Development and Validation of Markers for the Fertility Restorer Gene Rf1 in Sunflower


 ,
,  and
and
Abstract
1. Introduction
2. Results
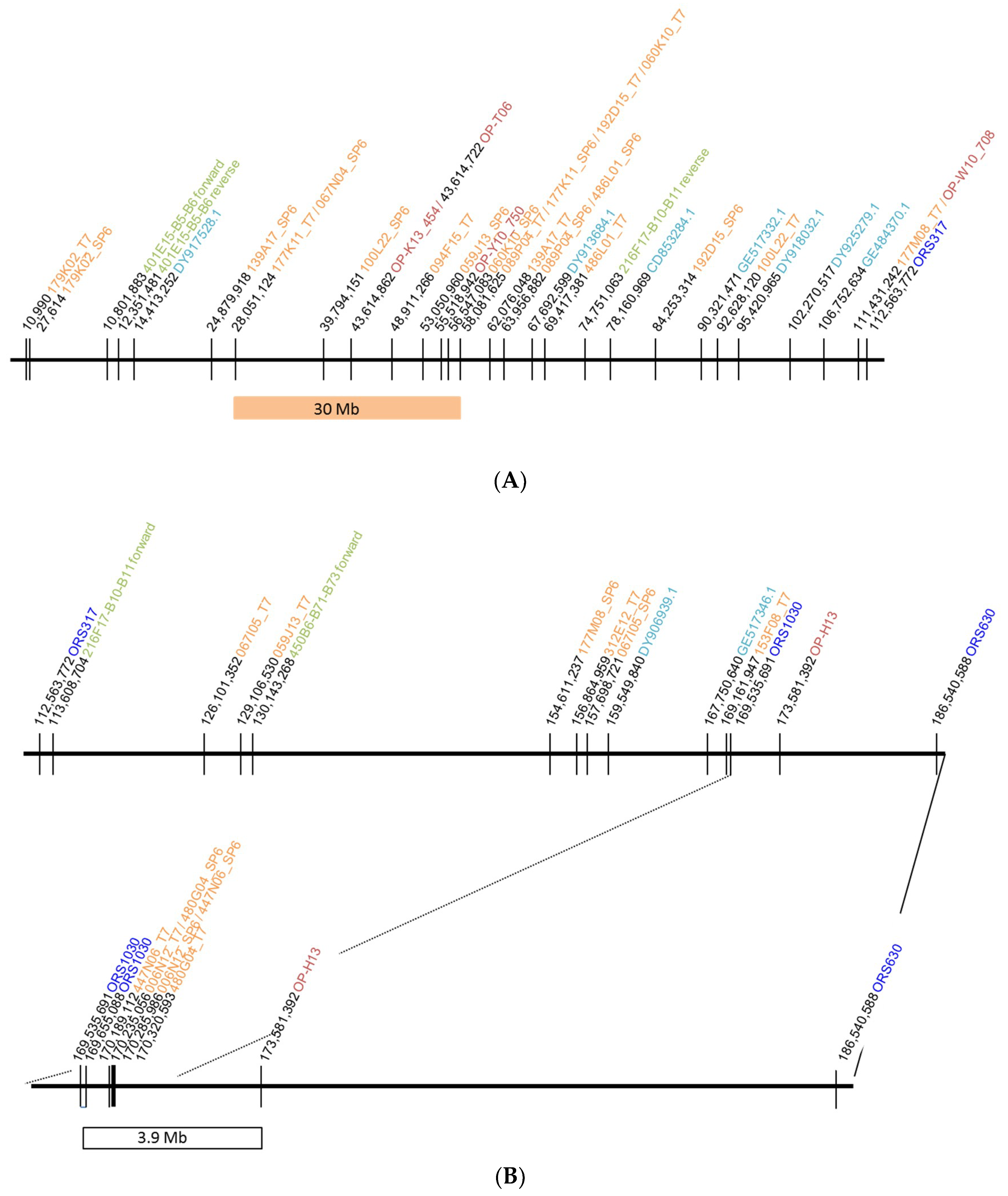
2.1. Localization of the Restorer Gene Rf1 in the Sunflower Reference Genome
2.1.1. Identification of BAC Clones by Hybridization with Cloned Markers for the Restorer Gene Rf1
2.1.2. Homology Search against the Reference Sunflower Genome
2.1.3. Potential Candidate Genes for Rf1 in the Annotated Sunflower Reference Genome
2.2. Association Studies for SNPs Linked to the Restorer Gene Rf1 Performed
2.2.1. SNP Detection in the Sunflower Association Panel
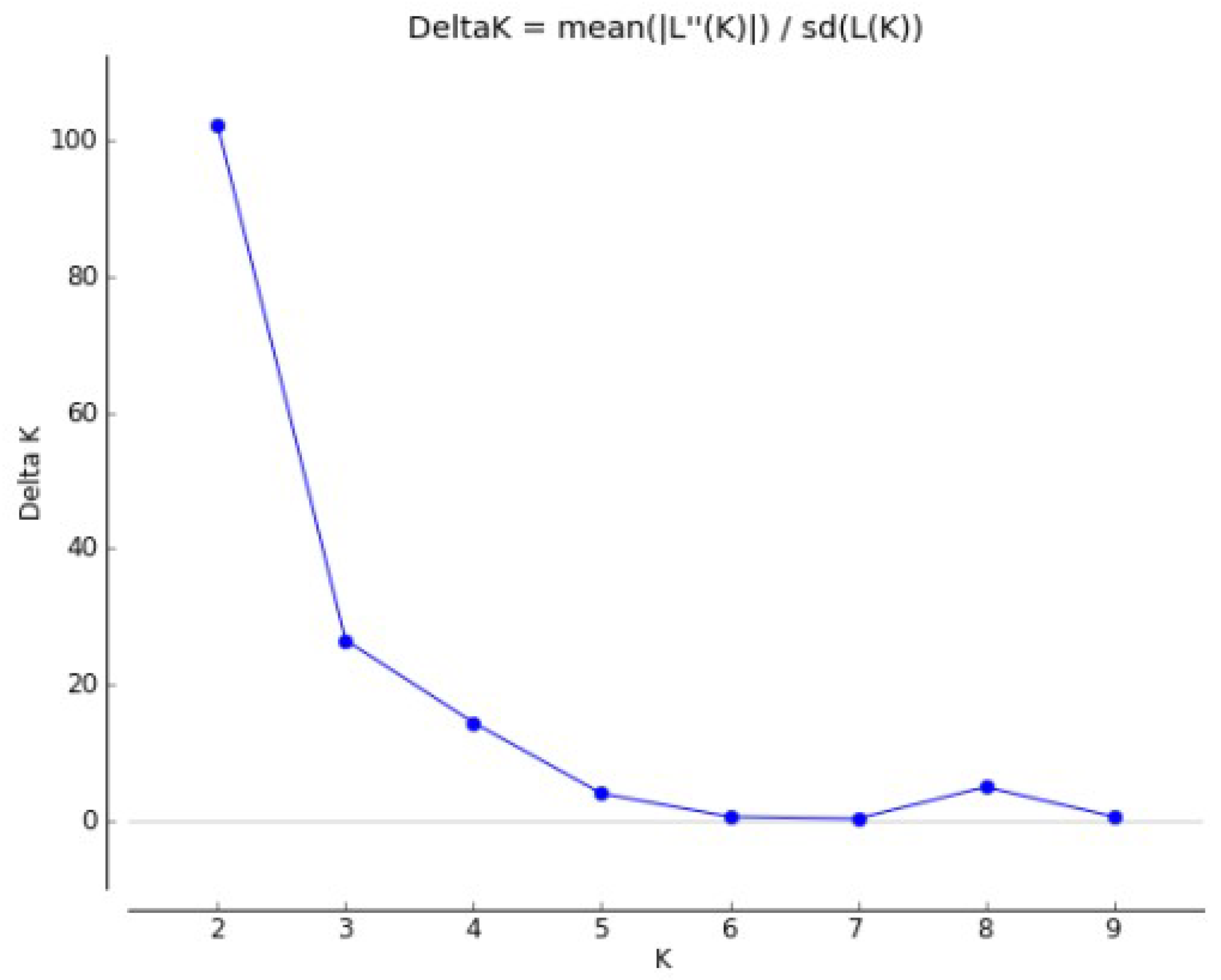
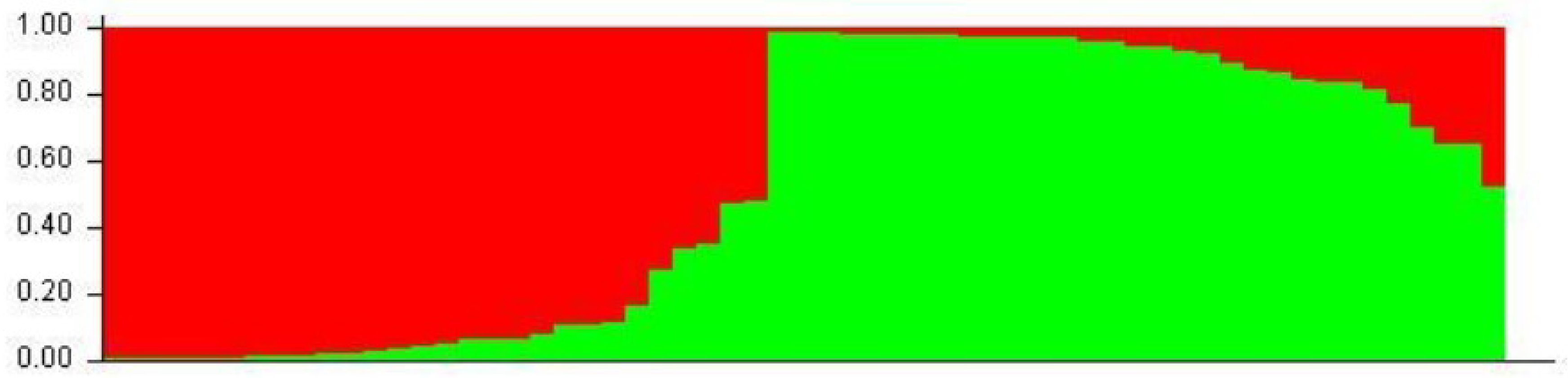
2.2.2. Populations Structure
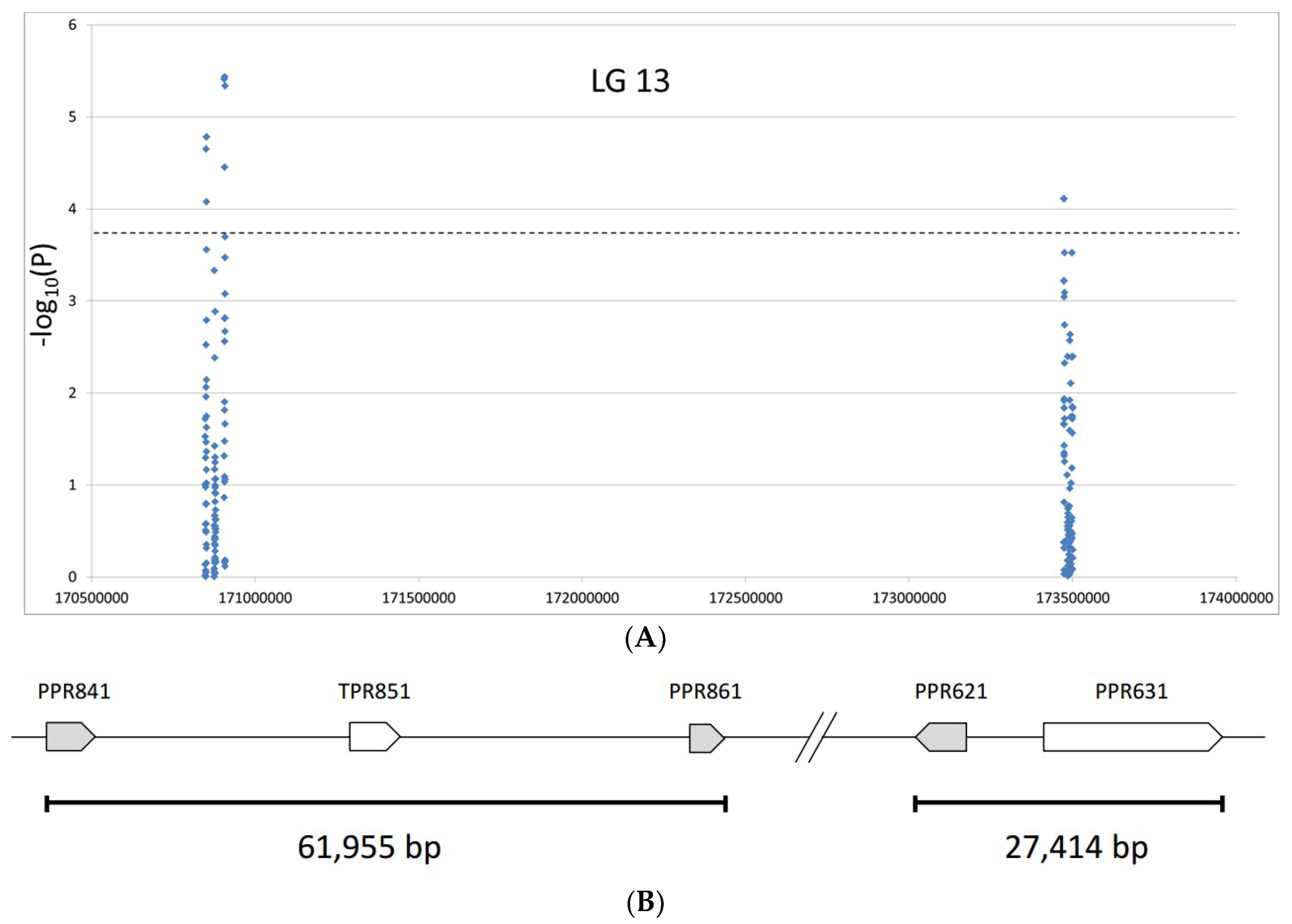
2.2.3. Association of SNPs with Fertility Restoration
2.3. Development of New SNP-Based Markers for the Restorer Gene Rf1
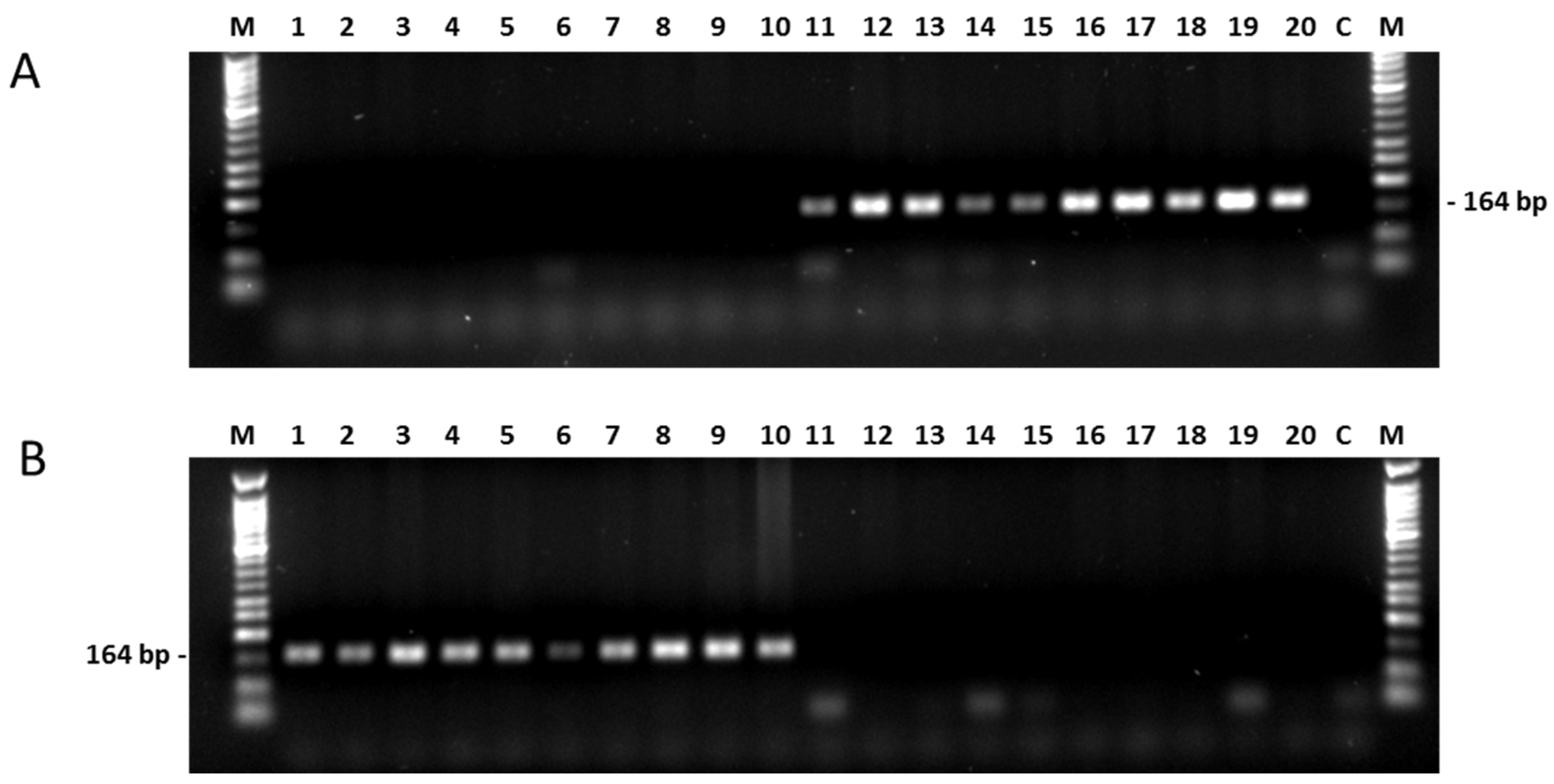
2.3.1. PAMSA Marker System
2.3.2. New SNP-Based Markers from the Association Analyses
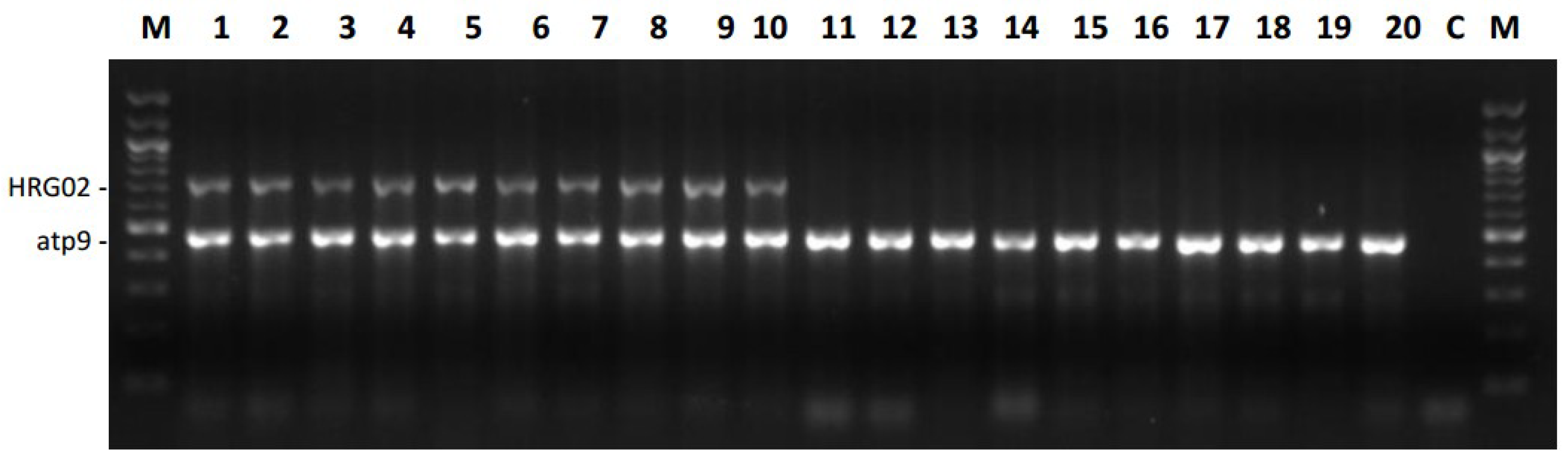
2.4. Versatility of Markers for the Restorer Gene Rf1
Testing Markers in A Large Association Panel for Their Efficiency
3. Discussion
4. Material and Methods
4.1. Plant Material for Association Mapping and Validation
4.2. Cloning and Sequencing of Markers Linked to the Restorer Gene Rf1
4.3. Microsatellite Markers
4.4. PAMSA Marker
4.5. SCAR Markers
4.6. Population Structure
4.7. Amplicon Sequencing
4.8. SNP/INDEL Detection
4.9. Association Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dimitrijevic, A.; Horn, R. Sunflower hybrid breeding: From markers to genomic selection. Front. Plant Sci. 2018, 8, 2238. [Google Scholar] [CrossRef] [PubMed]
- Vear, F. Changes in sunflower breeding over the last fifty years. OCL 2016, 23, D202. [Google Scholar] [CrossRef]
- Leclercq, P. Une sterilite male chez le tourneso1l. Ann. Amelior. Plant. 1969, 19, 99–106. [Google Scholar]
- Bohra, A.; Jha, U.C.; Adhimoolam, P.; Bisht, D.; Singh, N.P. Cytoplasmic male sterility (CMS) in hybrid breeding in field crops. Plant Cell Rep. 2016, 35, 967–993. [Google Scholar] [CrossRef] [PubMed]
- Kinman, M.L. New developments in the USDA and state experiment station sunflower breeding programs. In Proceedings of the 4th International Sunflower Conference, Memphis, TN, USA, 23 June 1970; pp. 181–183. [Google Scholar]
- Korell, M.; Mösges, G.; Friedt, W. Construction of a sunflower pedigree map. Helia 1992, 15, 7–16. [Google Scholar]
- Serieys, H. Identification, study, utilization in breeding programs of new CMS sources. In Proceedings of the Sunflower Subnetwork Progress Report, Novi Sad, Serbia, 17–20 July 2005; FAO: Rome, Italy, 2005; pp. 47–53. [Google Scholar]
- Ben-Ari, G.; Lavi, U. Marker-assisted selection in plant breeding. In Plant Biotechnology and Agriculture; Altman, A., Hasegawa, P.M., Eds.; Academic Press: Cambridge, MA, USA, 2012; Chapter 11; pp. 163–184. [Google Scholar]
