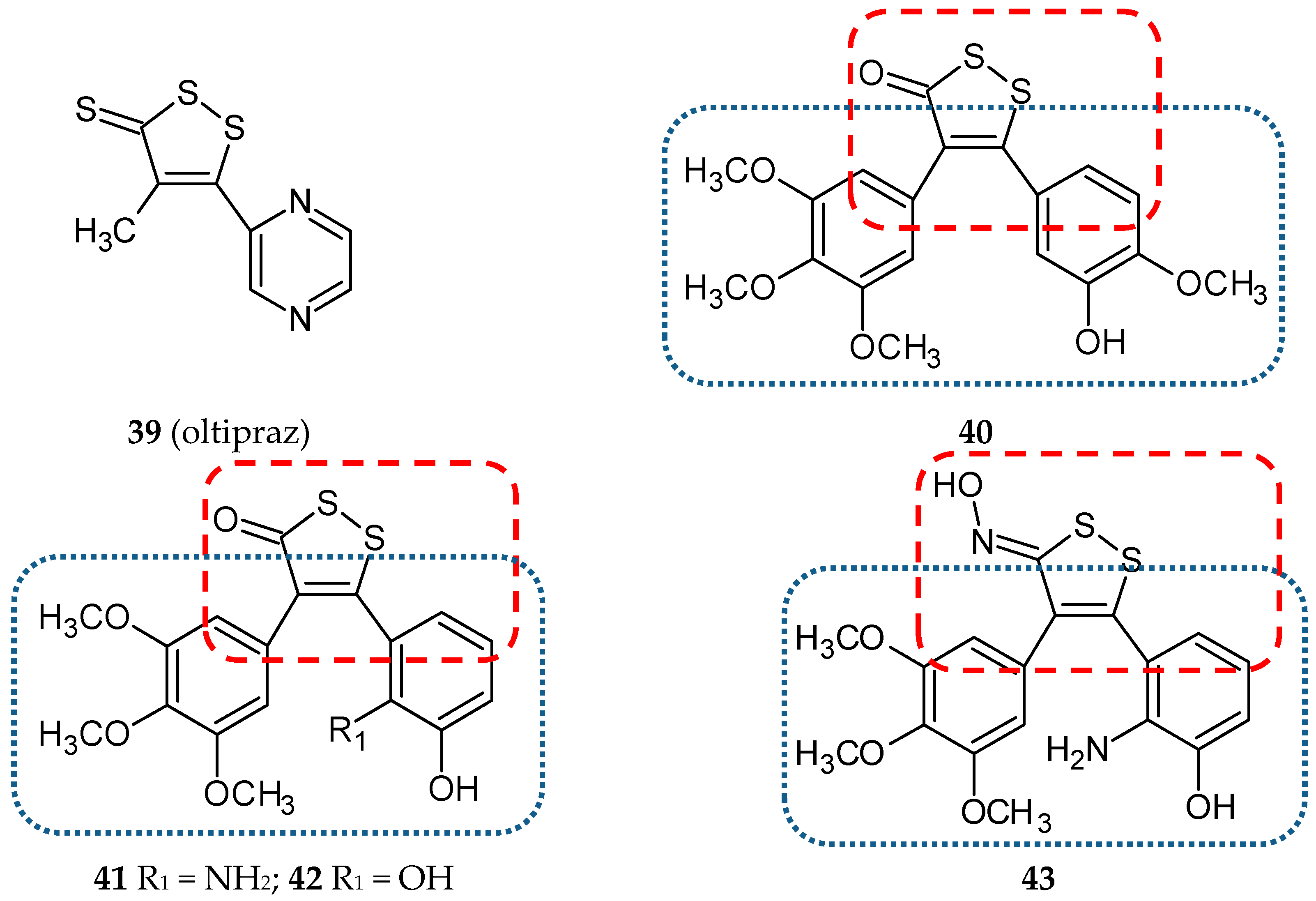
- Weerachayaphorn, J.; Luo, Y.; Mennone, A.; Soroka, C.J.; Harry, K.; Boyer, J.L. Deleterious effect of oltipraz on extrahepatic cholestasis in bile duct-ligated mice. J. Hepatol. 2014, 60, 160–166. [Google Scholar] [CrossRef]
- Qi, H.; Zuo, D.Y.; Bai, Z.S.; Xu, J.W.; Li, Z.Q.; Shen, Q.R.; Wang, Z.W.; Zhang, W.G.; Wu, Y.L. COH-203, a novel microtubule inhibitor, exhibits potent anti-tumor activity via p53-dependent senescence in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2014, 455, 262–268. [Google Scholar] [CrossRef]
- Wang, Z.; Qi, H.; Shen, Q.; Lu, G.; Li, M.; Bao, K.; Wu, Y.; Zhang, W. 4,5-Diaryl-3H-1,2-dithiole-3-thiones and related compounds as combretastatin A-4/oltipraz hybrids: Synthesis, molecular modelling and evaluation as antiproliferative agents and inhibitors of tubulin. Eur. J. Med. Chem. 2016, 122, 520–529. [Google Scholar] [CrossRef]
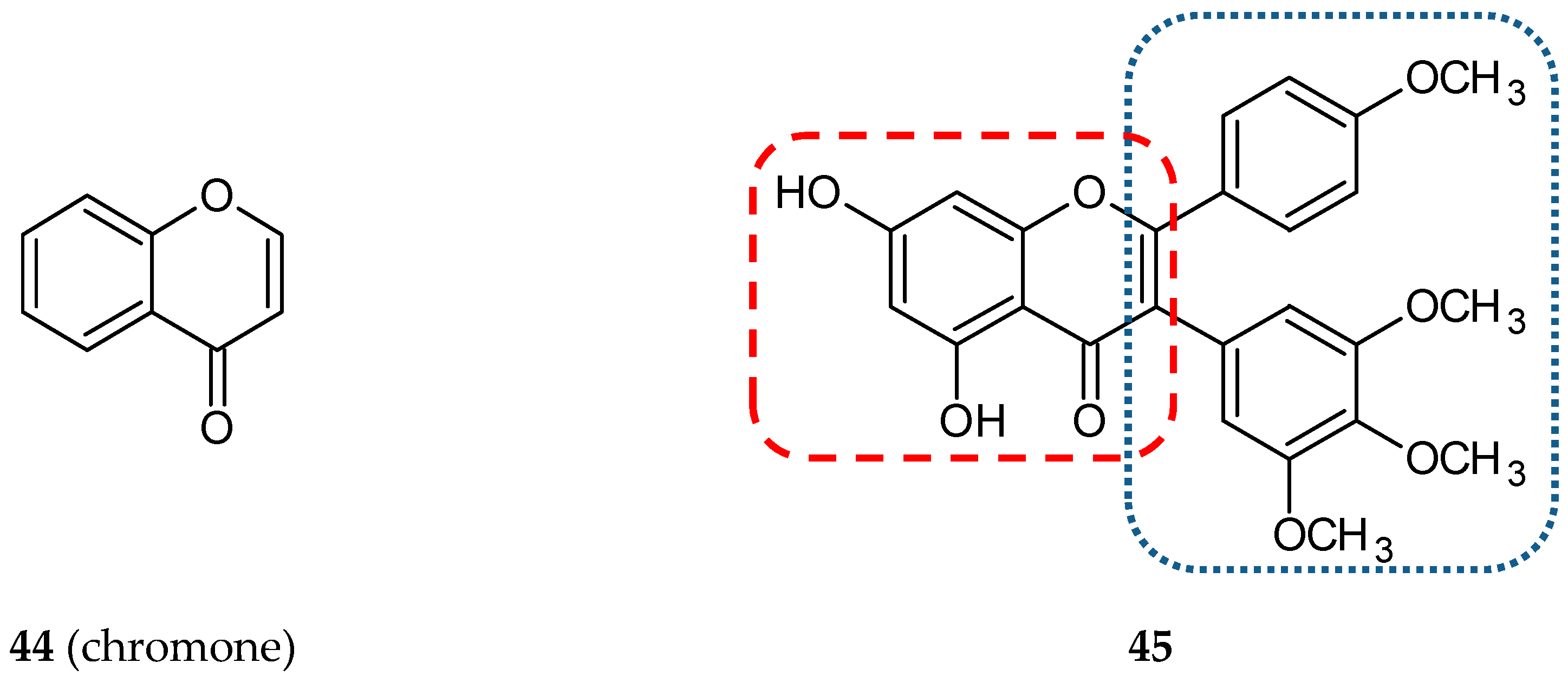
- Quintin, J.; Roullier, C.; Thoret, S.; Lewin, G. Synthesis and anti-tubulin evaluation of chromone-based analogues of combretastatins. Tetrahedron 2006, 62, 4038–4051. [Google Scholar] [CrossRef]
- Reis, J.; Gaspar, A.; Milhazes, N.; Borges, F. Chromone as a Privileged Scaffold in Drug Discovery: Recent Advances. J. Med. Chem. 2017, 60, 7941–7957. [Google Scholar] [CrossRef]
