KCa3.1 Transgene Induction in Murine Intestinal Epithelium Causes Duodenal Chyme Accumulation and Impairs Duodenal Contractility

 ,
,  , , ,
, , ,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
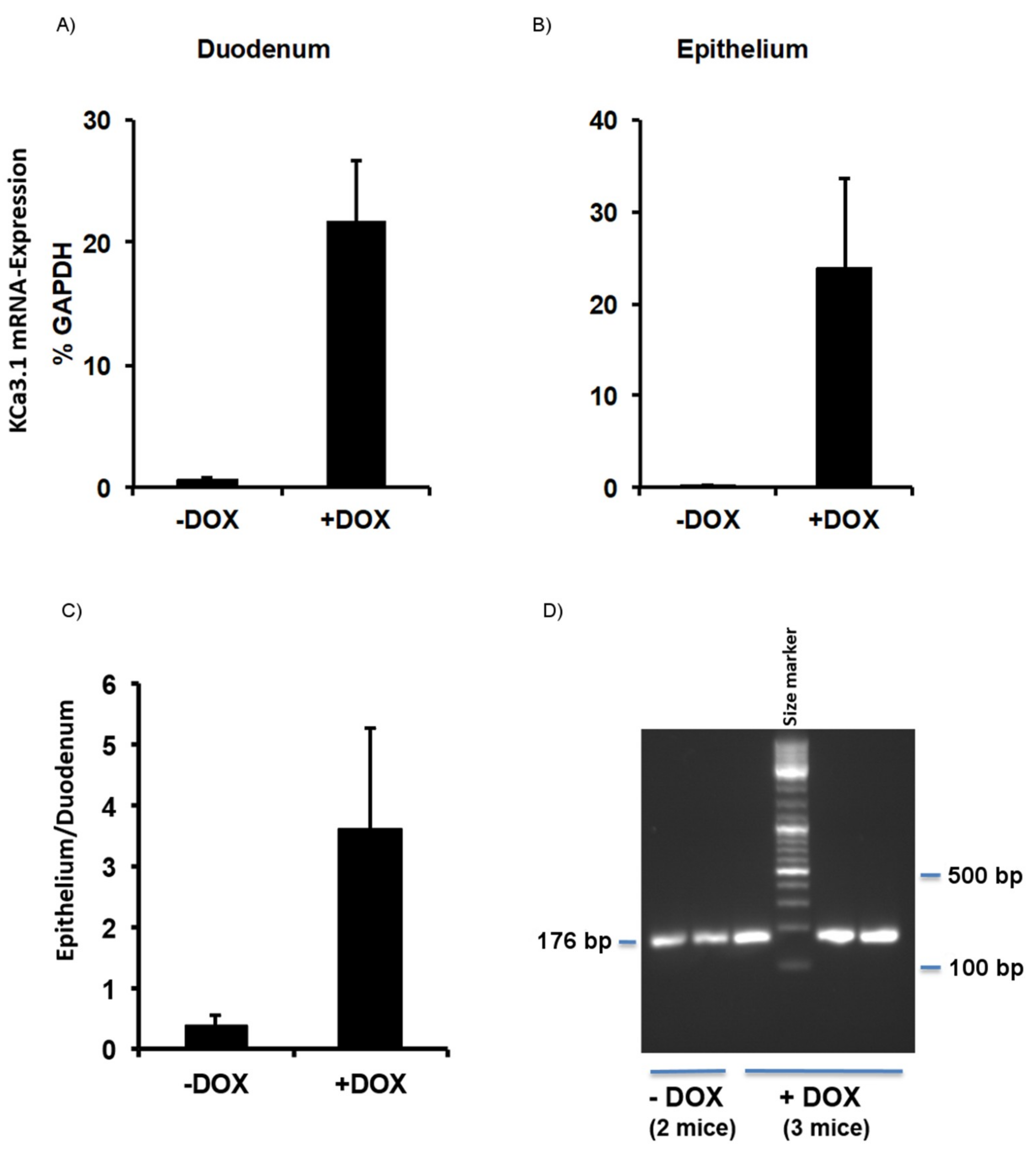
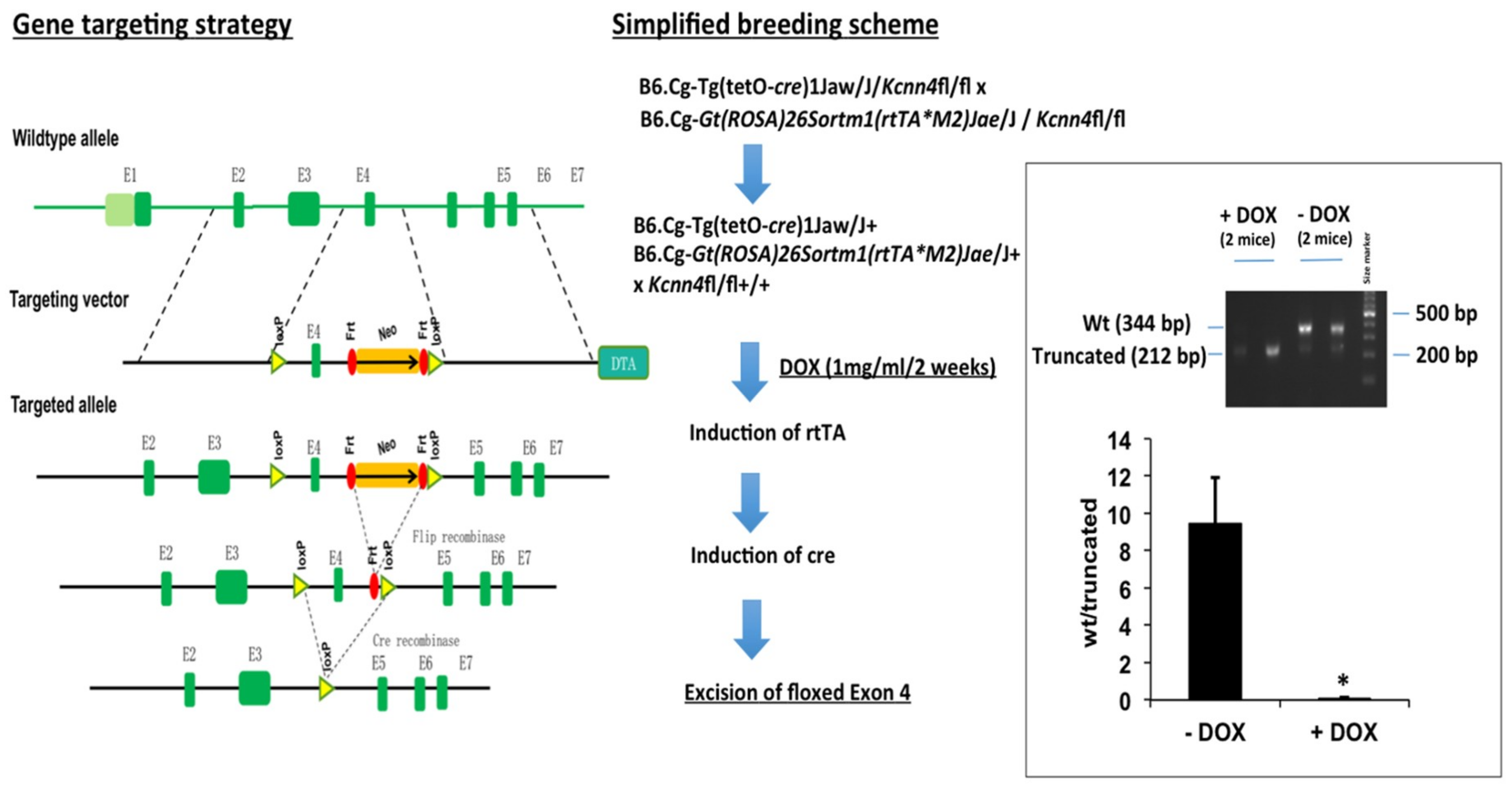
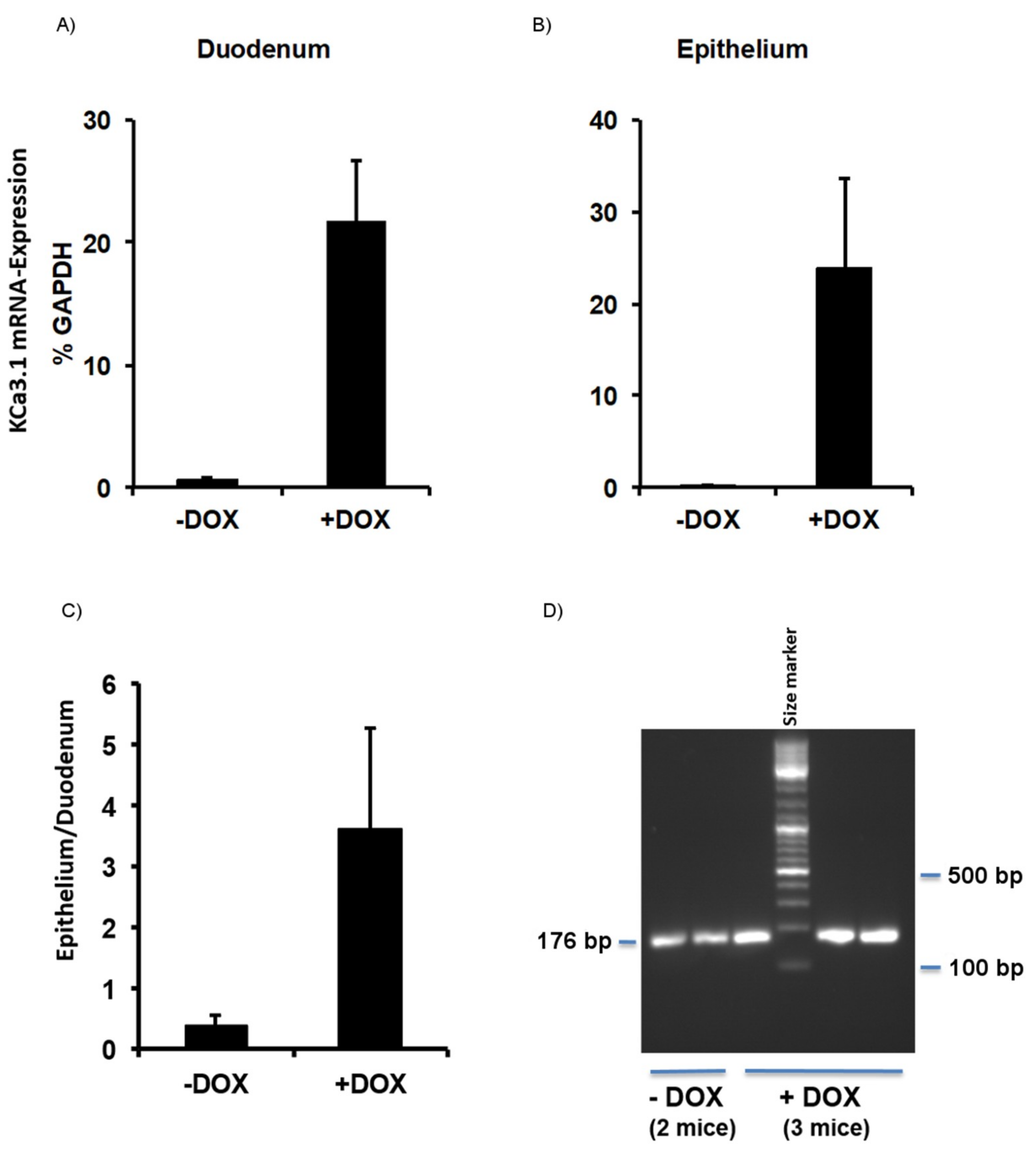
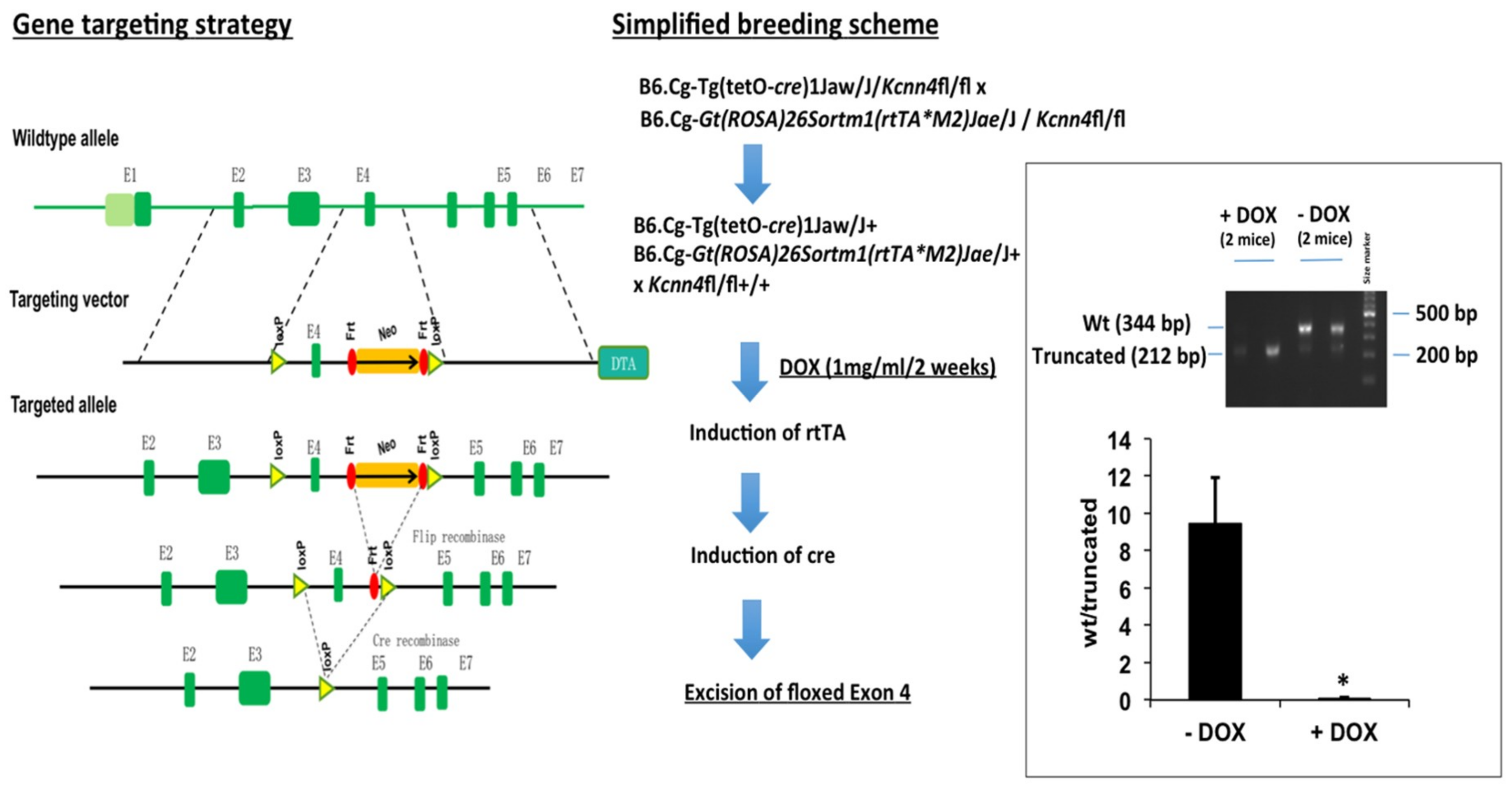
2.1. Murine Model of KCa3.1-Trangene Induction in the Intestinal Epithelium
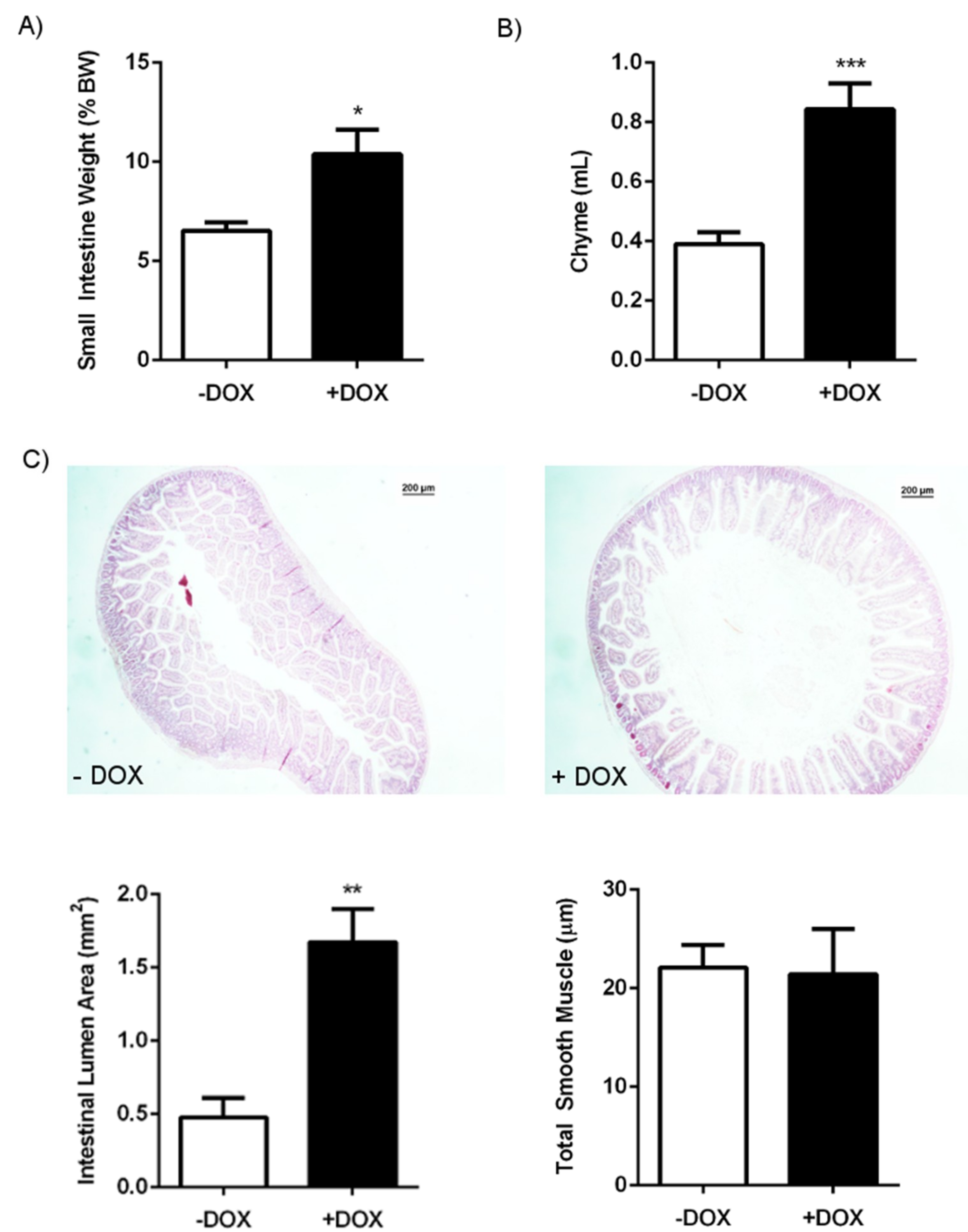