- Horn, R.; Kusterer, B.; Lazarescu, E.; Prüfe, M.; Friedt, W. Molecular mapping of the Rf1 gene restoring pollen fertility in PET1-based F1-hybrids in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 2003, 106, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Markin, N.; Usatov, A.; Makarenko, M.; Azarin, K.; Gorbachenko, O.; Kolokolova, N.; Usatenko, T.; Markina, O.; Gavrilova, V. Study of informative DNA markers of the Rf1 gene in sunflower for breeding practice. Plant Breed 2017, 53, 69–75. [Google Scholar] [CrossRef]
- Kusterer, B.; Horn, R.; Friedt, W. Molecular mapping of the fertility restoration locus Rf in sunflower and development of diagnostic markers for the restorer gene. Euphytica 2005, 143, 35–43. [Google Scholar] [CrossRef]
- Yue, B.; Vick, B.A.; Cai, X.; Hu, J. Genetic mapping for the Rf1 (fertility restoration) gene in sunflower (Helianthus annuus L.) by SSR and TRAP markers. Plant Breed 2010, 129, 24–28. [Google Scholar] [CrossRef]
- Hamrit, S.; Kusterer, B.; Friedt, W.; Horn, R. Verification of positive BAC clones near the Rf1 gene restoring pollen fertility in the presence of the PET1 cytoplasm in sunflower (Helianthus annuus L.) and direct isolation of BAC ends. In Proceedings of the 17th International Sunflower Conference, Córdoba, Spain, 8–12 June 2008; pp. 623–628. [Google Scholar]
- Hamrit, S. Chromosome walking at the fertility restorer locus Rf1 in sunflower (Helianthus annuus L.). Ph.D. Thesis, Mathematisch-Naturwissenschaftlichen Fakultät der Universität Rostock, Rostock, Germany, 2009. [Google Scholar]
- Badouin, H.; Gouzy, J.; Grassa, C.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef]
- Horn, R.; Gupta, J.K.; Colombo, N. Mitochondrion role in molecular basis of cytoplasmic male sterility. Mitochondrion 2014, 19, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Lurin, C.; Andres, C.; Aubourg, S.; Bellaoui, M.; Bitton, F.; Bruyere, C.; Caboche, M.; Debast, C.; Gualberto, J.; Hoffmann, B.; et al. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 2004, 16, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Linneweber, C.; Small, I. Pentatricopeptide repeat proteins: A socket set for organelle gene expression. Trends Plant Sci. 2008, 13, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Gaborieau, L.; Brown, G.G.; Mireau, H. The propensity of pentatricopeptide repeat genes to evolve into restorers of cytoplasmic male sterility. Front. Plant Sci. 2016, 7, 1816. [Google Scholar] [CrossRef] [PubMed]
- Bentolila, S.; Alfonso, A.A.; Hanson, M.R. A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male-sterile plants. Proc. Natl. Acad. Sci. USA 2002, 99, 10887–10892. [Google Scholar] [CrossRef]
