Ginsenoside Rh2 Ameliorates Doxorubicin-Induced Senescence Bystander Effect in Breast Carcinoma Cell MDA-MB-231 and Normal Epithelial Cell MCF-10A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
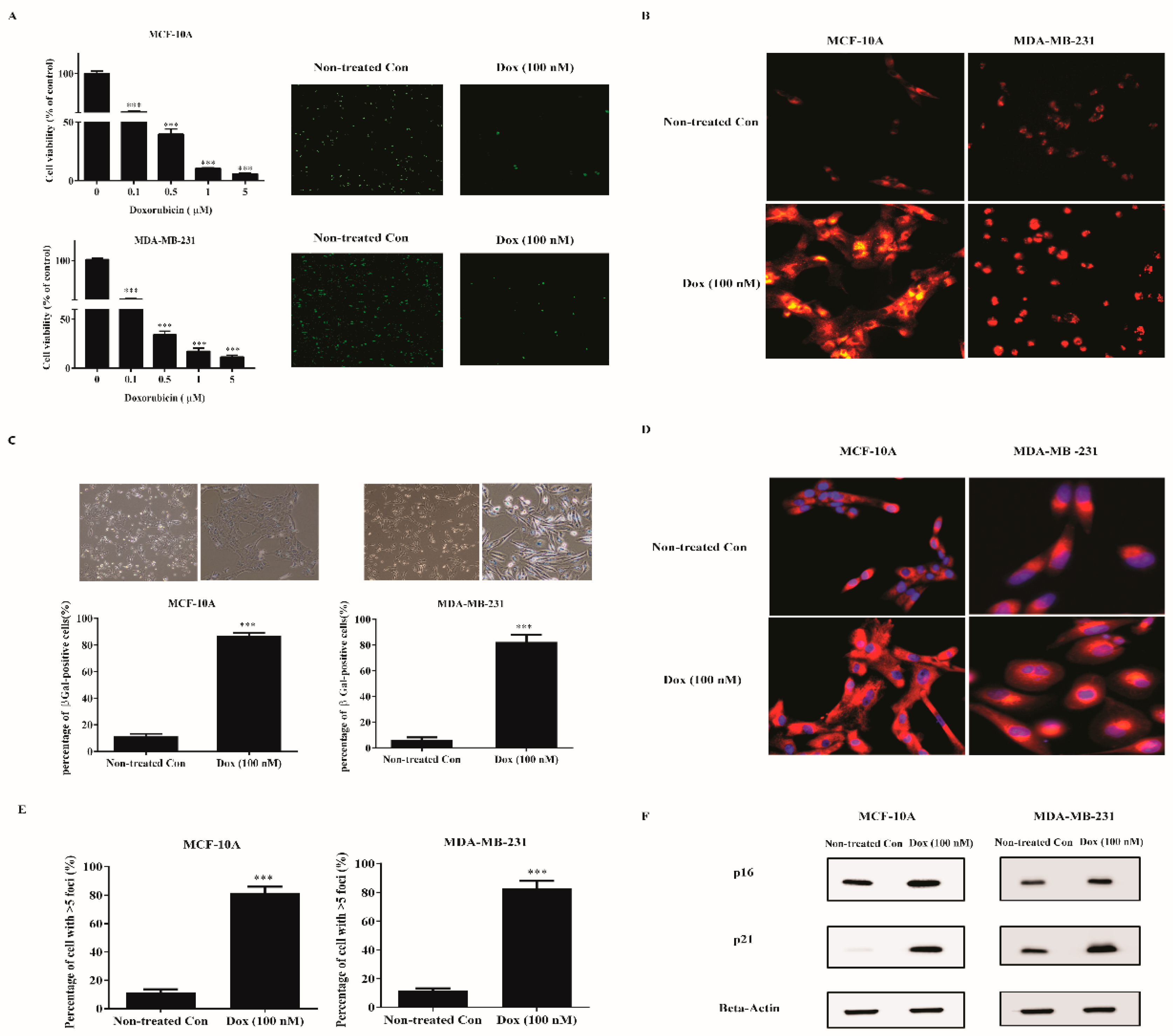
2.1. Low Dose Doxorubicin Induces Senescence of Human Breast Cells
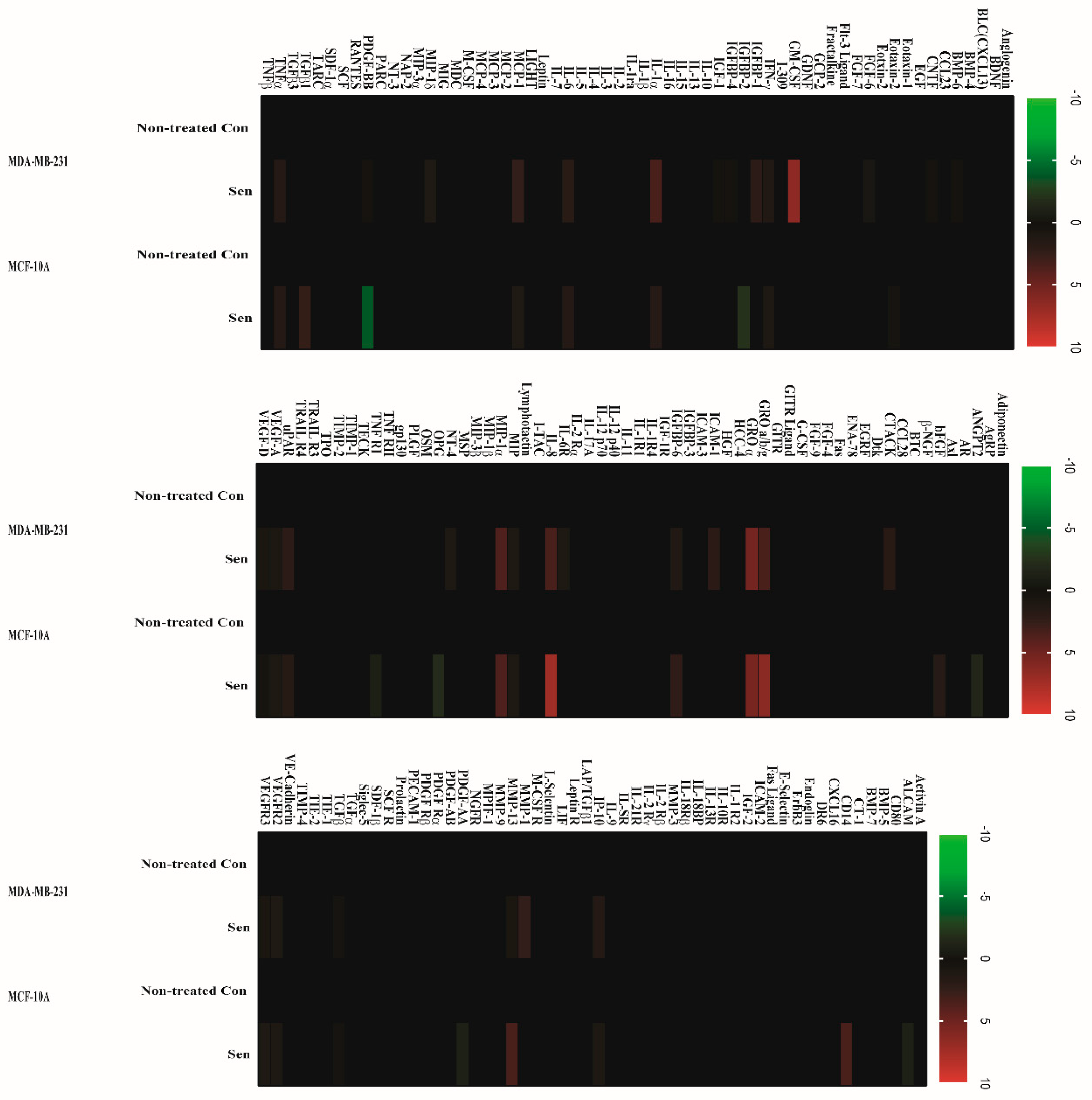
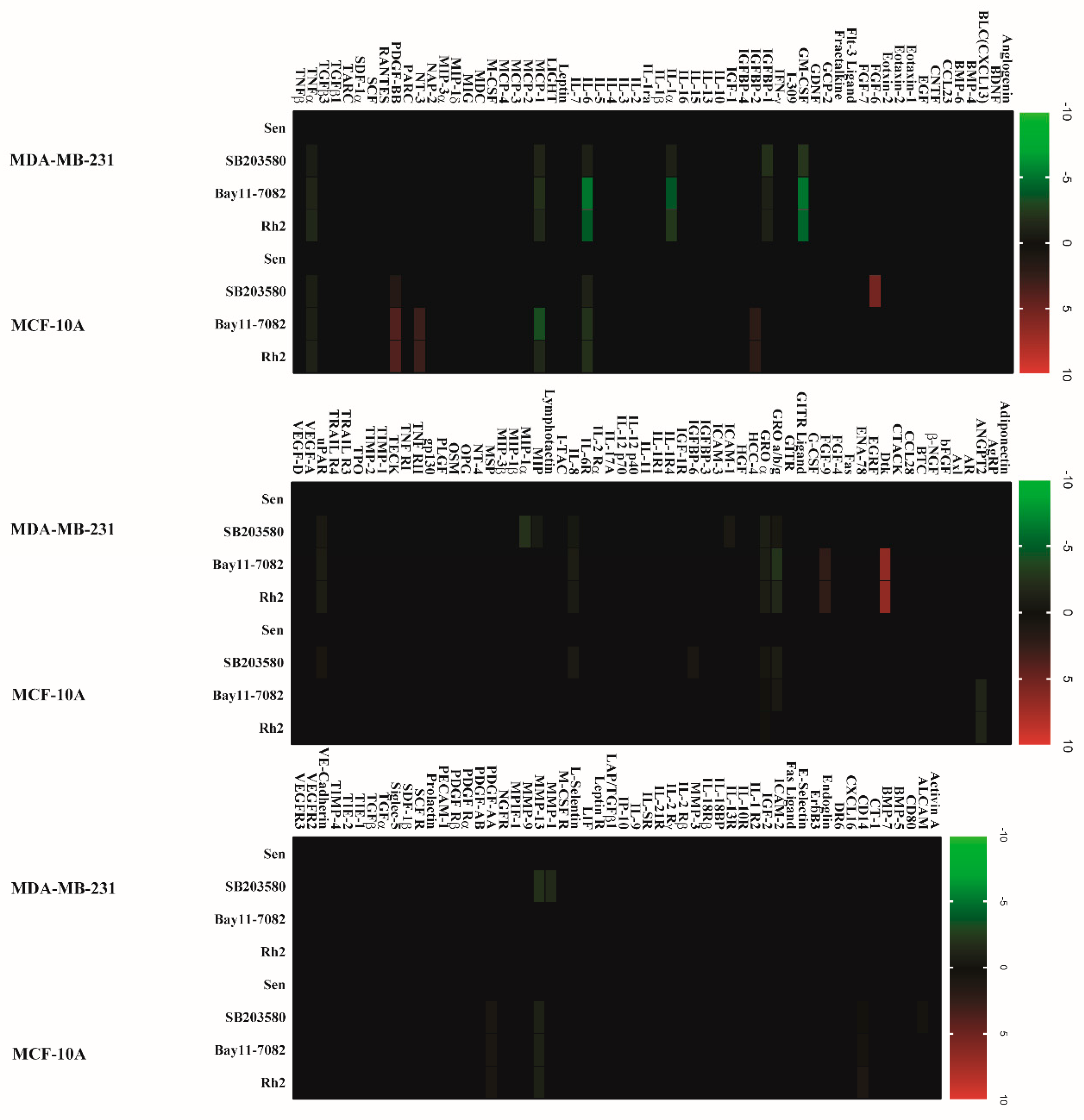
2.2. Doxorubicin-Induced SASP in Human Breast Cell Lines
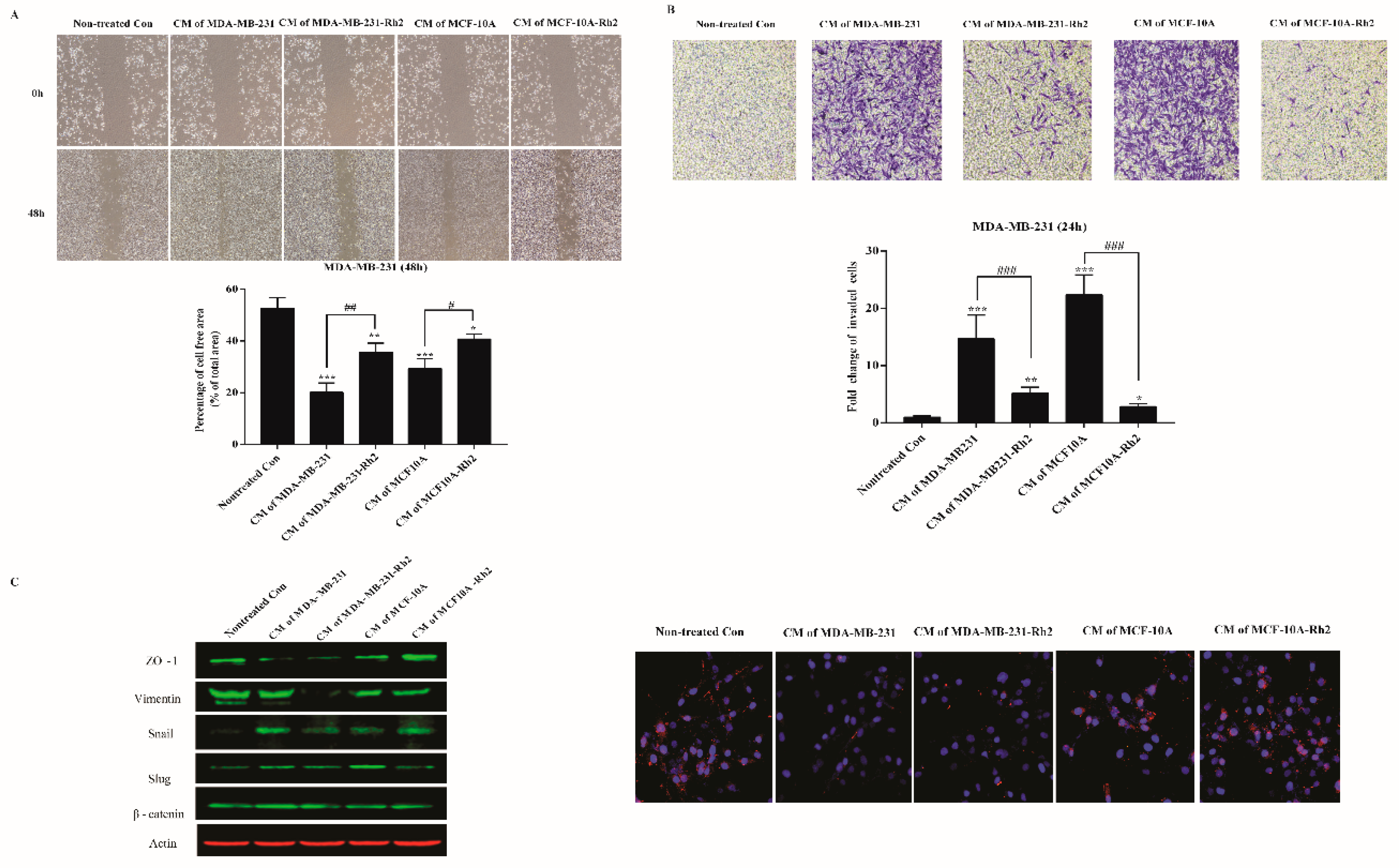
2.3. SASP Stimulates Migration and Invasion of Breast Cancer Cells
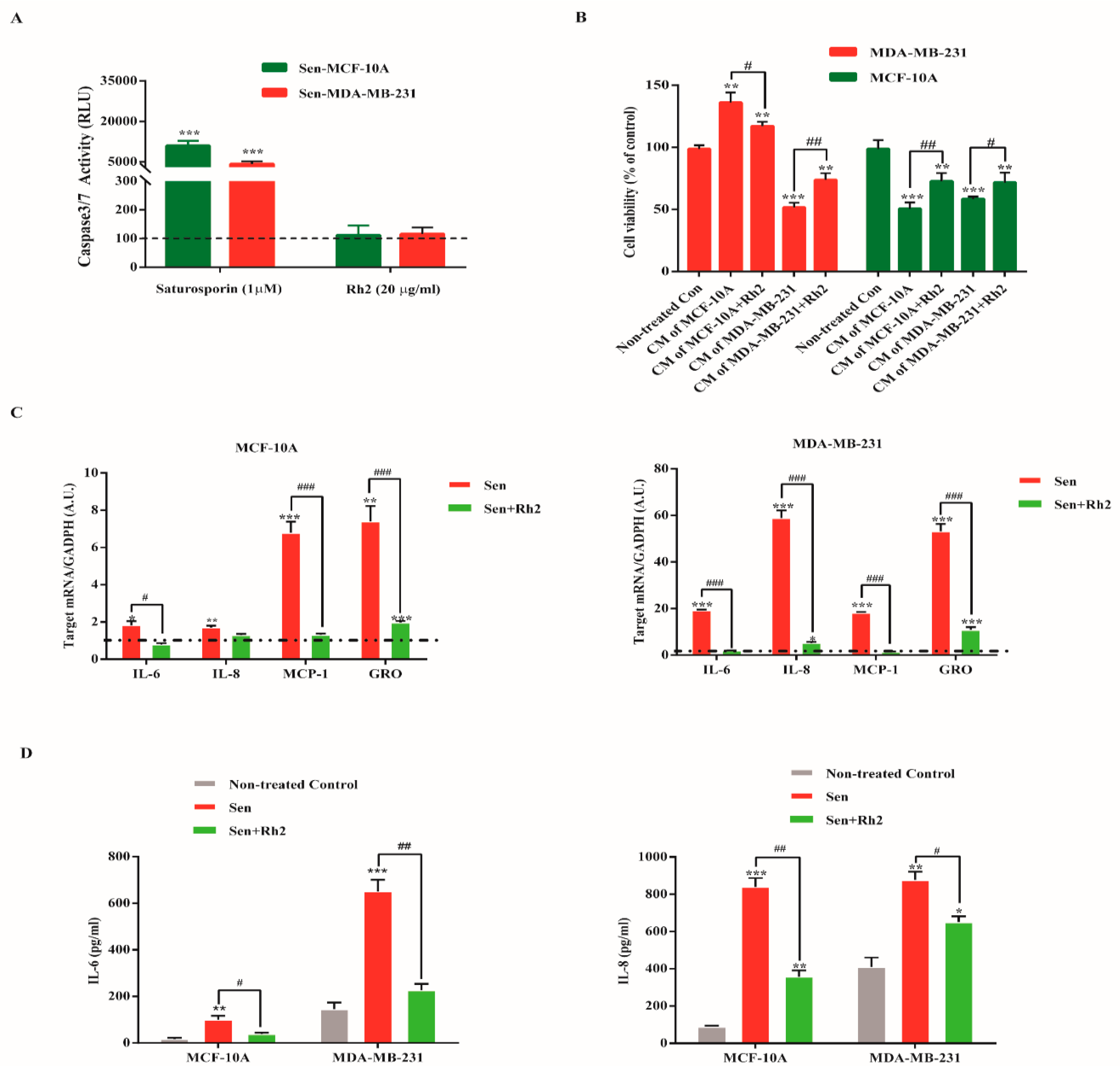
2.4. Ginsenoside Rh2 Does Not Exert Senolytic but Suppress the Paracrine Effects of Sasp in Human Breast Cell Lines
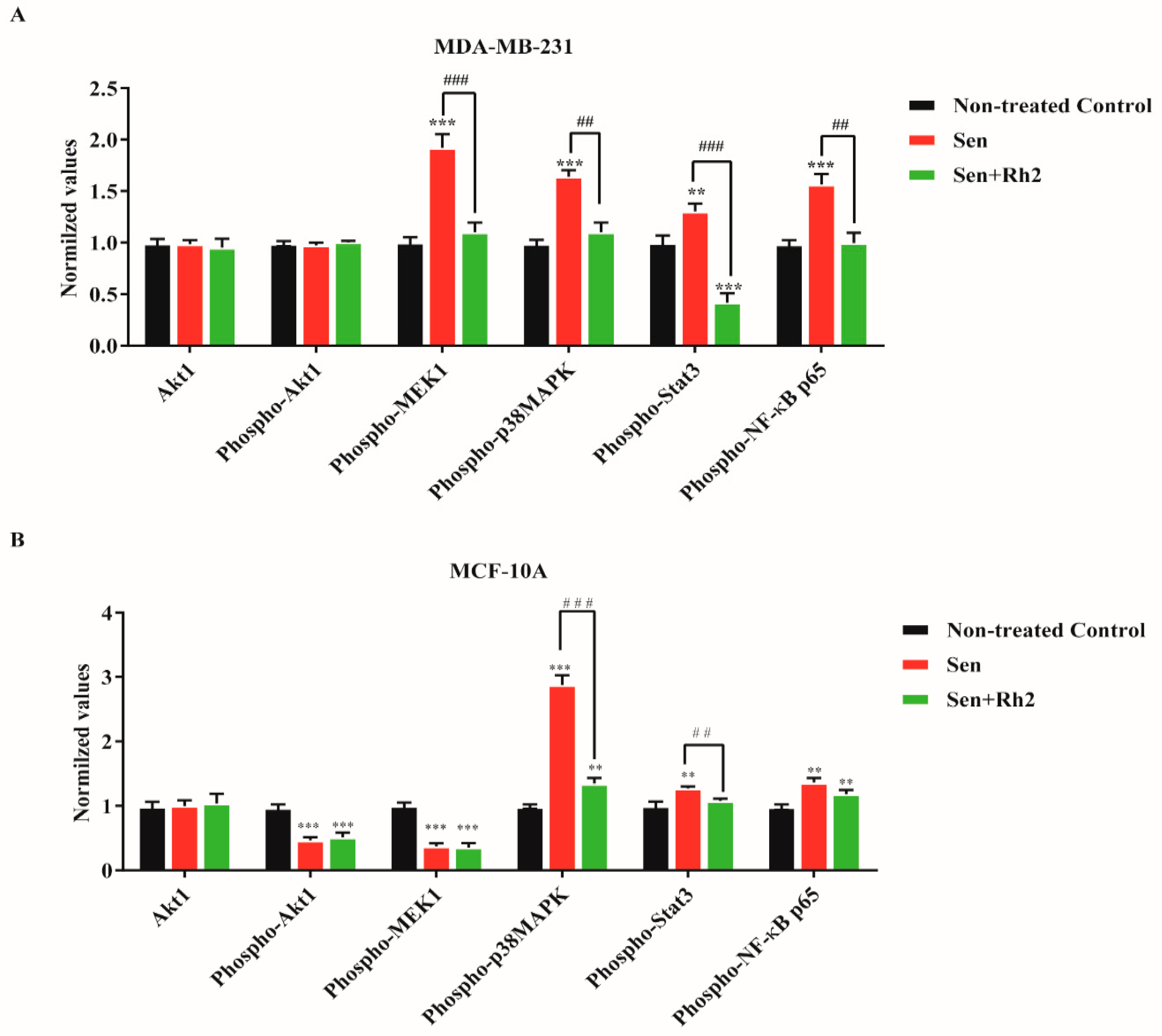
2.5. Ginsenoside Rh2 Suppresses Potential Signaling Pathways Inferred SASP Secretion in Human Breast Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Senescence Induction and Assessment
4.4. Cell Proliferation Assay
4.5. Assay of Caspase 3/7 Activation