2.1.1. Phenotype of KCa3.1 Overexpression Mice
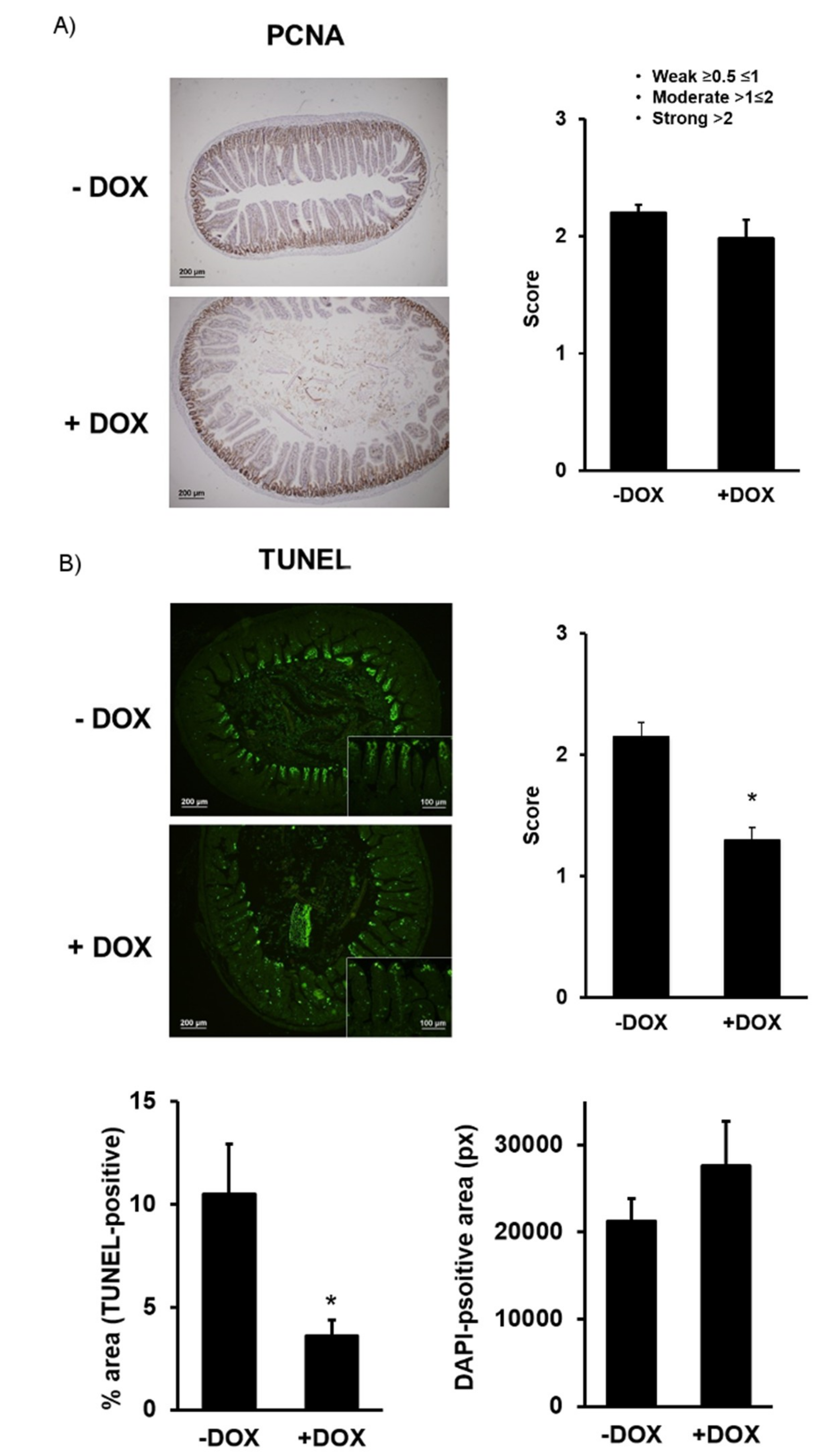
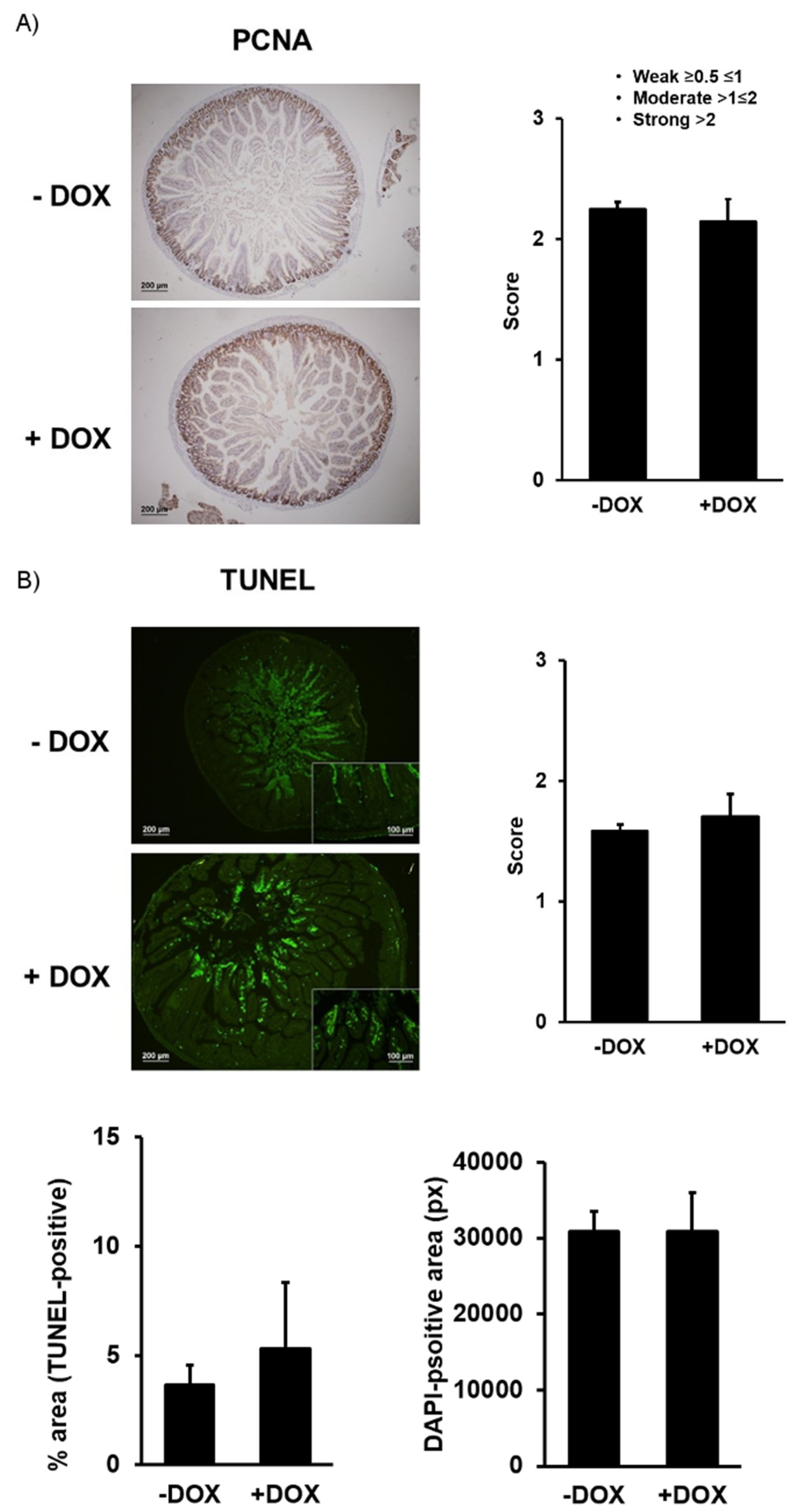
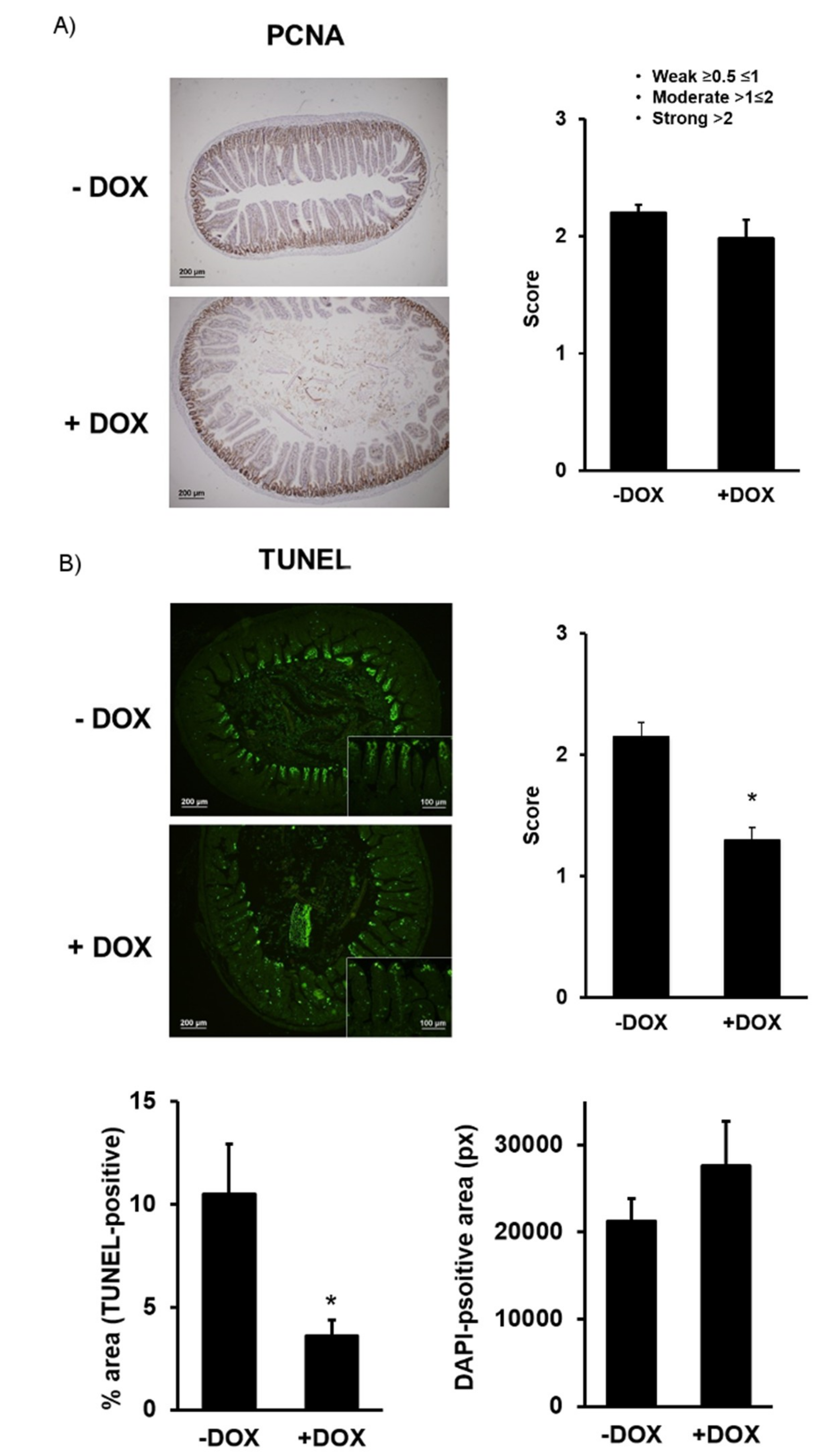
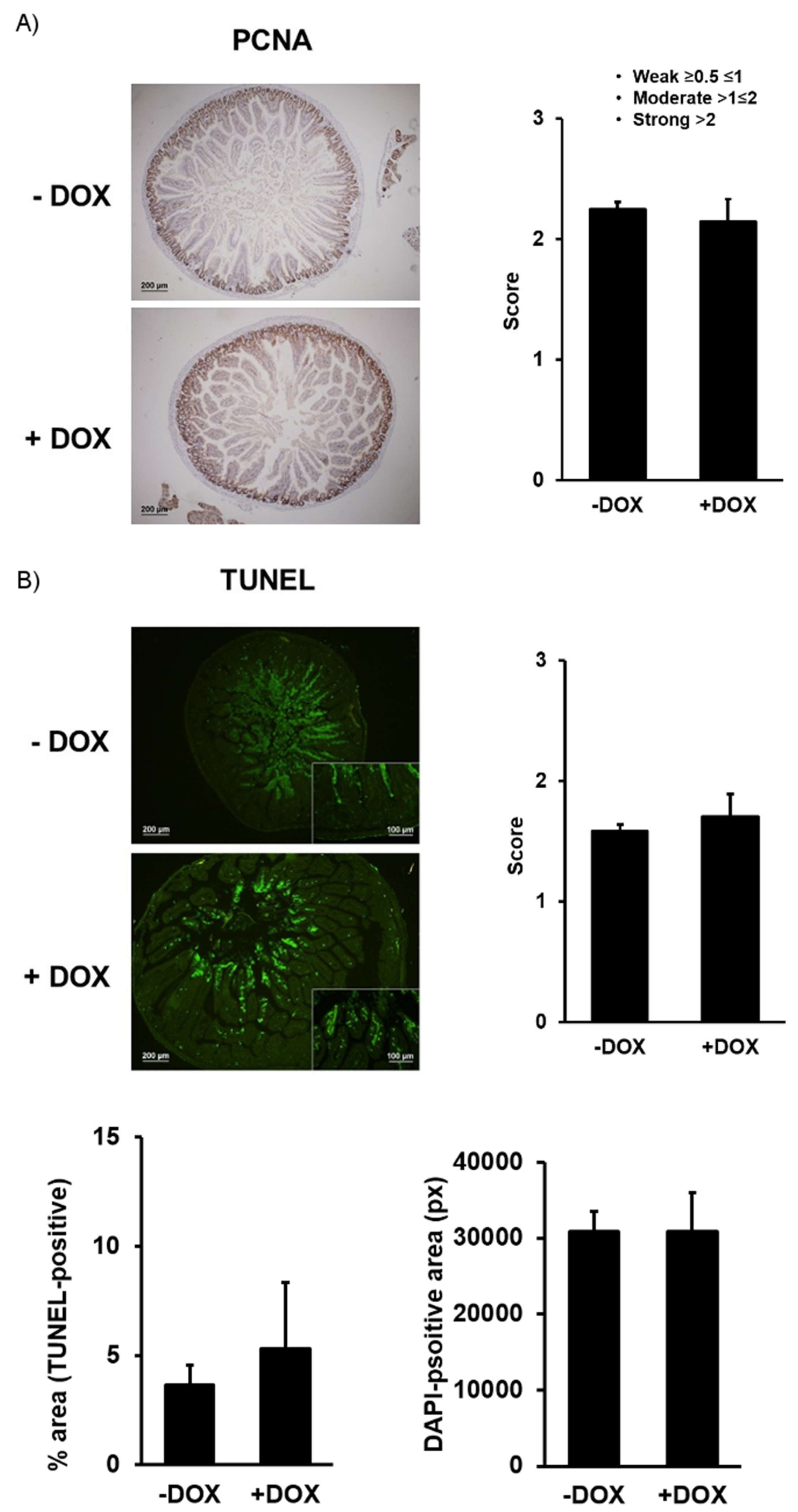
2.1.2. Epithelial Homeostasis of the Duodenum
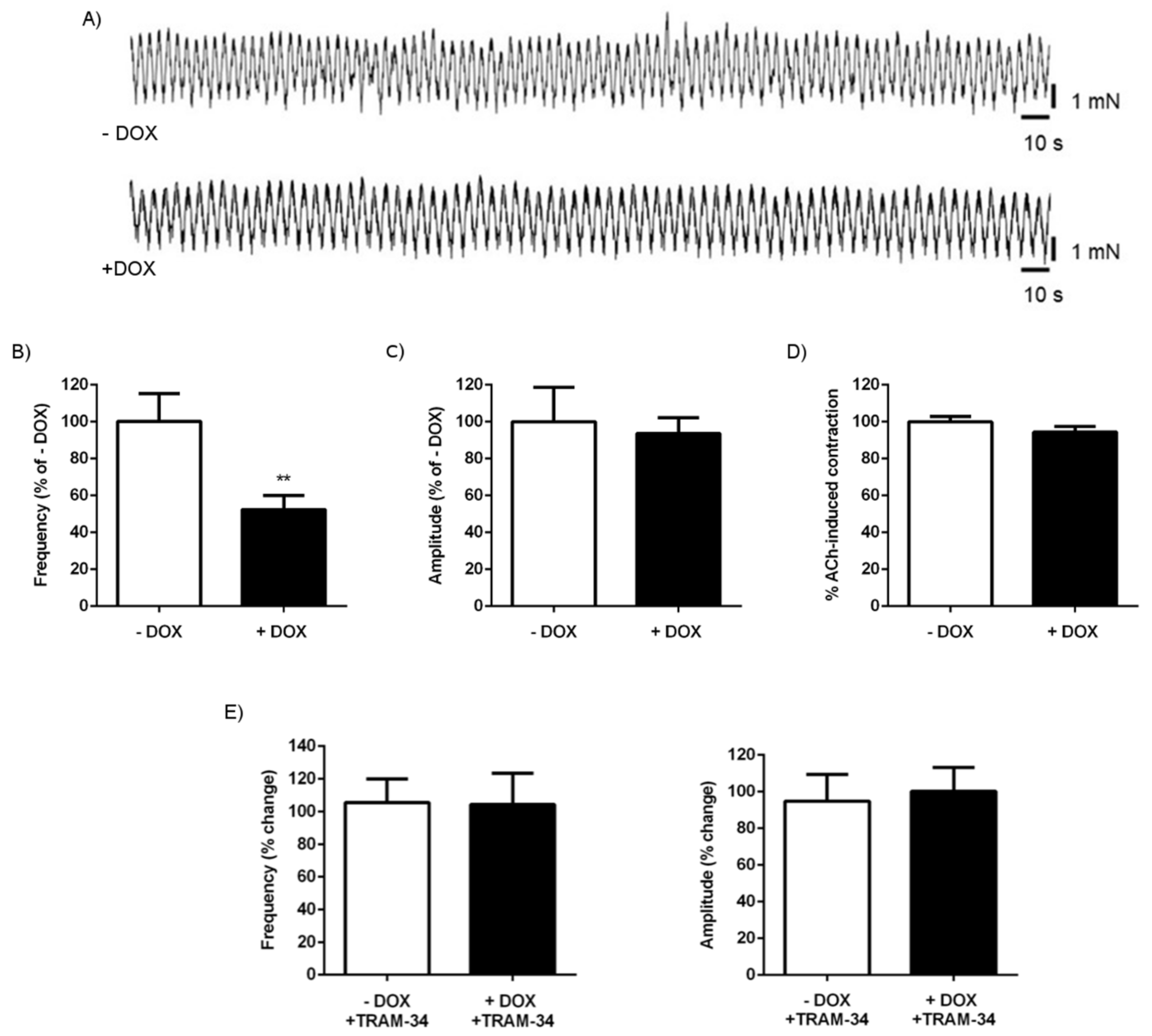
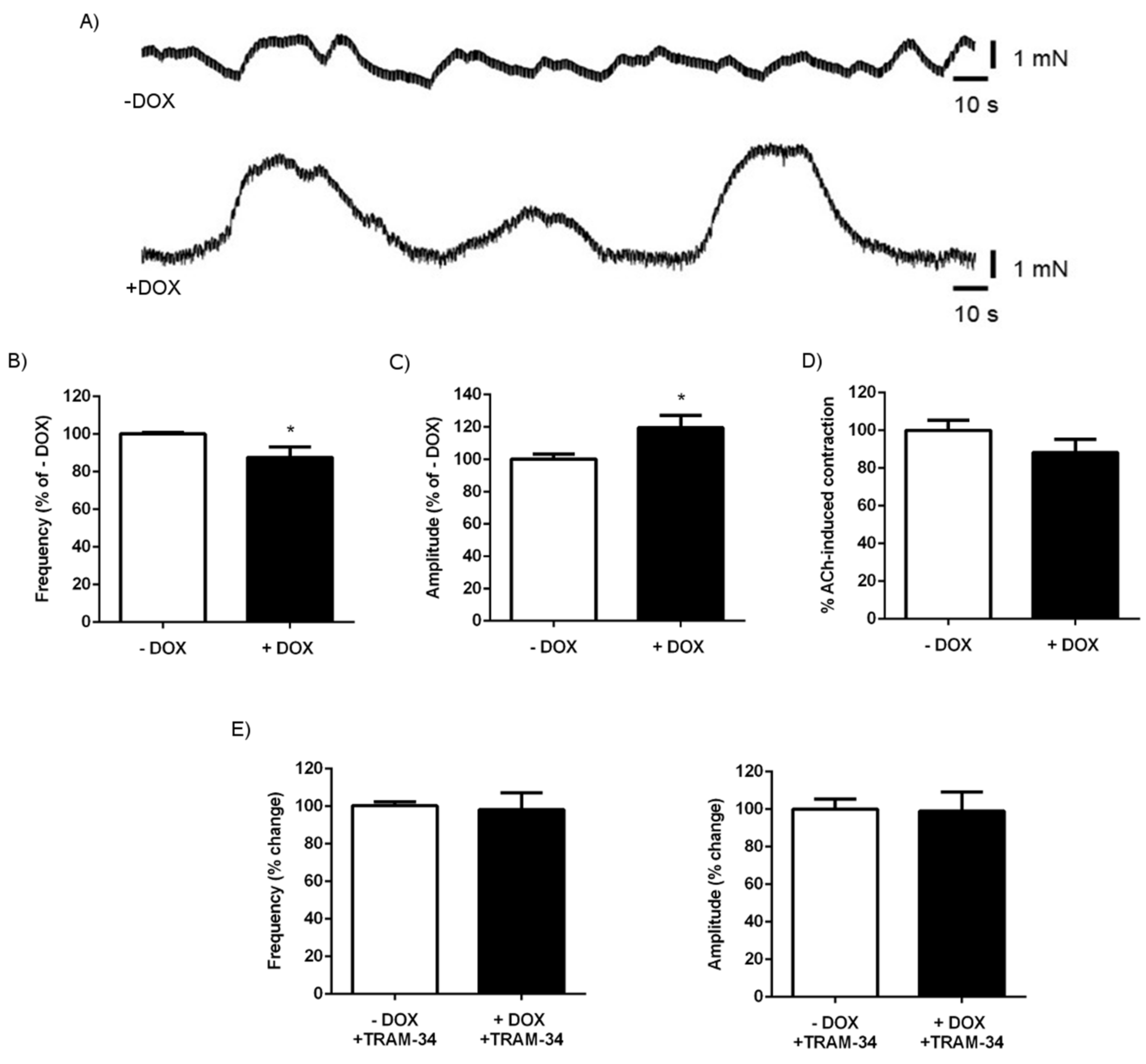
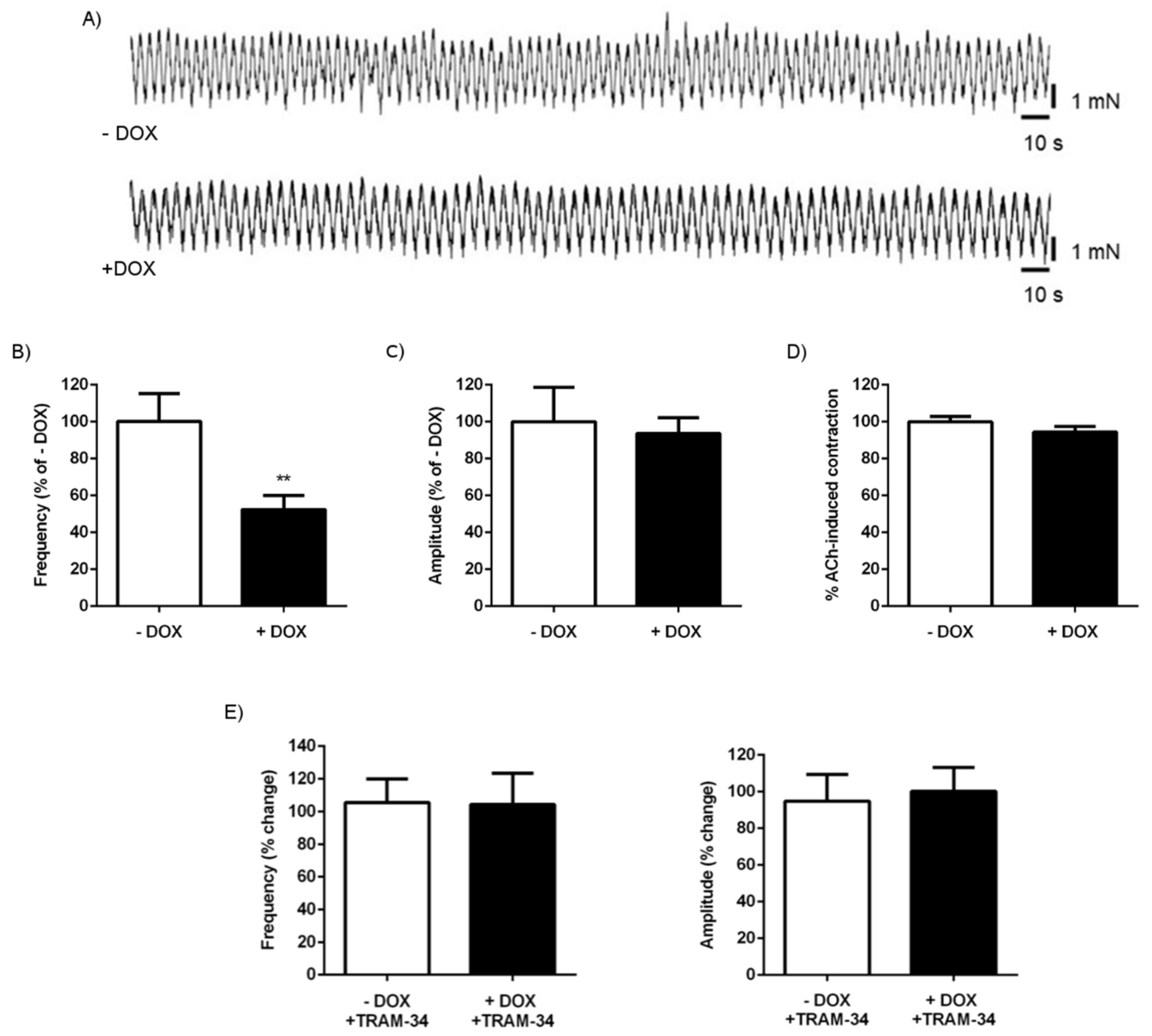
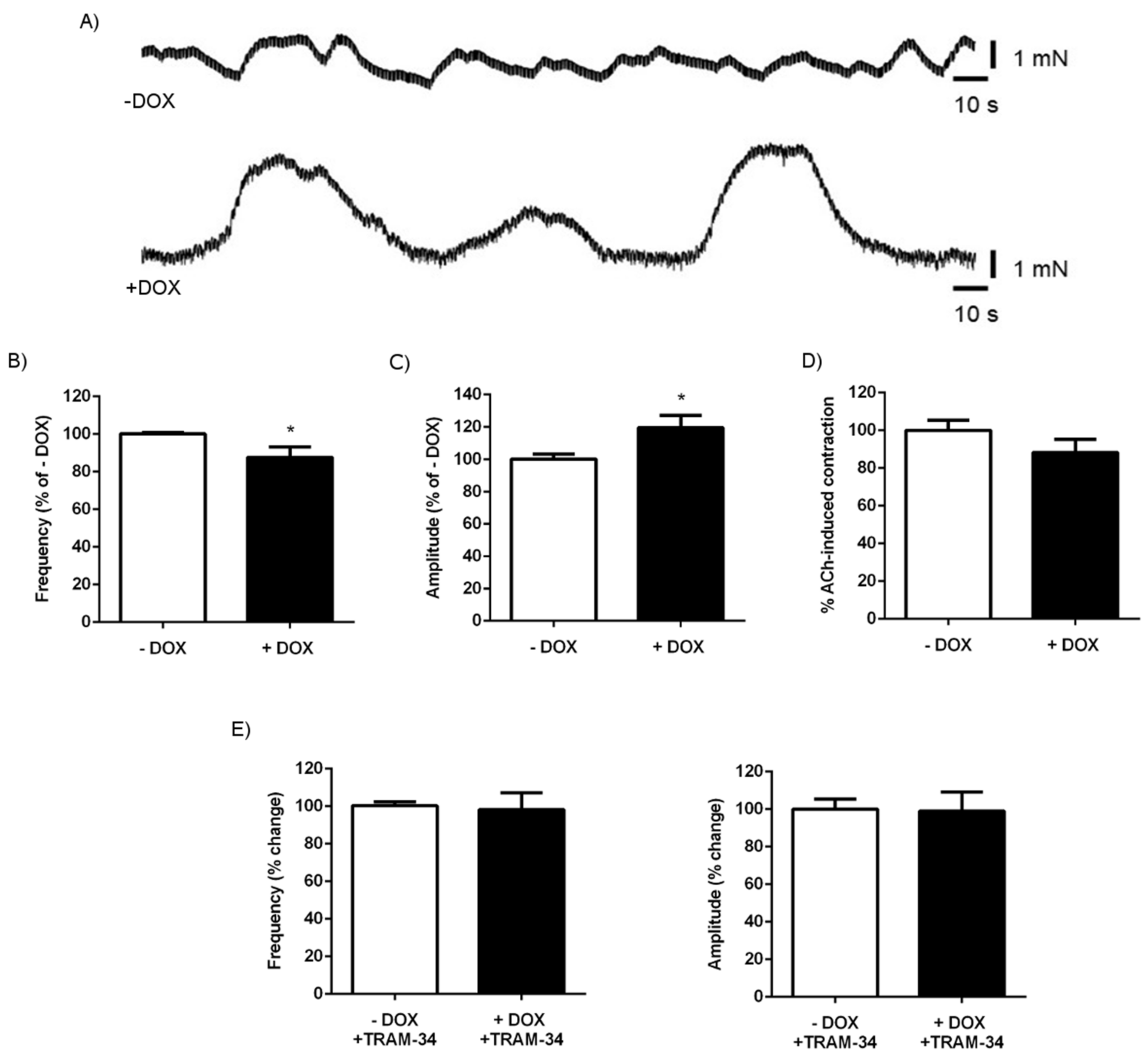
2.1.3. Alterations of Intestinal Contractility in KCa3.1+ Mice
2.1.4. Senicapoc Treatment
