Peanut Sprout Extracts Attenuate Triglyceride Accumulation by Promoting Mitochondrial Fatty Acid Oxidation in Adipocytes


 ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Total Polyphenol, Flavonoid, and Resveratrol Contents of PS Extracts
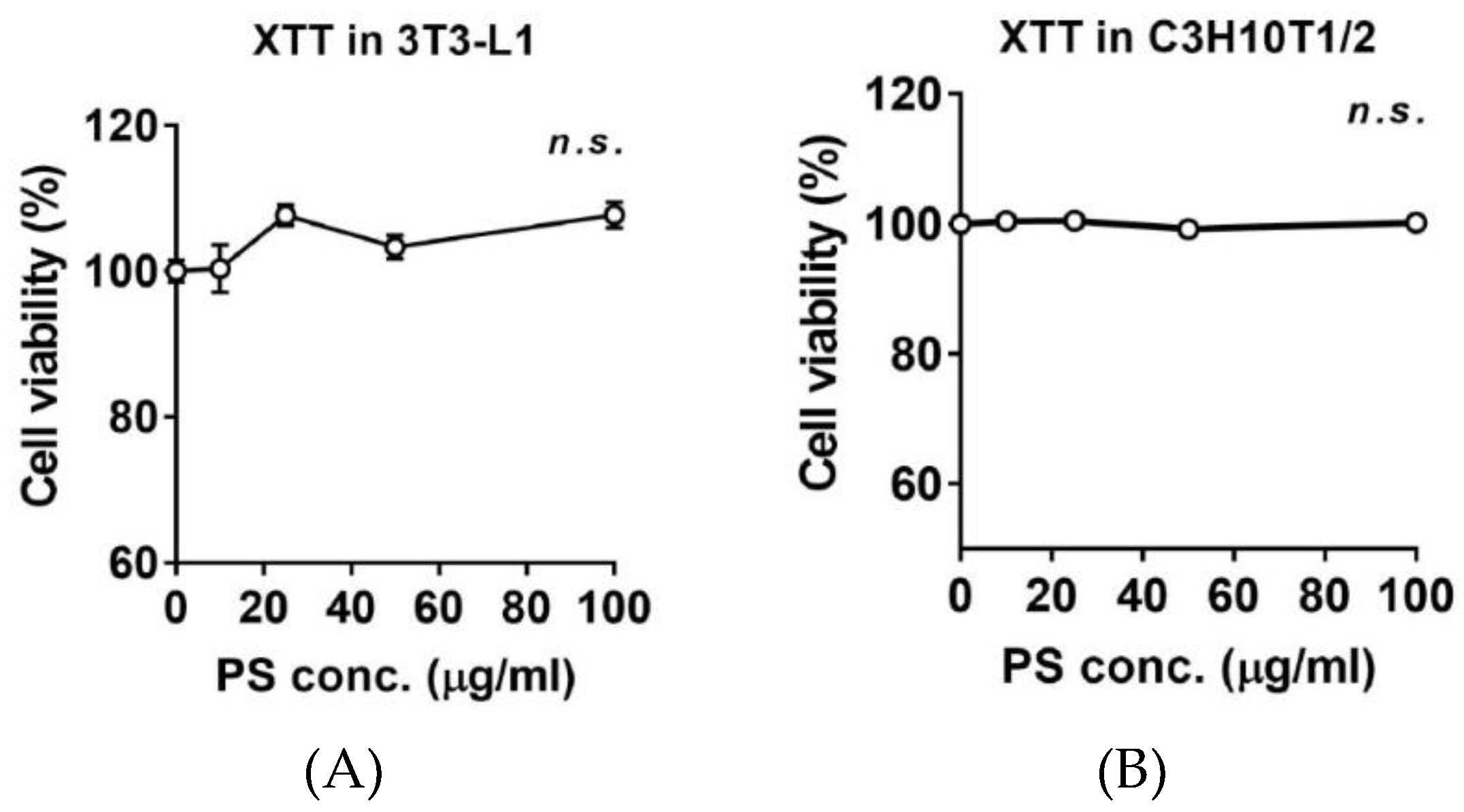
2.2. Effects of PSE on Cell Viability 3T3-L1 and C3H10T1/2 Preadipocytes
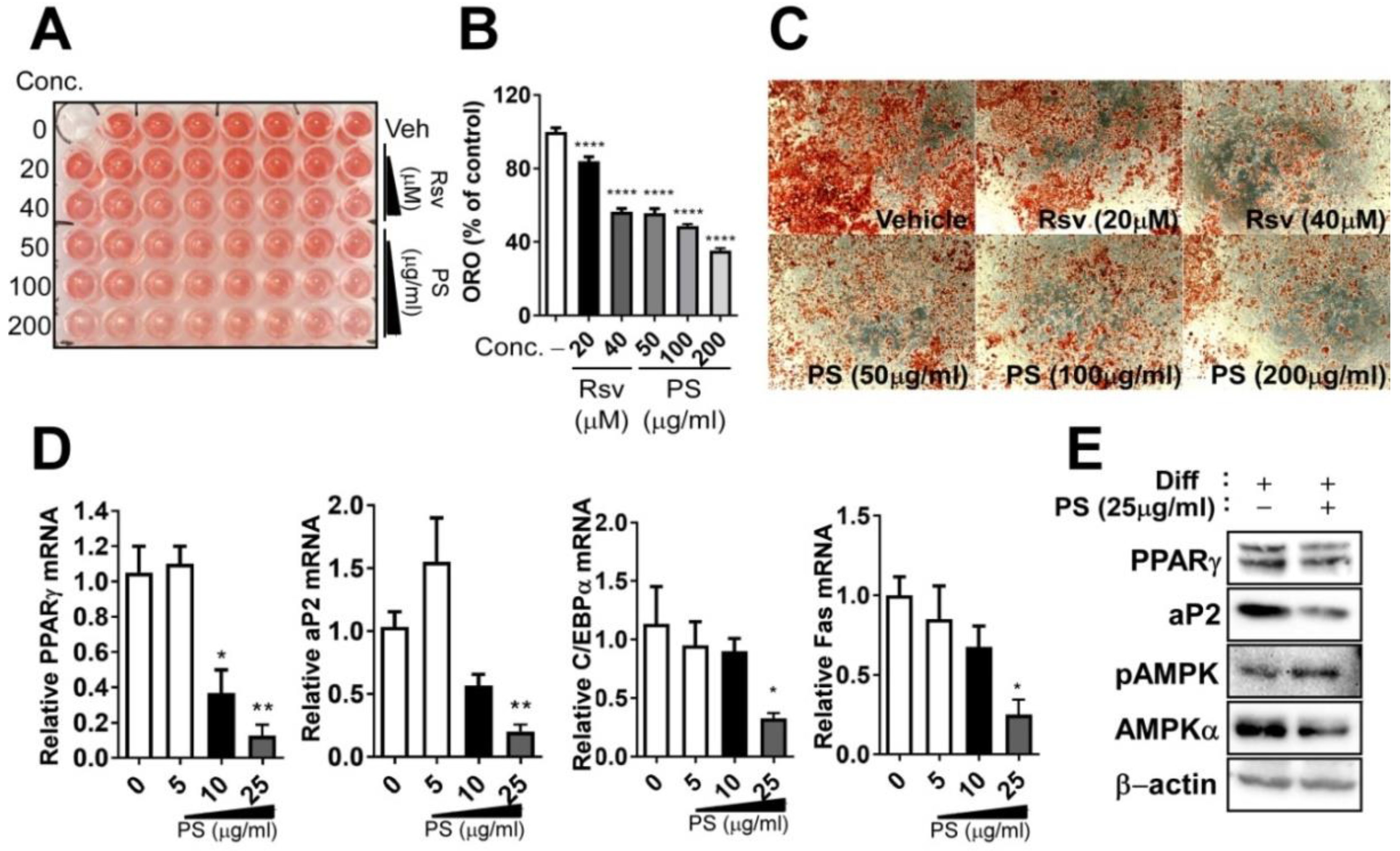
2.3. PSE Inhibits Adipogenesis in 3T3-L1 Adipocytes
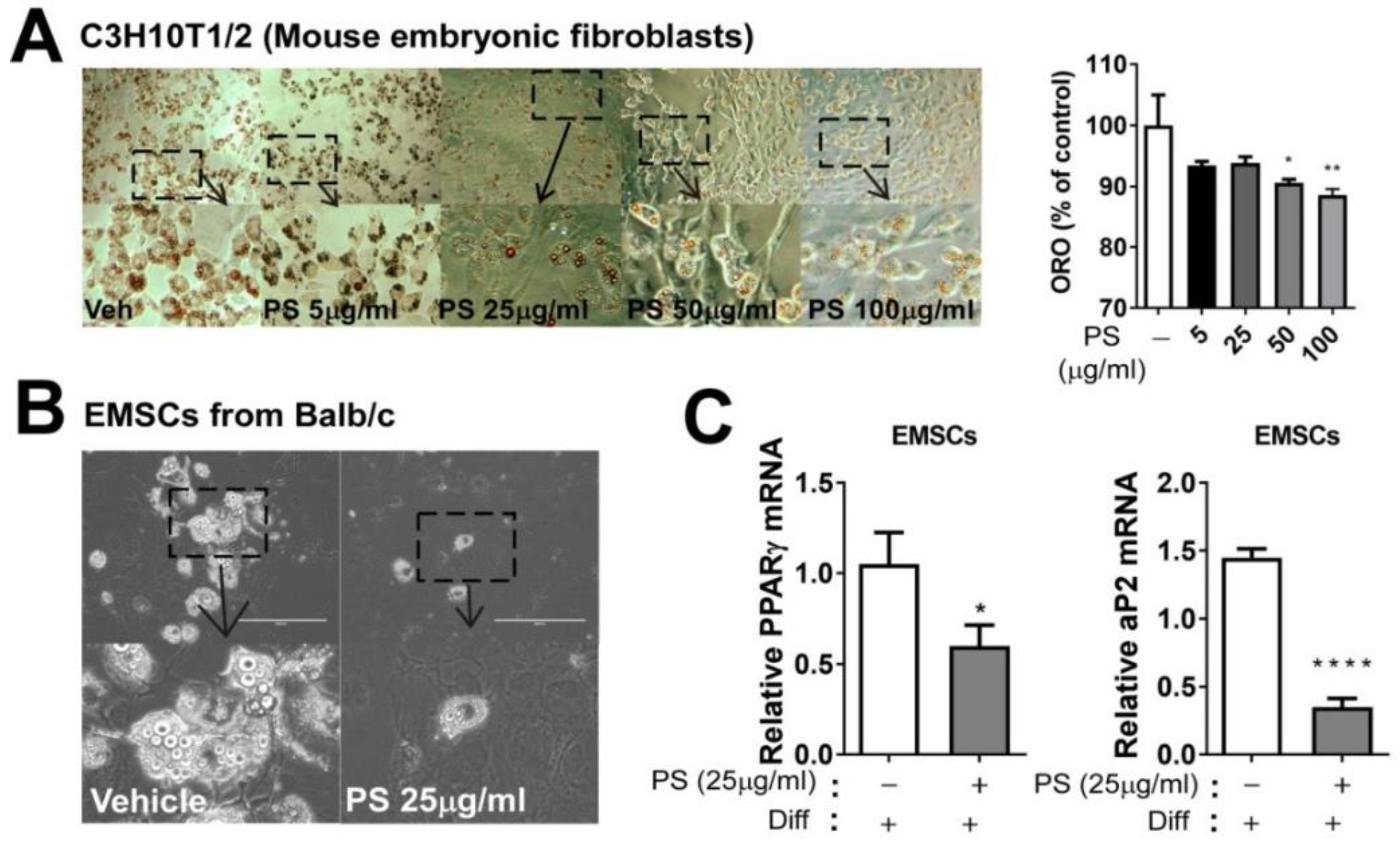
2.4. PSE Inhibits Adipogenesis in C3H10T1/2 Mouse Embryonic Fibroblasts Adipocytes and EMSCs
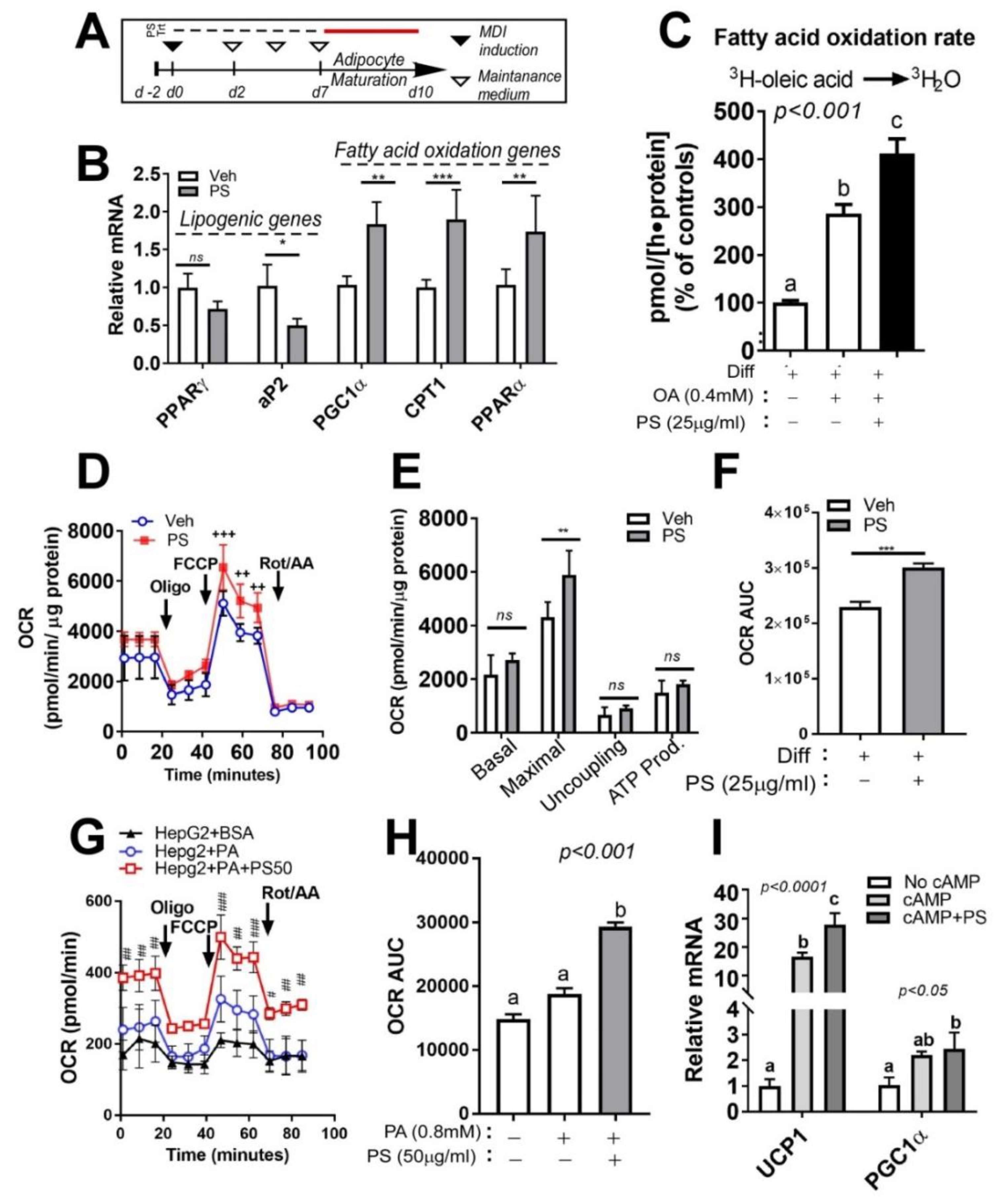
2.5. PSE Attenuates Lipid Accumulation in Cultures of Adipocytes by Upregulating Fatty Acid Oxidation and Mitochondrial Oxygen Consumption
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Sample Preparation
4.3. Total Polyphenol and Flavonoid Contents of PS Extracts
4.4. LC/MS Analyses of PS Extracts
4.5. Cell Culture
4.6. Cell Viability Assay
4.7. Lipid Accumulation
4.8. Total RNA Extraction and qPCR
4.9. Western Blot Analysis
4.10. Fatty Oxidation Rate Using [3H]-OA
4.11. Oxygen Consumption Rate (OCR) by Seahorse
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| aP2, FABP4 | fatty acid binding protein 4 |
| AUC | area under the curve |
| cAMP | Cyclic adenosine monophosphate |
| CPT1 | Carnitine palmitoyltransferase I |
| EMSCs | Ear mesenchymal stem cells |
| FA | fatty acid |
| MDI | methylisobutylxanthine, dexamethasone, and insulin |
| OA | oleic acid |
| OCR | oxygen consumption rate |
| ORO | oil red O |
| PA | Palmitic acid |
| PGC1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PS | Peanut sprouts |
| PSE | Peanut sprout extracts |
| SIRT1 | sirtuin 1 |
| TFC | total flavonoid contents |