4.6. Cytokine Antibody Array
4.7. Migration and Invasion Assay
4.8. Quantification of SASP Major Factors
4.9. PathScan® Signaling Nodes Multitarget Sandwich ELISA Assay
4.10. Western Blotting
4.11. Fluorescence Microscopy
4.12. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Widmer, N.; Bardin, C.; Chatelut, E.; Paci, A.; Beijnen, J.; Levêque, D.; Veal, G.; Astier, A. Review of therapeutic drug monitoring of anticancer drugs part two-targeted therapies. Eur. J. Cancer 2014, 50, 2020–2036. [Google Scholar] [CrossRef] [PubMed]
- Hickman, J.A. Apoptosis induced by anticancer drugs. Cancer Metastasis Rev. 1992, 11, 121–139. [Google Scholar] [CrossRef] [PubMed]
- Pietenpol, J.A.; Stewart, Z.A. Cell cycle checkpoint signaling: Cell cycle arrest versus apoptosis. Toxicology 2002, 181, 475–481. [Google Scholar] [CrossRef]
- Chang, B.D.; Xuan, Y.Z.; Broude, E.V.; Zhu, H.M.; Schott, B.; Fang, J.; Roninson, I.B. Role of p53 and p21(waf1/cip1) in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 1999, 18, 4808–4818. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Tao, J.J.; Visvanathan, K.; Wolff, A.C. Long term side effects of adjuvant chemotherapy in patients with early breast cancer. Breast 2015, 24, S149–S153. [Google Scholar] [CrossRef]
- Scuric, Z.; Carroll, J.E.; Bower, J.E.; Ramos-Perlberg, S.; Petersen, L.; Esquivel, S.; Hogan, M.; Chapman, A.M.; Irwin, M.R.; Breen, E.C.; et al. Biomarkers of aging associated with past treatments in breast cancer survivors. NPJ Breast Cancer 2017, 3, 50. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Aging of the immune system. Mechanisms and therapeutic targets. Ann. Am. Thorac. Soc. 2016, 13, S422–S428. [Google Scholar] [CrossRef]
- Park, E.Y.; Kim, M.H.; Kim, E.H.; Lee, E.K.; Park, I.S.; Yang, D.C.; Jun, H.S. Efficacy comparison of Korean ginseng and American ginseng on body temperature and metabolic parameters. Am. J. Chin. Med. 2014, 42, 173–187. [Google Scholar] [CrossRef]
- Yang, Y.; Ren, C.; Zhang, Y.; Wu, X. Ginseng: An Nonnegligible Natural Remedy for Healthy Aging. Aging Dis. 2017, 8, 708–720. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, E.H.; Ko, S.R.; Choi, K.J.; Park, J.H.; Im, D.S. Effects of ginsenosides Rg(3) and Rh-2 on the proliferation of prostate cancer cells. Arch. Pharm. Res. 2004, 27, 429–435. [Google Scholar] [CrossRef]
- Chen, W.; Qiu, Y. Ginsenoside Rh2 Targets EGFR by Up-Regulation of miR-491 to Enhance Anti-tumor Activity in Hepatitis B Virus-Related Hepatocellular Carcinoma. Cell Biochem. Biophys. 2015, 72, 325–331. [Google Scholar] [CrossRef]
- Ge, G.; Yan, Y.; Cai, H. Ginsenoside Rh2 Inhibited Proliferation by Inducing ROS Mediated ER Stress Dependent Apoptosis in Lung Cancer Cells. Biol. Pharm. Bull. 2017, 40, 2117–2124. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, J.S. Effect of ginsenoside Rh-2 via activation of caspase-3 and Bcl-2-insensitive pathway in ovarian cancer cells. Physiol. Res. 2016, 65, 1031–1037. [Google Scholar]