- Brown, G.G.; Formanova, N.; Jin, H.; Wargachuk, R.; Dendy, C.; Patil, P.; Laforest, M.; Zhang, J.; Cheung, W.Y.; Landry, B.S. The radish Rfo restorer gene of Ogura cytoplasmic male sterility encodes a protein with multiple pentatricopeptide repeats. Plant J. 2003, 35, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Desloire, S.; Gherbi, H.; Laloui, W.; Marhadour, S.; Clouet, V.; Cattolico, L.; Falentin, C.; Giancola, S.; Renard, M.; Budar, F.; et al. Identification of the fertility restoration locus, Rfo, in radish, as a member of the pentatricopeptide-repeat protein family. EMBO Rep. 2003, 4, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Koizuka, N.; Imai, R.; Fujimoto, H.; Hayakawa, T.; Kimuraa, Y.; Kohno-Murase, J.; Sakai, T.; Kawasaki, S.; Imamura, J. Genetic characterization of a pentatricopeptide repeat protein gene, orf687, that restores fertility in the cytoplasmic male-sterile Kosena radish. Plant J. 2003, 34, 407–415. [Google Scholar] [CrossRef]
- Wang, Z.; Zou, Y.; Li, X.; Zhang, Q.; Chen, L.; Wu, H.; Su, D.; Chen, Y.; Guo, J.; Luo, D.; et al. Cytoplasmic male sterility of rice with boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell 2006, 18, 676–687. [Google Scholar] [CrossRef]
- Klein, R.R.; Klein, P.E.; Mullet, J.E.; Minx, P.; Rooney, W.L.; Schertz, K.F. Fertility restorer locus Rf1 of sorghum (Sorghum bicolor L.) encodes a pentatricopeptide repeat protein not present in the colinear region of rice chromosome 12. Theor. Appl. Genet. 2005, 111, 994–1012. [Google Scholar] [CrossRef]
- Hu, J.; Wang, K.; Huang, W.; Liu, G.; Gao, Y.; Wang, J.; Huang, Q.; Ji, Y.; Qin, X.; Wan, L. The rice pentatricopeptide repeat protein RF5 restores fertility in hong-lian cytoplasmic male–sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell 2012, 24, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yu, C.; Hu, J.; Wang, L.; Dan, Z.; Zhou, W.; He, C.; Zeng, Y.; Yao, G.; Qi, J.; et al. Pentatricopeptide-repeat family protein RF6 function with hexokinase 6 to rescue rice cytoplasmic male sterility. Proc. Natl. Acad. Sci. USA 2015, 112, 14984–14989. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Dong, F.; Wang, X.; Wang, T.; Su, R.; Hong, D.; Yang, G. A pentatriopeptide repeat protein restores nap cytoplasmic male sterility in Brassica napus. J. Exp. Bot. 2017, 68, 4115–4123. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Wise, R.P.; Schnable, P.S. The rf2 nuclear restorer gene of male-sterile T-cytoplasm in maize. Science 1996, 272, 1334–1336. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cui, X.; Horner, H.T.; Winer, H.; Schnable, P.S. Mitochondrial aldehyde dehydrogenase activity is required for male fertility in maize. Plant Cell 2001, 13, 1063–1078. [Google Scholar] [CrossRef]
- Fujii, S.; Toriyama, K. Suppressed expression of RETROGRADE_REGULATED MALE STERILITY restores pollen fertility in cytoplasmic male sterile rice plants. Proc. Natl. Acad. Sci. USA 2009, 106, 9513–9518. [Google Scholar] [CrossRef] [PubMed]
- Itabashi, E.; Iwata, N.; Fujii, S.; Kazama, T.; Toriyama, K. The fertility restorer gene, Rf2, for Lead Rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine-rich protein. Plant J. 2011, 65, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Matsuhira, H.; Kagami, H.; Kurata, M.; Kitazaki, K.; Matsunaga, M.; Hamaguchi, Y.; Hagihara, E.; Ueda, M.; Harada, M.; Muramatsu, A.; et al. Unusual and typical features of a novel restorer-of-fertility gene in sugar beet (Beta vulgaris L.). Genetics 