- Pettit, G.R.; Grealish, M.P.; Herald, D.L.; Boyd, M.R.; Hamel, E.; Pettit, R.K. Antineoplastic agents. 443. Synthesis of the cancer cell growth inhibitor hydroxyphenstatin and its sodium diphosphate prodrug. J. Med. Chem. 2000, 43, 2731–2737. [Google Scholar] [CrossRef]
- Pettit, G.R.; Toki, B.; Herald, D.L.; Verdier-Pinard, P.; Boyd, M.R.; Hamel, E.; Pettit, R.K. Antineoplastic agents. 379. Synthesis of phenstatin phosphate. J. Med. Chem. 1998, 41, 1688–1695. [Google Scholar] [CrossRef]
- Kamal, A.; Reddy Ch, R.; Vishnuvardhan, M.V.; Mahesh, R.; Lakshma Nayak, V.; Prabhakar, S.; Reddy, C.S. Synthesis and biological evaluation of cinnamido linked benzophenone hybrids as tubulin polymerization inhibitors and apoptosis inducing agents. Bioorg. Med. Chem. Lett. 2014, 24, 2309–2314. [Google Scholar] [CrossRef]
- Karimi-Sales, E.; Mohaddes, G.; Alipour, M.R. Chalcones as putative hepatoprotective agents: Preclinical evidence and molecular mechanisms. Pharmacol. Res. 2018, 129, 177–187. [Google Scholar] [CrossRef]
- Chen, J.; Brown, D.P.; Wang, Y.J.; Chen, Z.S. New phenstatin-fatty acid conjugates: Synthesis and evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 5119–5122. [Google Scholar] [CrossRef]
- Parihar, S.; Kumar, A.; Chaturvedi, A.K.; Sachan, N.K.; Luqman, S.; Changkija, B.; Manohar, M.; Prakash, O.; Chanda, D.; Khan, F.; et al. Synthesis of combretastatin A4 analogues on steroidal framework and their anti-breast cancer activity. J. Steroid. Biochem. Mol. Biol. 2013, 137, 332–344. [Google Scholar] [CrossRef]