| TPC | total polyphenol contents |
| UCP1 | uncoupling protein 1 |
References
- World Health Organization. Obesity: Preventing and Managing the Global Epidemic: Report of a WHO Consultation; World Health Organization: Geneva, Switzerland, 2000; p. 253. [Google Scholar]
- Imes, C.C.; Burke, L.E. The Obesity Epidemic: The United States as a Cautionary Tale for the Rest of the World. Curr. Epidemiol. Rep. 2014, 1, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or Hyperplasia: Dynamics of Adipose Tissue Growth. PLoS Comput. Biol. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Despres, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Griel, A.E.; Eissenstat, B.; Juturu, V.; Hsieh, G.; Kris-Etherton, P.M. Improved diet quality with peanut consumption. J. Am. Coll. Nutr. 2004, 23, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.H.; Lai, Y.H.; Chang, J.C.; Ko, T.F.; Shyu, S.L.; Chiou, R.Y. Germination of peanut kernels to enhance resveratrol biosynthesis and prepare sprouts as a functional vegetable. J. Agric. Food Chem. 2005, 53, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Choi, D.I.; Lee, J.B.; Yun, S.J.; Lee, D.H.; Eun, J.B.; Lee, S.C. Ethanol extract of peanut sprout induces Nrf2 activation and expression of antioxidant and detoxifying enzymes in human dermal fibroblasts: Implication for its protection against UVB-irradiated oxidative stress. Photochem. Photobiol. 2013, 89, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Limmongkon, A.; Nopprang, P.; Chaikeandee, P.; Somboon, T.; Wongshaya, P.; Pilaisangsuree, V. LC-MS/MS profiles and interrelationships between the anti-inflammatory activity, total phenolic content and antioxidant potential of Kalasin 2 cultivar peanut sprout crude extract. Food Chem. 