2012, 192, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Kitazaki, K.; Arakawa, T.; Matsunaga, M.; Yui-Kurino, R.; Matsuhira, H.; Mikami, T.; Kubo, T. Post-translational mechanisms are associated with fertility restoration of cytoplasmic male sterility in sugar beet. Plant J. 2015, 83, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Köhler, R.H.; Horn, R.; Lössl, A.; Zetsche, K. Cytoplasmic male sterility in sunflower is correlated with the co-transcription of a new open reading frame with the atpA gene. Mol. Gen. Genet. 1991, 227, 369–376. [Google Scholar] [CrossRef]
- Horn, R.; Köhler, R.H.; Zetsche, K. A mitochondrial 16 kDa protein is associated with cytoplasmic male sterility in sunflower. Plant Mol. Biol. 1991, 17, 29–36. [Google Scholar] [CrossRef]
- Laver, H.K.; Reynolds, S.J.; Monéger, F.; Leaver, C.J. Mitochondrial genome organization and expression associated with cytoplasmic male sterility in sunflower (Helianthus annuus). Plant J. 1991, 1, 185–193. [Google Scholar] [CrossRef]
- Monéger, F.; Smart, C.J.; Leaver, C.J. Nuclear restoration of cytoplasmic male sterility in sunflower is associated with the tissue-specific regulation of a novel mitochondrial gene. EMBO J. 1994, 13, 8–17. [Google Scholar] [CrossRef]
- Smart, C.J.; Moneger, F.; Leaver, C.J. Cell-specific regulation of gene expression in mitochondria during anther development in sunflower. Plant Cell 1994, 6, 811–825. [Google Scholar] [CrossRef]
- Lange, H.; Sement, F.M.; Canaday, J.; Gagliardi, D. Polyadenylation-assisted RNA degradation processes in plants. Trends Plant Sci. 2009, 14, 497–504. [Google Scholar] [CrossRef]
- Vi, S.L.; Trost, G.; Lange, P.; Czesnick, H.; Rao, N.; Lieber, D.; Laux, T.; Gray, W.M.; Manley, J.L.; Groth, D.; et al. Target specific among canonical nuclear poly(A) polymerases in plants modulates organ growth and pathogen response. Proc. Natl. Acad. Sci. USA 2013, 110, 13994–13999. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, N.; Horn, R.; Friedt, W. Construction and characterization of a BAC library for sunflower (Helianthus annuus L.). Euphytica 2004, 138, 177–183. [Google Scholar] [CrossRef]
- Livaja, M.; Unterseer, S.; Erath, W.; Lehermeier, C.; Wieseke, R.; Ploeske, J.; Polley, A.; Luerßen, H.; Wieckhorst, S.; Mascher, M.; et al. Diversity analysis and genomic prediction of Sclerotinia resistance in sunflower using a new 25 K SNP genotyping array. Theor. Appl. Genet. 2016, 129, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Kishore, V.K.; Knapp, S.J. PCR-multiplexes for a genome-wide framework of simple sequence repeat marker loci in cultivated sunflower. Theor. Appl. Genet. 2003, 107, 6–19. [Google Scholar] [CrossRef]
- Gaudet, M.; Fara, A.G.; Sabatti, M.; Kuzminsky, E.; Mugnozza, G.S. Single-reaction for SNP genotyping on agarose gel by allele-specific PCR in black poplar (Populus nigra L.). Plant Mol. Biol. Rep. 2007, 25, 1–9. [Google Scholar] [CrossRef]