- Pakravan, P.; Kashanian, S.; Khodaei, M.M.; Harding, F.J. Biochemical and pharmacological characterization of isatin and its derivatives: From structure to activity. Pharmacol. Rep. 2013, 65, 313–335. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, L.; Zhao, X.; Zhang, Y. A Hybrid Chalcone Combining the Trimethoxyphenyl and Isatinyl Groups Targets Multiple Oncogenic Proteins and Pathways in Hepatocellular Carcinoma Cells. PLoS ONE 2016, 11, e0161025. [Google Scholar] [CrossRef]
- Jain, S.; Chandra, V.; Kumar Jain, P.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive review on current developments of quinoline-based anticancer agents. Arab. J. Chem. 2016. [Google Scholar] [CrossRef]
- Srivastava, V.; Lee, H. Synthesis and bio-evaluation of novel quinolino-stilbene derivatives as potential anticancer agents. Bioorg. Med. Chem. 2015, 23, 7629–7640. [Google Scholar] [CrossRef]
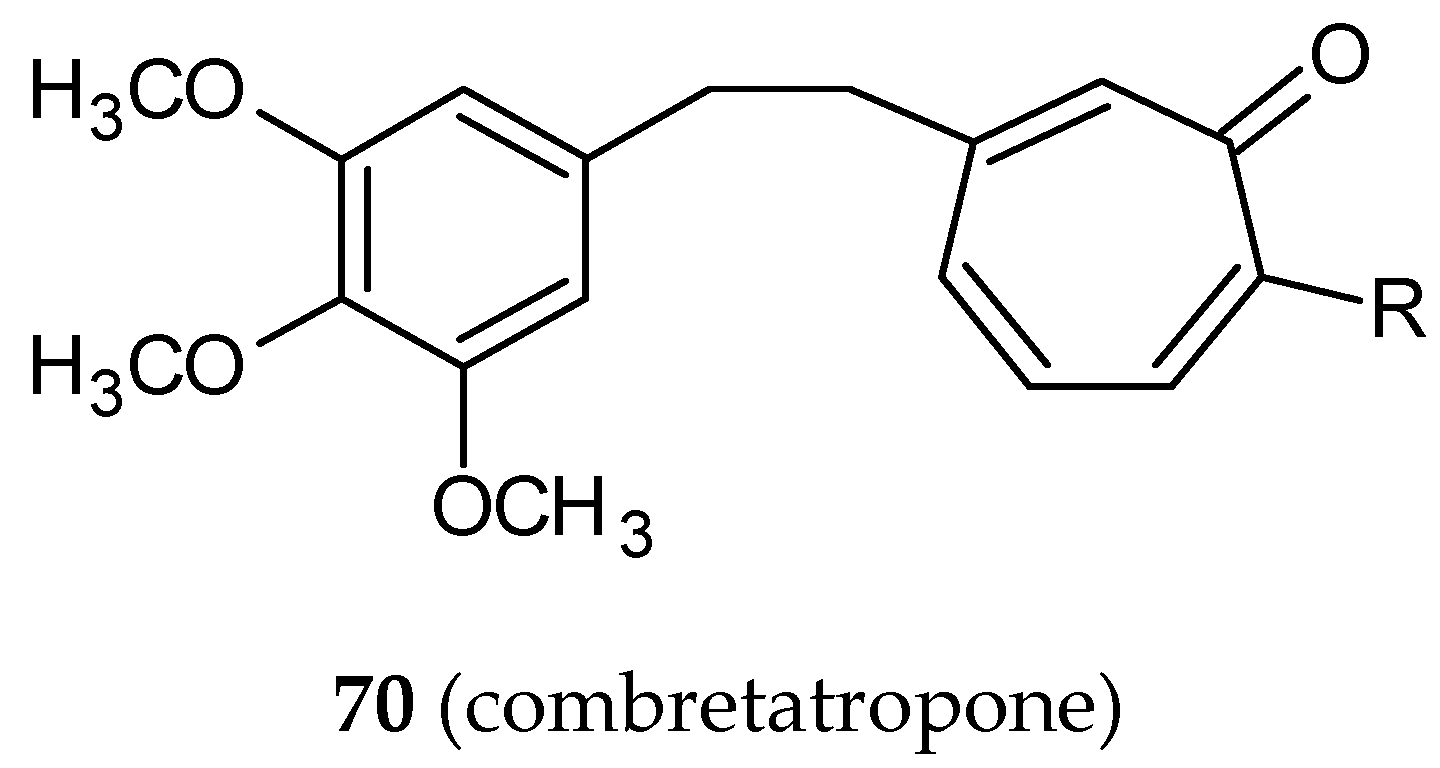
- Andres, C.J.; Bernardo, J.E.; Yan, Q.; Hastie, S.B.; Macdonald, T.L. “Combretatropones”—hybrids of combretastatin and colchicine. Synthesis and biochemical evaluation. Bioorg. Med. Chem. Lett. 1993, 3, 565–570. [Google Scholar] [CrossRef]
- Bentley, R. A fresh look at natural tropolonoids. Nat. Prod. Rep. 2008, 25, 118–138. [Google Scholar] [CrossRef]