- Xie, X.; Eberding, A.; Madera, C.; Fazli, L.; Jia, W.; Goldenberg, L.; Gleave, M.; Guns, E.S. Rh2 synergistically enhances paclitaxel or mitoxantrone in prostate cancer models. J. Urol. 2006, 175, 1926–1931. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, Q.; Liu, K.; Li, G.; Zheng, R. Ginsenoside Rh2 enhances antitumour activity and decreases genotoxic effect of cyclophosphamide. Basic Clin. Pharmacol. Toxicol. 2006, 98, 411–415. [Google Scholar] [CrossRef]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef]
- Eom, Y.W.; Kim, M.A.; Park, S.S.; Goo, M.J.; Kwon, H.J.; Sohn, S.; Kim, W.H.; Yoon, G.; Choi, K.S. Two distinct modes of cell death induced by doxorubicin: Apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005, 24, 4765–4777. [Google Scholar] [CrossRef]
- Legha, S.S.; Benjamin, R.S.; Mackay, B.; Ewer, M.; Wallace, S.; Valdivieso, M.; Rasmussen, S.L.; Blumenschein, G.R.; Freireich, E.J. Reduction of doxorubicin cardiotoxicity by prolonged continuous intravenous infusion. Ann. Intern. Med. 1982, 96, 133–139. [Google Scholar] [CrossRef]