- Collard, B.C.Y.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. 2007, 363, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Reif, J.C.; Mallauer, A.R.; Melchinger, A.E. Heterosis and heterotic patterns in maize. Maydica 2005, 50, 215–223. [Google Scholar]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and prospect of association mapping in plants. Plant Genome 2008, 1, 5–20. [Google Scholar] [CrossRef]
- Mandel, J.R.; Nambeesan, S.; Bowers, J.E.; Marek, L.; Ebert, D.; Rieseberg, L.H.; Knapp, S.J.; Burke, J.M. Association mapping and the genomic consequences of selection in sunflower. PLoS Genet. 2013, 9, e1003378. [Google Scholar] [CrossRef] [PubMed]
- Nambeesan, S.U.; Mandel, J.R.; Bowers, J.E.; Marek, L.F.; Ebert, D.; Corbi, J.; Rieseberg, L.H.; Knapp, S.J.; Burke, J.M. Association mapping in sunflower (Helianthus annuus L.) reveals independent control of apical vs. basal branching. BMC Plant Biol. 2015, 15, 84. [Google Scholar] [CrossRef]
- Cadic, E.; Coque, M.; Vear, F.; Grezes-Besset, B.; Puaquet, J.; Piquemal, J.; Lippi, Y.; Blanchard, P.; Romestant, M.; Pouilly, N.; et al. Combined linkage and association mapping of flowering time in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 2013, 126, 1337–1356. [Google Scholar] [CrossRef] [PubMed]
- McAssey, E.; Corbi, J.; Burke, J.M. Range-wide phenotypic and genetic differentiation in wild sunflower. BMC Plant Biol. 2016, 16, 249. [Google Scholar] [CrossRef] [PubMed]
- Talukder, Z.I.; Hulke, B.S.; Qi, L.; Scheffler, B.E.; Pegadaraju, V.; McPhee, K.; Gulya, T.J. Candidate gene association of Sclerotinia rot resistance in sunflower (Helianthus annuus L.) uncovers the importance of COI1 homologs. Theor. Appl. Genet. 2014, 127, 193–209. [Google Scholar] [CrossRef]
- Fujii, S.; Bond, C.S.; Small, I.D. Selection patterns on restorer-like genes reveal a conflict between nuclear and mitochondrial genomes throughout angiosperm evolution. Proc. Natl. Acad. Sci. USA 2011, 108, 1723–1728. [Google Scholar] [CrossRef]
- Geddy, R.; Brown, G.G. Genes encoding pentatricopeptide repeat (PPR) proteins are not conserved in location in plant genomes and may be subject to diversifying selection. BMC Genom. 2007, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, N.; Hattori, M.; Andres, C.; Iida, K.; Lurin, C.; Schmitz-Linneweber, C.; Sugita, M.; Small, I. On the expansion of the pentatricopeptide repeat gene family in plants. Mol. Biol. Evol. 2008, 25, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Stiti, N.; Misshoun, T.D.; Kotchoni, S.O.; Kirch, H.-H.; Bartels, D. Aldehyde dehydrogenase in Arabidopsis thaliana: Biochemical requirements, metabolic pathways, and functional analysis. Front. Plant Sci. 2011, 2, 65. [Google Scholar] [CrossRef] [PubMed]
- Meeks, L.R.; Addepalli, B.; Hunt, A.G. Characterization of genes encoding poly(A) polymerase in plants: Evidence for duplication and functional specialization. PLoS ONE 2009, 4, e8082. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.L.; Baute, G.J.; Hubner, S.; Rieseberg, L.H. Genomic sequence and copy number evolution during hybrid crop development in sunflower. Evol. Appl. 2019, 12, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Semagn, K.; Babu, R.; Hearne, S.; Olsen, M. Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): Overview of the technology and its application in crop improvement. Mol Breed. 2014, 33, 1–14. [Google Scholar] [CrossRef]