- Liu, N.; Song, W.; Schienebeck, C.M.; Zhang, M.; Tang, W. Synthesis of Naturally Occurring Tropones and Tropolones. Tetrahedron 2014, 70, 9281–9305. [Google Scholar] [CrossRef]
- Janik, M.E.; Bane, S.L. Synthesis and antimicrotubule activity of combretatropone derivatives. Bioorg. Med. Chem. 2002, 10, 1895–1903. [Google Scholar] [CrossRef]
- Hahn, K.M.; Humphreys, W.G.; Helms, A.M.; Hastie, S.B.; Macdonald, T.L. Structural requirements for the binding of colchicine analogs to tubulin: The role of the C-10 substituent. Bioorg. Med. Chem. Lett. 1991, 1, 471–476. [Google Scholar] [CrossRef]
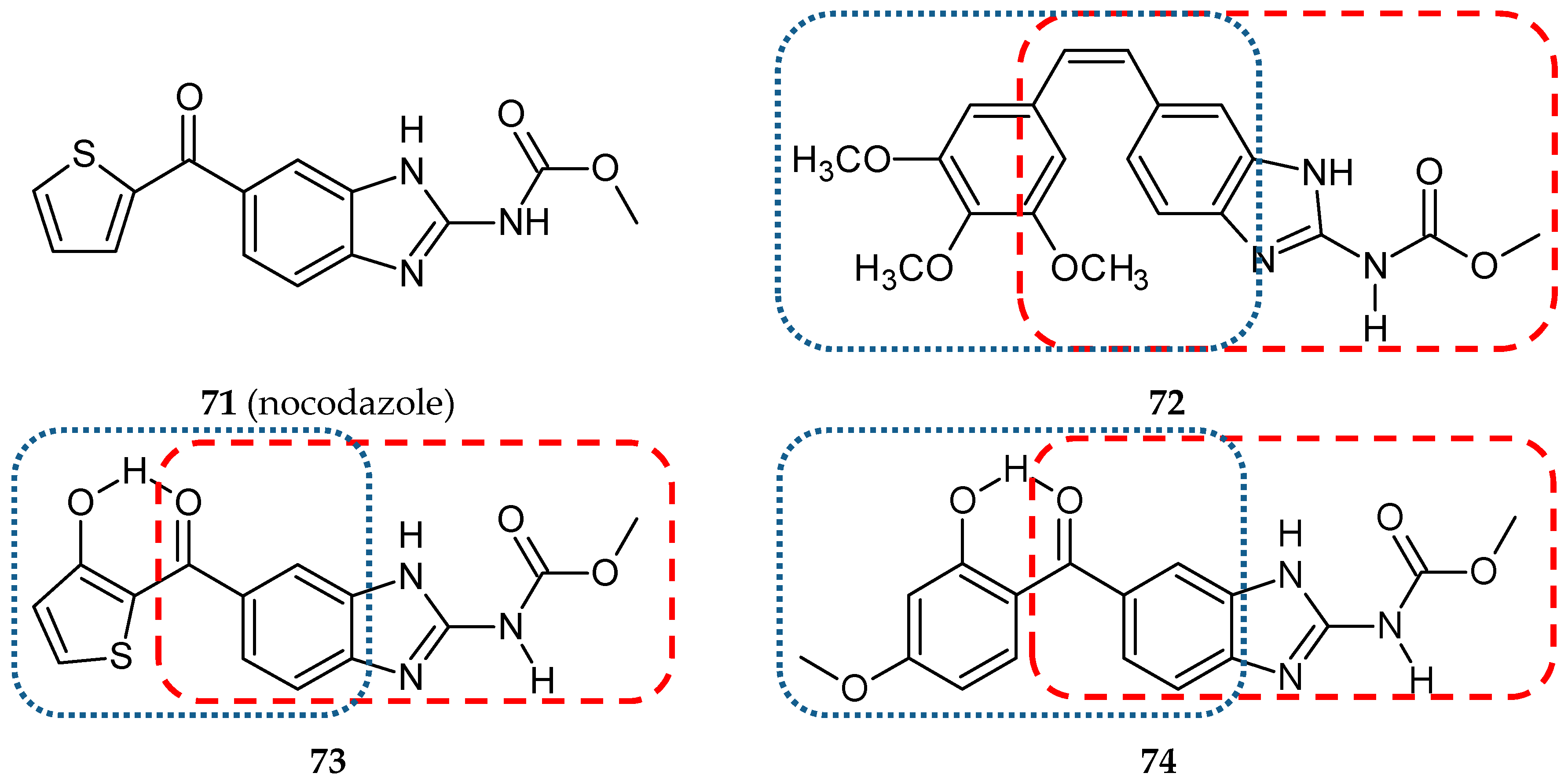
- Kamal, A.; Shaik, A.B.; Polepalli, S.; Kumar, G.B.; Reddy, V.S.; Mahesh, R.; Garimella, S.; Jain, N. Synthesis of arylpyrazole linked benzimidazole conjugates as potential microtubule disruptors. Bioorg. Med. Chem. 2015, 23, 1082–1095. [Google Scholar] [CrossRef]
- Isshiki, K.; Hirase, T.; Matsuda, S.; Miyamoto, K.; Tsuji, A.; Yuasa, K. Death-associated protein kinase 2 mediates nocodazole-induced apoptosis through interaction with tubulin. Biochem. Biophys. Res. Commun. 2015, 468, 113–118. [Google Scholar] [CrossRef]
- Kale, S.S.; Jedhe, G.S.; Meshram, S.N.; Santra, M.K.; Hamel, E.; Sanjayan, G.J. Novel hybrid nocodazole analogues as tubulin polymerization inhibitors and their antiproliferative activity. Bioorg. Med. Chem. Lett. 2015, 25, 1982–1985. [Google Scholar] [CrossRef]