2018, 239, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.E.; Ha, A.W.; Woo, H.W.; Kim, W.K. Peanut sprouts extract (Arachis hypogaea L.) has anti-obesity effects by controlling the protein expressions of PPARgamma and adiponectin of adipose tissue in rats fed high-fat diet. Nutr. Res. Pract. 2014, 8, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Ha, A.W.; Kang, N.E.; Kim, W.K. Ethanol Extract of Peanut Sprout Lowers Blood Triglyceride Levels, Possibly Through a Pathway Involving SREBP-1c in Rats Fed a High-Fat Diet. J. Med. Food 2015, 18, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; Kang, N.E.; Kim, M.H.; Ha, A.W. Peanut sprout ethanol extract inhibits the adipocyte proliferation, differentiation, and matrix metalloproteinases activities in mouse fibroblast 3T3-L1 preadipocytes. Nutr. Res. Pract. 2013, 7, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Jefcoate, C.R.; Wang, S.; Liu, X. Methods that resolve different contributions of clonal expansion to adipogenesis in 3T3-L1 and C3H10T1/2 cells. Methods Mol. Biol. 2008, 456, 173–193. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Jeon, H.J.; Lee, O.H.; Lee, B.Y. Dieckol, a major phlorotannin in Ecklonia cava, suppresses lipid accumulation in the adipocytes of high-fat diet-fed zebrafish and mice: Inhibition of early adipogenesis via cell-cycle arrest and AMPKalpha activation. Mol. Nutr. Food Res. 2015, 59, 1458–1471. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Kim, Y.; Tomas-Barberan, F.A.; Espin, J.C.; Chung, S. Urolithin A, C, and D, but not iso-urolithin A and urolithin B, attenuate triglyceride accumulation in human cultures of adipocytes and hepatocytes. Mol. Nutr. Food Res. 2016, 60, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.T.; Park, I.J.; Shin, J.I.; Lee, Y.K.; Lee, S.K.; Baik, H.W.; Ha, J.; Park, O.J. Genistein, EGCG, and capsaicin inhibit adipocyte differentiation process via activating AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 2005, 338, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xiao, X.; Feng, X.; Li, W.; Zhou, N.; Zheng, L.; Sun, Y.; Zhang, Z.; Zhu, W. Resveratrol induces Sirt1-dependent apoptosis in 3T3-L1 preadipocytes by activating AMPK and suppressing AKT activity and survivin expression. J. Nutr. Biochem. 2012, 23, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, E.K.; Gonzalez, A.; Garcia, C.; Tadros, J.H.; Chakraborty, G.; Toney, J.H. Oleic acid and peanut oil high in oleic acid reverse the inhibitory effect of insulin production of the inflammatory cytokine TNF-alpha both in vitro and in vivo systems. Lipids Health Dis. 2009, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Manson, J.E.; Stampfer, M.J.; Liu, S.; Willett, W.C.; Hu, F.B. Nut and peanut butter consumption and risk of type 2 diabetes in women. JAMA 2002, 288, 2554–2560. [Google Scholar] [CrossRef] [PubMed]
- Alper, C.M.; Mattes, R.D. Peanut consumption improves indices of cardiovascular disease risk in healthy adults. J. Am. Coll. Nutr. 2003, 22, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Langcake, P.; Pryce, R.J. The production of resveratrol and the viniferins by grapevines in response to ultraviolet irradiation. Phytochemistry 1977, 16, 1193–1196. [Google Scholar] [CrossRef]
- Jeandet, P.; Bessis, R.; Gautheron, B. The Production of Resveratrol (3,5,4′-trihydroxystilbene) by Grape Berries in Different Developmental Stages. Am. J. Enol. Vitic. 1991, 42, 41–46. [Google Scholar]