- Patterson, E.L.; Fleming, M.B.; Kessler, K.C.; Nissen, S.J.; Gaines, T.A. A KASP genotyping method to identify Northern watermilfoil, Eurasian watermilfoil, and their interspecific hybrids. Front. Plant Sci. 2017, 8, 752. [Google Scholar] [CrossRef] [PubMed]
- Broccanello, C.; Chiodi, C.; Funk, A.; McGrath, J.M.; Panella, L.; Stevanato, P. Comparison of three PCR-based assays for SNP genotyping in plants. Plant Methods 2018, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Jan, C.C.; Vick, B.A. Inheritance and allelic relationship of fertility restoration genes for seven new sources of male-sterile cytoplasm. Plant Breed. 2007, 126, 213–217. [Google Scholar] [CrossRef]
- Liu, Z.; Mulpuri, S.; Feng, J.; Vick, B.A.; Jan, C.C. Molecular mapping of the Rf3 fertility restoration gene to facilitate its utilization in breeding confection sunflower. Mol. Breed. 2012, 29, 275–284. [Google Scholar] [CrossRef]
- Qi, L.L.; Seiler, G.J.; Vick, B.A.; Gulya, T.J. Genetics and mapping of the R11 gene conferring resistance to recently emerged rust races, tightly linked to male fertility restoration, in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 2012, 125, 921–932. [Google Scholar] [CrossRef]
- Seiler, G.J.; Qi, L.L.; Marek, L.F. Utilization of sunflower crop wild relatives for cultivated sunflower improvement. Crop Sci. 2017, 57, 1083–1101. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Reddemann, R.; Horn, R. Recombination events involving the atp9 gene are associated with male sterility of CMS PET2 in sunflower. Int. J. Mol. Sci. 2018, 19, 806. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, N.; Horn, R.; Friedt, W. Isolation of HMW DNA from sunflower (Helianthus annuus L.) for BAC cloning. Plant Mol. Biol. Rep. 2002, 20, 239–249. [Google Scholar] [CrossRef]
- Chapman, M.A.; Pashley, C.H.; Wenzler, J.; Hvala, J.; Tang, S.; Knapp, S.J.; Burke, J.M. A genomic scan for selection reveals candidates for genes involved in the evolution of cultivated sunflower (Helianthus annuus L.). Plant Cell 2008, 20, 2931–2945. [Google Scholar] [CrossRef] [PubMed]
- Mandel, J.R.; Dechaine, J.M.; Marek, L.F.; Burke, J.M. Genetic diversity and population structure in cultivated sunflower and a comparison to its wild progenitor, Helianthus annuus L. Theor. Appl. Genet. 2011, 123, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Sajer, O.; Scorza, R.; Dardick, C.; Zhebentyayeva, T.; Abbott, A.G.; Horn, R. Development of sequence-tagged site markers linked to the pillar growth type in peach (Prunus persica). Plant Breed. 2012, 131, 186–192. [Google Scholar] [CrossRef]
- Qi, L.L.; Foley, M.E.; Cai, X.W.; Gulya, T.J. Genetics and mapping of a novel downy mildew resistance gene, Pl 18, introgressed from wild Helianthus argophyllus into cultivated sunflower (Helianthus annuus L.). Theor. Appl. Genet. 2016, 129, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe | Library | BAC Clone | Probe | Library | BAC Clone |