- Yan, J.; Guo, Y.; Wang, Y.; Mao, F.; Huang, L.; Li, X. Design, synthesis, and biological evaluation of benzoselenazole-stilbene hybrids as multi-target-directed anti-cancer agents. Eur. J. Med. Chem. 2015, 95, 220–229. [Google Scholar] [CrossRef]
- Huang, X.; Huang, R.; Gou, S.; Wang, Z.; Liao, Z.; Wang, H. Combretastatin A-4 Analogue: A Dual-Targeting and Tubulin Inhibitor Containing Antitumor Pt(IV) Moiety with a Unique Mode of Action. Bioconj. Chem. 2016, 27, 2132–2148. [Google Scholar] [CrossRef]
- Schmitt, F.; Gosch, L.C.; Dittmer, A.; Rothemund, M.; Mueller, T.; Schobert, R.; Biersack, B.; Volkamer, A.; Hopfner, M. Oxazole-Bridged Combretastatin A-4 Derivatives with Tethered Hydroxamic Acids: Structure(-)Activity Relations of New Inhibitors of HDAC and/or Tubulin Function. Int. J. Mol. Sci. 2019, 20, 383. [Google Scholar] [CrossRef]
- Kelly, P.M.; Keely, N.O.; Bright, S.A.; Yassin, B.; Ana, G.; Fayne, D.; Zisterer, D.M.; Meegan, M.J. Novel Selective Estrogen Receptor Ligand Conjugates Incorporating Endoxifen-Combretastatin and Cyclofenil-Combretastatin Hybrid Scaffolds: Synthesis and Biochemical Evaluation. Molecules 2017, 22, 1440. [Google Scholar] [CrossRef]
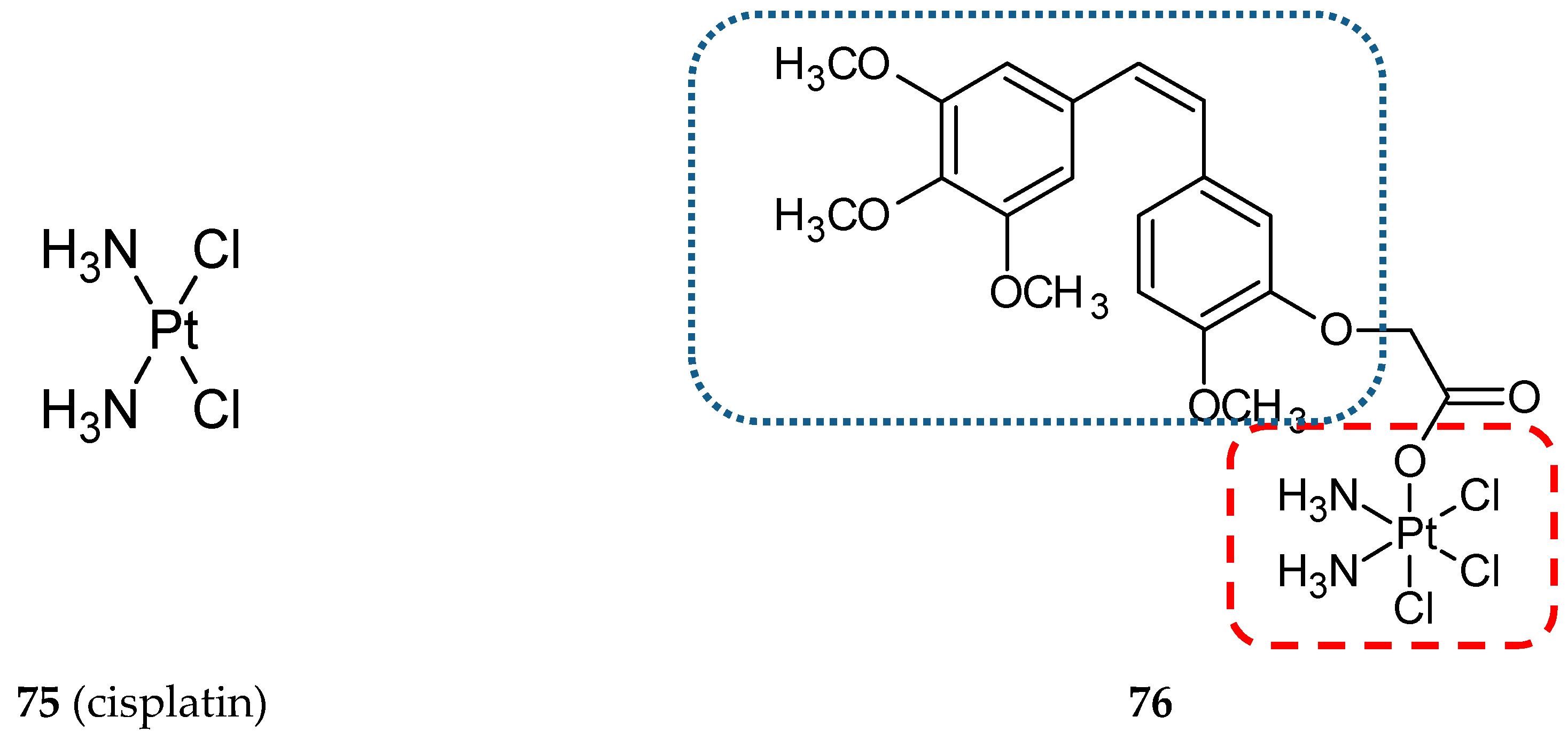
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Huang, X.; Hua, S.; Huang, R.; Liu, Z.; Gou, S.; Wang, Z.; Liao, Z.; Wang, H. Dual-targeting antitumor hybrids derived from Pt(IV) species and millepachine analogues. Eur. J. Med. Chem. 2018, 148, 1–25. [Google Scholar] [CrossRef]