2.1.5. KCa3.1 Suppression in Intestinal Epithelium
3. Discussion
4. Materials and Methods
4.1. Epithelium-Specific Inducible KCa3.1+ and Inducible KCa3.1− Mice
4.2. Tissue Preparation
4.3. Histology
4.4. Intestinal Contractility Studies
4.5. Hydroelectrolytic Determination in Serum, Chyme, and Stools
4.6. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DAPI | 4′,6-Diamidine-2′-phenylindole dihydrochloride |
| DOX | Doxycycline |
| KCa3.1 | Intermediate-conductance calcium-activated potassium channel Type 4 |
| KCa3.1+ | Conditional KCa3.1-overexpressing mice |
| KCa3.1− | Conditional KCa3.1-suppressing mice |
| Kcnn4 | Intermediate-conductance calcium-activated potassium channel Type 4 gene |
| PCNA | proliferating cell nuclear antigen |
| TUNEL | terminal deoxynucleotide transferase mediated X-dUTP nick end labeling |
References
- Wei, A.D.; Gutman, G.A.; Aldrich, R.; Chandy, K.G.; Grissmer, S.; Wulff, H. International Union of Pharmacology. LII. Nomenclature and Molecular Relationships of Calcium-Activated Potassium Channels. Pharmacol. Rev. 2005, 57, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.M.; Silvia, C.; Hirschberg, B.; Bond, C.T.; Adelman, J.P.; Maylie, J. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA 1997, 94, 11651–11656. [Google Scholar] [CrossRef] [PubMed]
- Devor, D.C.; Singh, A.K.; Frizzell, R.A.; Bridges, R.J. Modulation of Cl- secretion by benzimidazolones. I. Direct activation of a Ca2+-dependent K+ channel. Am. J. Physiol. Cell. Mol. Physiol. 1996, 271, L775–L784. [Google Scholar] [CrossRef] [PubMed]