|---|---|---|---|---|---|
| OP-K13_454A | RHA 325 | 067N04 | 67N04-BAC end | RHA 325 | 139A17 |
| 059J13 | 179K02 | ||||
| 067I05 | HA 383 | 060K10 | |||
| HA 383 | 216F17 | 089P04 | |||
| 225D09 | 153F08 | ||||
| 401E15 | 177K11 | ||||
| E41M48_113A | RHA 325 | 094F15 | 177M08 | ||
| HA 383 | 100L22 | 178G06 | |||
| 147A03 | 180A24 | ||||
| 233O05 | 192D15 | ||||
| 482D10 | 312E12 | ||||
| E62M52-249A | HA 383 | 006N12 | 467E16 | ||
| 447N06 | 486L01 | ||||
| 480G04 | E44M70_275A | HA383 | 450B06 |
| Gene ID | Gene Annotation | Orientation | chrom Start | chrom End | cds Start | cds Stop | Length Total |
|---|---|---|---|---|---|---|---|
| HanXRQChr13g0392791 | Putative pentatricopeptide repeat | + | 41353989 | 41357247 | 41355989 | 41356747 | 3258 |
| HanXRQChr13g0393411 | Probable aldehyde dehydrogenase 22A1 | + | 45380421 | 45389613 | 45382421 | 45389113 | 9192 |
| HanXRQChr13g0394161 | Probable pentatricopeptide repeat (PPR) superfamily protein | - | 51625496 | 51621365 | 51623496 | 51621865 | 4131 |
| HanXRQChr13g0394751 | Probable poly(A) polymerase 3 (PAPS3) | - | 56435098 | 56428877 | 56433098 | 56429377 | 6221 |
| HanXRQChr13g0418841 | Putative pentatricopeptide repeat | + | 170848155 | 170852502 | 170850155 | 170852002 | 4347 |
| HanXRQChr13g0418851 | Putative tetratricopeptide-like helical domain | + | 170875322 | 170879807 | 170877322 | 170879307 | 4485 |
| HanXRQChr13g0418861 | Putative pentatricopeptide repeat | + | 170906019 | 170909610 | 170908019 | 170909110 | 3591 |
| HanXRQChr13g0419621 | PP198, Pentatricopeptide repeat | - | 173477525 | 173472987 | 173475525 | 173473487 | 4538 |
| HanXRQChr13g0419631 | Putative pentatricopeptide repeat | + | 173482455 | 173500901 | 173484455 | 173500401 | 18446 |
| Gene ID | Short Name | Total Variants | Whole Genome Area (Including 2000 bp Upstream and 500 bp Downstream of the Gene) | Exon | ||
|---|---|---|---|---|---|---|
| No. SNPs | No. INDELs | No. SNPs | No. INDELs | |||
| HanXRQChr13g0392791 | PPR791 | 7 | - | 7 | - | - |
| HanXRQChr13g0393411 | ALD22A1 | 9 | - | 9 | - | 6 |
| HanXRQChr13g0394161 | PPR161 | - | - | - | - | - |
| HanXRQChr13g0394751 | PAPS3 | 7 | 4 | 3 | 2 | 2 |
| HanXRQChr13g0418841 | PPR841 | 46 | 36 | 10 | 22 | 3 |
| HanXRQChr13g0418851 | TPR851 | 56 | 45 | 11 | 24 | 4 |
| HanXRQChr13g0418861 | PPR861 | 30 | 24 | 6 | 1 | - |
| HanXRQChr13g0419621 | PPR621 | 30 | 26 | 4 | 22 | 1 |
| HanXRQChr13g0419631 | PPR631 | 92 | 75 | 17 | 7 | - |
| SNP | Chromosome * | Position of SNP * | SNP_F | p-Value |
|---|---|---|---|---|
| PPR861.11 | 13 | 170,907,603 | 26.37 | 3.68 × 10−6 |
| PPR861.3 | 13 | 170,906,233 | 15.83 | 3.88 × 10−6 |
| PPR861.19 | 13 | 170,908,139 | 25.94 | 4.61 × 10−6 |
| PPR841.29 | 13 | 170,851,469 | 22.23 | 1.65 × 10−5 |
| PPR841.38 | 13 | 170,851,758 | 22.23 | 1.65 × 10−5 |
| PPR841.26 | 13 | 170,851,288 | 21.5 | 2.22 × 10−5 |