- Poulsen, M.M.; Vestergaard, P.F.; Clasen, B.F.; Radko, Y.; Christensen, L.P.; Stodkilde-Jorgensen, H.; Moller, N.; Jessen, N.; Pedersen, S.B.; Jorgensen, J.O. High-dose resveratrol supplementation in obese men: An investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 2013, 62, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Youn, C.K.; Jo, E.R.; Sim, J.H.; Cho, S.I. Peanut sprout extract attenuates cisplatin-induced ototoxicity by induction of the Akt/Nrf2-mediated redox pathway. Int. J. Pediatr. Otorhinolaryngol. 2017, 92, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.I.; Choi, J.Y.; Kim, Y.J.; Lee, J.B.; Kim, S.O.; Shin, H.T.; Lee, S.C. Ethanol Extract of Peanut Sprout Exhibits a Potent Anti-Inflammatory Activity in Both an Oxazolone-Induced Contact Dermatitis Mouse Model and Compound 48/80-Treated HaCaT Cells. Ann. Dermatol. 2015, 27, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Chavey, C.; Mari, B.; Monthouel, M.N.; Bonnafous, S.; Anglard, P.; Van Obberghen, E.; Tartare-Deckert, S. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. J. Biol. Chem. 2003, 278, 11888–11896. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, H.; Park, D.; Ahn, J.; Shin, S.S.; Yoon, M. Ginseng and Its Active Components Ginsenosides Inhibit Adipogenesis in 3T3-L1 Cells by Regulating MMP-2 and MMP-9. Evid. Complement. Alternat. Med. 2012, 2012, 265023. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liang, X.; Yang, Q.; Fu, X.; Zhu, M.; Rodgers, B.D.; Jiang, Q.; Dodson, M.V.; Du, M. Resveratrol enhances brown adipocyte formation and function by activating AMP-activated protein kinase (AMPK) alpha1 in mice fed high-fat diet. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhao, W.; Zhao, D.; Wang, C.; Yu, N.; An, T.; Mo, F.; Liu, J.; Miao, J.; Lv, B.; et al. Salvianolic Acid B Improves Mitochondrial Function in 3T3-L1 Adipocytes Through a Pathway Involving PPARgamma Coactivator-1alpha (PGC-1alpha). Front. Pharmacol. 2018, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Skates, E.; Overall, J.; DeZego, K.; Wilson, M.; Esposito, D.; Lila, M.A.; Komarnytsky, S. Berries containing anthocyanins with enhanced methylation profiles are more effective at ameliorating high fat diet-induced metabolic damage. Food Chem. Toxicol. 2018, 111, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Wilkening, S.; Stahl, F.; Bader, A. Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metab. Dispos. 2003, 31, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Zorita, S.; Fernandez-Quintela, A.; Macarulla, M.T.; Aguirre, L.; Hijona, E.; Bujanda, L.; Milagro, F.; Martinez, J.A.; Portillo, M.P. Resveratrol attenuates steatosis in obese Zucker rats by decreasing fatty acid availability and reducing oxidative stress. Br. J. Nutr. 2012, 107, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Kim, M.B.; Lim, S.B. Enhancement of Phenolic Production and Antioxidant Activity from Buckwheat Leaves by Subcritical Water Extraction. Prev. Nutr. Food Sci. 2017, 22, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Singleton, V.L.; Rossi, J.A. Colorimetry of Total Phenolics with Phosphomolybdic-Phosphotungstic Acid Reagents. Am. J. Enol. Vitic. 1965, 16, 144–158. [Google Scholar]
- Moreno, M.I.; Isla, M.I.; Sampietro, A.R.; Vattuone, M.A. Comparison of the free radical-scavenging activity of propolis from several regions of Argentina. J. Ethnopharmacol. 2000, 71, 109–114. [Google Scholar] [CrossRef]