- Liu, W.; Liang, L.; Zhao, L.; Tan, H.; Wu, J.; Qin, Q.; Gou, X.; Sun, X. Synthesis and characterization of a photoresponsive doxorubicin/combretastatin A4 hybrid prodrug. Bioorg. Med. Chem. Lett. 2019, 29, 487–490. [Google Scholar] [CrossRef]
- Hura, N.; Naaz, A.; Prassanawar, S.S.; Guchhait, S.K.; Panda, D. Drug-Clinical Agent Molecular Hybrid: Synthesis of Diaryl(trifluoromethyl)pyrazoles as Tubulin Targeting Anticancer Agents. ACS Omega 2018, 3, 1955–1969. [Google Scholar] [CrossRef]
- Regulski, M.; Regulska, K.; Prukala, W.; Piotrowska, H.; Stanisz, B.; Murias, M. COX-2 inhibitors: A novel strategy in the management of breast cancer. Drug Discov. Today 2016, 21, 598–615. [Google Scholar] [CrossRef]





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid Molecule | Entity I | Entity II | Cell Line | Ratio # | Ref. |
|---|---|---|---|---|---|
| 9 | Tamoxifen | CA-4 | MCF-7 | 0.625 *a | [32] |
| 11 | Chlorambucil | CA-4 | SH-SY5Y | 0.427 *a | [34] |
| 13 | Pironetin | CA-4 | HT-29 | 0.095 *a | [35] |
| 21 | Amidobenzothiazole | CA-4 | EKVX | 0.481 **a | [36] |
| 25 | Phenylcinnamide | CA-4 | MCF-7 | 0.939 **a | [37] |
| DU145 | 0.978 **a | ||||
| 29 | 2-Azetidinone | CA-4 | CT-26 | 0.746 *a | [38] |
| Caco-2 | 0.371 *a | ||||
| HT-29 | 0.006 *a | ||||