| PPR861.9 | 13 | 170,907,279 | 12.42 | 3.53 × 10−5 |
| PPR621.5 | 13 | 173,473,513 | 18.25 | 7.73 × 10−5 |
| PPR621.11 | 13 | 173,473,976 | 18.25 | 7.73 × 10−5 |
| PPR841.39 | 13 | 170,851,781 | 18.01 | 8.34 × 10−5 |
| PPR861.20 | 13 | 170,908,143 | 10.02 | 2.00 × 10−4 |
| PPR841.40 | 13 | 170,851,807 | 10.07 | 2.76 × 10−4 |
| PPR621.27 | 13 | 173,475,377 | 14.88 | 2.98 × 10−4 |
| PPR631.79 | 13 | 173,498,360 | 14.88 | 2.98 × 10−4 |
| PPR861.26 | 13 | 170,908,690 | 9.28 | 3.36 × 10−4 |
| TPR851.2 | 13 | 170,875,633 | 8.93 | 4.65 × 10−4 |
| PPR621.10 | 13 | 173,473,934 | 13.25 | 6.02 × 10−4 |
| PPR621.4 | 13 | 173,473,493 | 13.25 | 6.02 × 10−4 |
| PPR621.30 | 13 | 173,475,599 | 8.64 | 8.08 × 10−4 |
| PPR861.27 | 13 | 170,909,093 | 8.36 | 8.39 × 10−4 |
| PPR621.9 | 13 | 173,473,774 | 12.38 | 9.01 × 10−4 |
| Genotypes | PPR 841.26 | PPR 841.29 | PPR 841.38 | PPR 841.39 | PPR 861.3 | PPR 861.9 | PPR 861.11 | PPR 861.19 | PPR 621.5 | PPR 621.11 | Percentage |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HanXRQ | TT | GG | GG | CC | GG | AA | CC | GG | GG | GG | |
| Maintainer | TT | GG | GG | CC | GG | AA | CC | GG | GG | GG | 85.2% |
| Restorer | CC | GA | AA | CT | AA | TT | CT | CC | CC | -- | 56.3% |
| Restorer | CC | GA | AA | CY | RA | WT | CT | CC | CC | -- | 81.3% |
| Marker | Type of Marker | No. of Lines with Restorer Profile | No. of Lines with Maintainer Profile | No. of Heterozygous Lines | No. of Lines with Data |
|---|---|---|---|---|---|
| HRG01 (K13) | Dominant | 71 | 486 | - | 557 |
| HRG02 (Y10) | Dominant | 127 | 430 | - | 557 |
| orfH522-CMS | Dominant | 135 | 422 | - | 557 |
| H13 HinfI | Co-dominant | 71 | 332 | 6 | 409 |
| 67N04_P | Co-dominant | 130 | 426 | 19 | 556 |
| PPR621.5R | Dominant | 114 | 409 | 24 | 547 |
| PPR621.5M | Dominant | 114 | 409 | 24 | 547 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horn, R.; Radanovic, A.; Fuhrmann, L.; Sprycha, Y.; Hamrit, S.; Jockovic, M.; Miladinovic, D.; Jansen, C. Development and Validation of Markers for the Fertility Restorer Gene Rf1 in Sunflower. Int. J. Mol. Sci. 2019, 20, 1260. https://doi.org/10.3390/ijms20061260
Horn R, Radanovic A, Fuhrmann L, Sprycha Y, Hamrit S, Jockovic M, Miladinovic D, Jansen C. Development and Validation of Markers for the Fertility Restorer Gene Rf1 in Sunflower. International Journal of Molecular Sciences. 2019; 20(6):1260. https://doi.org/10.3390/ijms20061260
Chicago/Turabian StyleHorn, Renate, Aleksandra Radanovic, Lena Fuhrmann, Yves Sprycha, Sonia Hamrit, Milan Jockovic, Dragana Miladinovic, and Constantin Jansen. 2019. "Development and Validation of Markers for the Fertility Restorer Gene Rf1 in Sunflower" International Journal of Molecular Sciences 20, no. 6: 1260. https://doi.org/10.3390/ijms20061260
APA StyleHorn, R., Radanovic, A., Fuhrmann, L., Sprycha, Y., Hamrit, S., Jockovic, M., Miladinovic, D., & Jansen, C. (2019). Development and Validation of Markers for the Fertility Restorer Gene Rf1 in Sunflower. International Journal of Molecular Sciences, 20(6), 1260. https://doi.org/10.3390/ijms20061260