- Levina, V.; Marrangoni, A.M.; DeMarco, R.; Gorelik, E.; Lokshin, A.E. Drug-selected human lung cancer stem cells: Cytokine network, tumorigenic and metastatic properties. PLoS ONE 2008, 3, e3077. [Google Scholar] [CrossRef]
- Thorn, M.; Guha, P.; Cunetta, M.; Espat, N.J.; Miller, G.; Junghans, R.P.; Katz, S.C. Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther. 2016, 23, 188–198. [Google Scholar] [CrossRef]
- Takahashi, M.; Miyazaki, H.; Furihata, M.; Sakai, H.; Konakahara, T.; Watanabe, M.; Okada, T. Chemokine CCL2/MCP-1 negatively regulates metastasis in a highly bone marrow-metastatic mouse breast cancer model. Clin. Exp. Metastasis 2009, 26, 817–828. [Google Scholar] [CrossRef]
- Pellikainen, J.M.; Ropponen, K.M.; Kataja, V.V.; Kellokoski, J.K.; Eskelinen, M.J.; Kosma, V.M. Expression of matrix metalloproteinase (MMP)-2 and MMP-9 in breast cancer with a special reference to activator protein-2, HER2, and prognosis. Clin. Cancer Res. 2004, 10, 7621–7628. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Martens, J.W.; Dorssers, L.C.; Klijn, J.G.; Foekens, J.A. Differential effects of fibroblast growth factors on expression of genes of the plasminogen activator and insulin-like growth factor systems by human breast fibroblasts. Thromb. Haemost. 2002, 87, 674–683. [Google Scholar]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Tkach, M.; Coria, L.; Rosemblit, C.; Rivas, M.A.; Proietti, C.J.; Diaz Flaque, M.C.; Beguelin, W.; Frahm, I.; Charreau, E.H.; Cassataro, J.; et al. Targeting Stat3 induces senescence in tumor cells and elicits prophylactic and therapeutic immune responses against breast cancer growth mediated by NK cells and CD4+ T cells. J. Immunol. 2012, 189, 1162–1172. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, J.-G.; Jeon, B.-M.; Yun, Y.-J.; Cui, C.-H.; Kim, S.-C. Ginsenoside Rh2 Ameliorates Doxorubicin-Induced Senescence Bystander Effect in Breast Carcinoma Cell MDA-MB-231 and Normal Epithelial Cell MCF-10A. Int. J. Mol. Sci. 2019, 20, 1244. https://doi.org/10.3390/ijms20051244
Hou J-G, Jeon B-M, Yun Y-J, Cui C-H, Kim S-C. Ginsenoside Rh2 Ameliorates Doxorubicin-Induced Senescence Bystander Effect in Breast Carcinoma Cell MDA-MB-231 and Normal Epithelial Cell MCF-10A. International Journal of Molecular Sciences. 2019; 20(5):1244. https://doi.org/10.3390/ijms20051244
Chicago/Turabian StyleHou, Jin-Gang, Byeong-Min Jeon, Yee-Jin Yun, Chang-Hao Cui, and Sun-Chang Kim. 2019. "Ginsenoside Rh2 Ameliorates Doxorubicin-Induced Senescence Bystander Effect in Breast Carcinoma Cell MDA-MB-231 and Normal Epithelial Cell MCF-10A" International Journal of Molecular Sciences 20, no. 5: 1244. https://doi.org/10.3390/ijms20051244
APA StyleHou, J.-G., Jeon, B.-M., Yun, Y.-J., Cui, C.-H., & Kim, S.-C. (2019). Ginsenoside Rh2 Ameliorates Doxorubicin-Induced Senescence Bystander Effect in Breast Carcinoma Cell MDA-MB-231 and Normal Epithelial Cell MCF-10A. International Journal of Molecular Sciences, 20(5), 1244. https://doi.org/10.3390/ijms20051244