| 53 | Chalcone | Phenstatin | MCF-7 | 0.625 *b | [39] |
| MDA-MB-231 | 0.800 *b | ||||
| 59 | Estradiol | Phenstatin | MDA-MB-231 | 0.714*c | [40] |
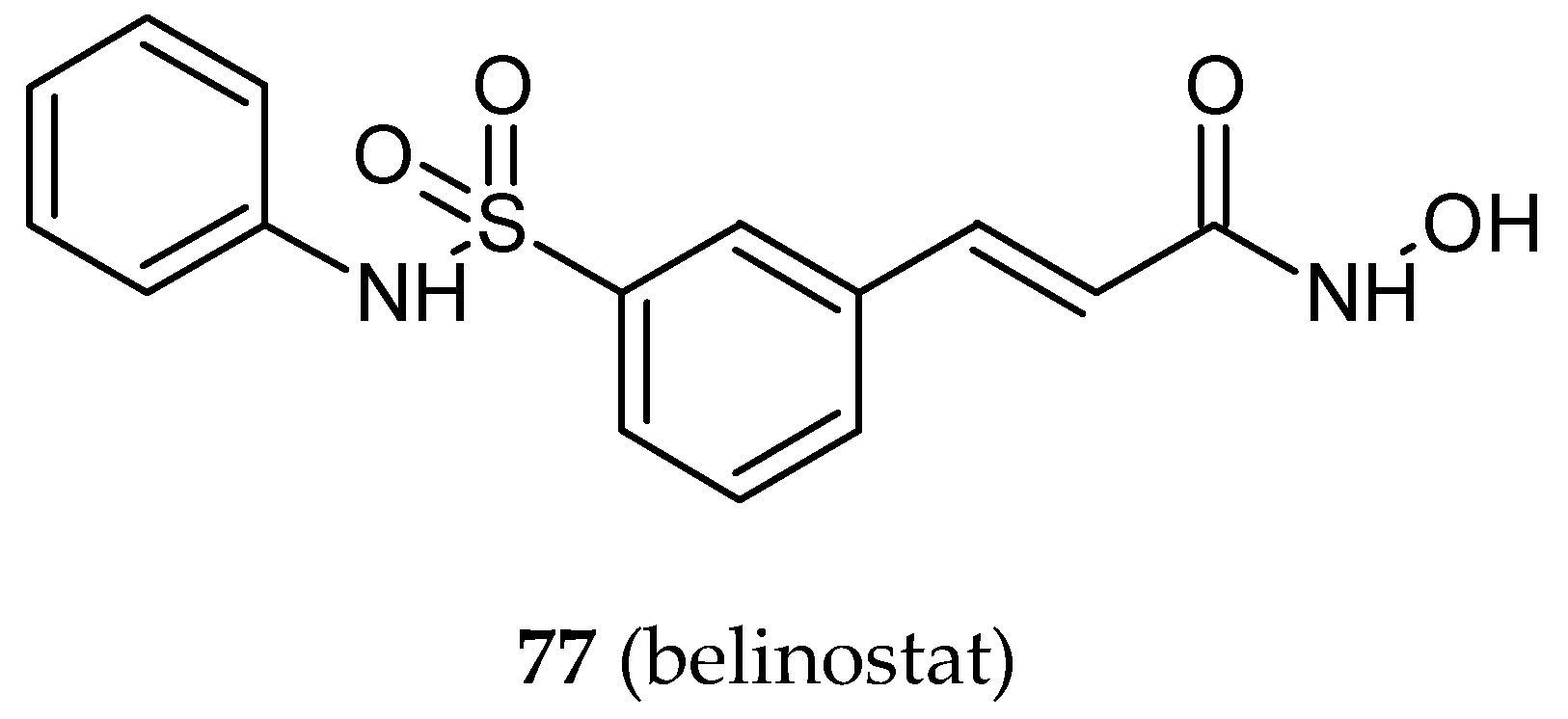
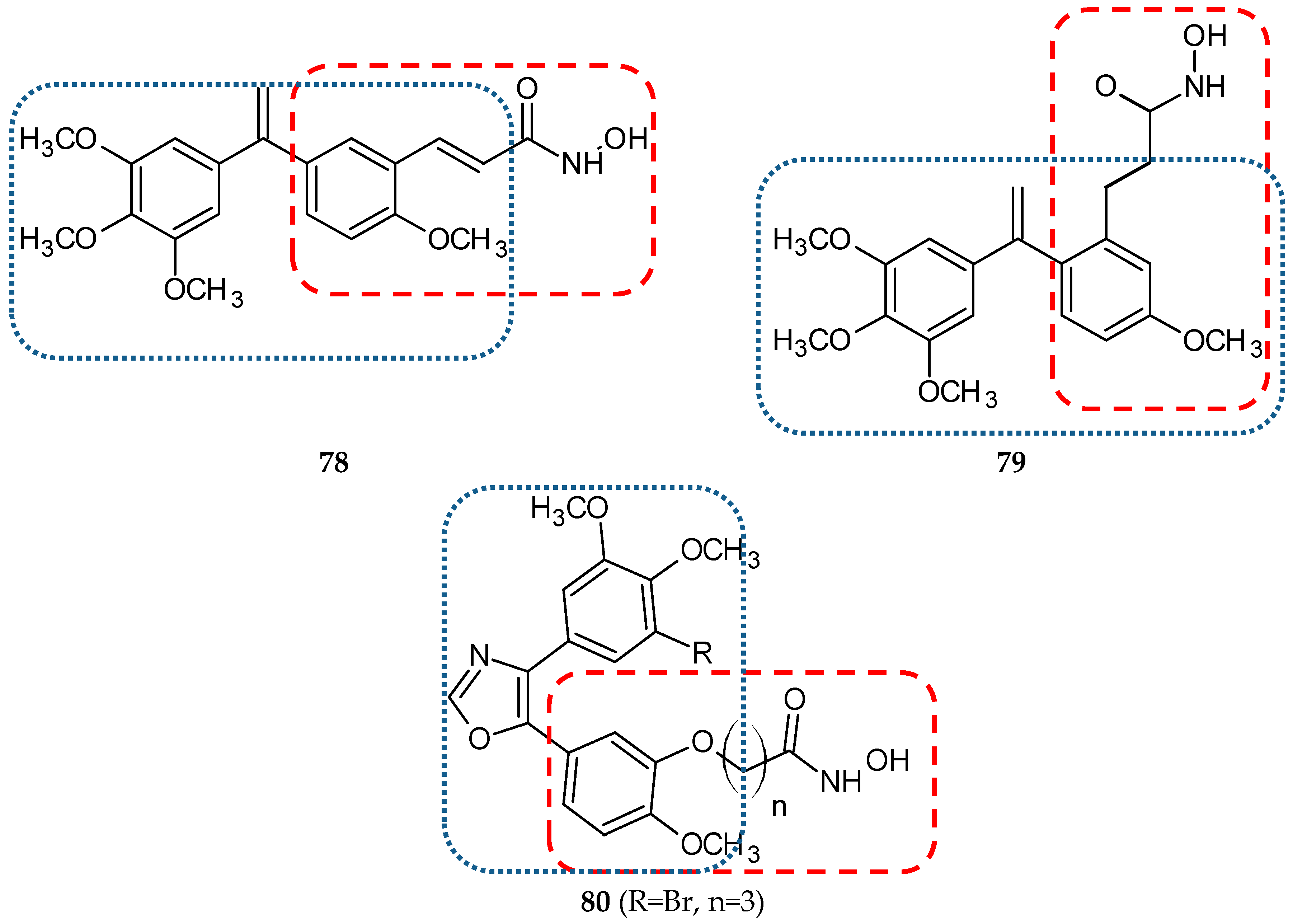
| 78 | Belinostat | isoCA-4 | HCT-116 | 0.750 **d | [41] |
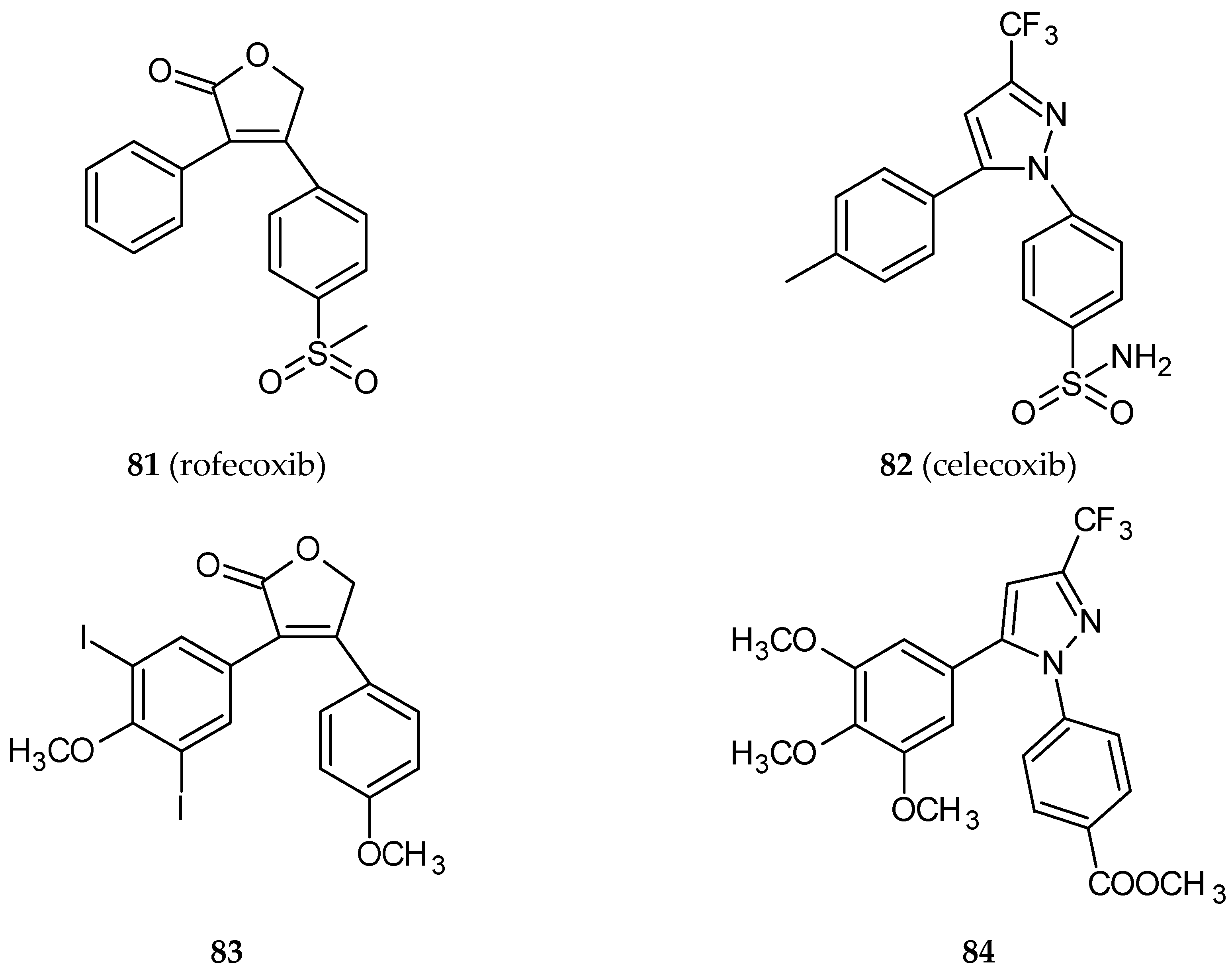
| 83 | Rofecoxib | CA-4 | HT-29 | 0.058 *a | [42] |
| LoVo | 0.676 *a |
| Hybrid Number | Dose | Model (Animal/Sex/Tumor Cell Line) | Main Outcomes (Cancer) | Pharmacological Effects/Toxicity/Adverse Effects | Ref. |
|---|---|---|---|---|---|
| 28 | 40 mg/kg (i.p.) single dose | Tumor xenograft (Mi/F/CT-26) | decrease tumor volume after 11 days | rough coat, diarrhoea, loss of appetite, mortality rate 1/5 | [38] |
| 40 | 25, 50, 100 mg/kg/2 days (i.p.) | Tumor xenograft (Mi/M/BEL-7402) | significantly lower size and weight of the tumors | no differences observed in body weight, heart, liver, spleen, lung, kidney, brain | [63] |
| 59 | 5, 50, 300 mg/kg (p.o.) single dose | (Mi/F and M) | − | no significant changes in tested haematological and biochemical parameters | [40] |
| 60 | 5, 50, 300, 1000 mg/kg (p.o.) single dose | (Mi/F and M) | − | no significant changes in tested haematological and biochemical parameters up to dose 300 mg/kg | [72] |
| 76 | 5, 10 mg/kg/week (i.v.) | Tumor xenograft (Mi/F/HepG-2) | high antitumor activity (better than CA-4) | lower body weight loss in comparison to cisplatin | [86] |