- Gawronska-Kozak, B. Preparation and differentiation of mesenchymal stem cells from ears of adult mice. Methods Enzymol. 2014, 538, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Olpin, S.E.; Manning, N.J.; Pollitt, R.J.; Clarke, S. Improved detection of long-chain fatty acid oxidation defects in intact cells using [9,10-3H] oleic acid. J. Inherit. Metab. Dis. 1997, 20, 415–419. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total polyphenols, Flavonoids, and Resveratrol Contents | Peanut Sprout Extract |
|---|---|
| Total polyphenols (mg Gallic acid/extract g) | 10.87 ± 0.11 |
| Total flavonoids (mg Catechin/extract g) | 3.79 ± 0.14 1 |
| Resveratrol (μg/g) | 18 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, S.H.; Jo, S.-M.; Kim, J.; Lee, M.; Lee, Y.; Kang, I. Peanut Sprout Extracts Attenuate Triglyceride Accumulation by Promoting Mitochondrial Fatty Acid Oxidation in Adipocytes. Int. J. Mol. Sci. 2019, 20, 1216. https://doi.org/10.3390/ijms20051216
Seo SH, Jo S-M, Kim J, Lee M, Lee Y, Kang I. Peanut Sprout Extracts Attenuate Triglyceride Accumulation by Promoting Mitochondrial Fatty Acid Oxidation in Adipocytes. International Journal of Molecular Sciences. 2019; 20(5):1216. https://doi.org/10.3390/ijms20051216
Chicago/Turabian StyleSeo, Seok Hee, Sang-Mi Jo, Jiyoung Kim, Myoungsook Lee, Yunkyoung Lee, and Inhae Kang. 2019. "Peanut Sprout Extracts Attenuate Triglyceride Accumulation by Promoting Mitochondrial Fatty Acid Oxidation in Adipocytes" International Journal of Molecular Sciences 20, no. 5: 1216. https://doi.org/10.3390/ijms20051216
APA StyleSeo, S. H., Jo, S.-M., Kim, J., Lee, M., Lee, Y., & Kang, I. (2019). Peanut Sprout Extracts Attenuate Triglyceride Accumulation by Promoting Mitochondrial Fatty Acid Oxidation in Adipocytes. International Journal of Molecular Sciences, 20(5), 1216. https://doi.org/10.3390/ijms20051216