| 80 | 1 × 100 mg/kg (i.p.) 1 × 200 mg/kg (p.o.) | (Nude Mice) | − | no significant toxicity (no weight loss, normal behaviour) | [87] |
| 83 | 25 mg/kg/day (i.p.) | Tumor xenograft (Mi/F/HT-29) | reduction of the overall tumor weight compared to CA-4 treatment | no remarkable changes in the average body weights; no significant changes in liver, kidney, heart, spleen, and brain tissues | [42] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piekuś-Słomka, N.; Mikstacka, R.; Ronowicz, J.; Sobiak, S. Hybrid cis-stilbene Molecules: Novel Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1300. https://doi.org/10.3390/ijms20061300
Piekuś-Słomka N, Mikstacka R, Ronowicz J, Sobiak S. Hybrid cis-stilbene Molecules: Novel Anticancer Agents. International Journal of Molecular Sciences. 2019; 20(6):1300. https://doi.org/10.3390/ijms20061300
Chicago/Turabian StylePiekuś-Słomka, Natalia, Renata Mikstacka, Joanna Ronowicz, and Stanisław Sobiak. 2019. "Hybrid cis-stilbene Molecules: Novel Anticancer Agents" International Journal of Molecular Sciences 20, no. 6: 1300. https://doi.org/10.3390/ijms20061300
APA StylePiekuś-Słomka, N., Mikstacka, R., Ronowicz, J., & Sobiak, S. (2019). Hybrid cis-stilbene Molecules: Novel Anticancer Agents. International Journal of Molecular Sciences, 20(6), 1300. https://doi.org/10.3390/ijms20061300