- Rufo, P.A.; Merlin, D.; Riegler, M.; Ferguson-Maltzman, M.H.; Dickinson, B.L.; Brugnara, C.; Alper, S.L.; Lencer, W.I. The antifungal antibiotic, clotrimazole, inhibits chloride secretion by human intestinal T84 cells via blockade of distinct basolateral K+ conductances. Demonstration of efficacy in intact rabbit colon and in an in vivo mouse model of cholera. J. Clin. Investig. 1997, 100, 3111–3120. [Google Scholar] [CrossRef] [PubMed]
- Al-Hazza, A.; Linley, J.E.; Aziz, Q.; MacLennan, K.A.; Hunter, M.; Sandle, G.I. Potential role of reduced basolateral potassium (IKCa3.1) channel expression in the pathogenesis of diarrhoea in ulcerative colitis. J. Pathol. 2012, 226, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Srivastava, S.; Zhdanova, O.; Ding, Y.; Li, Z.; Wulff, H.; Lafaille, M.; Skolnik, E.Y. Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis. Proc. Natl. Acad. Sci. USA 2010, 107, 1541–1546. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.K.; Sevelsted-Møller, L.; Rabjerg, M.; Larsen, D.; Hansen, T.P.; Wulff, H.; Knudsen, T.; Kjeldsen, J.; Köhler, R. Expression of T-cell K V 1.3 potassium channel correlates with pro-inflammatory cytokines and disease activity in ulcerative colitis. J. Crohns Colitis 2014, 8, 1378–1391. [Google Scholar] [CrossRef]
- Strøbaek, D.; Brown, D.; Jenkins, D.; Chen, Y.-J.; Coleman, N.; Ando, Y.; Chiu, P.; Jørgensen, S.; Demnitz, J.; Wulff, H.; et al. NS6180, a new KCa3.1 channel inhibitor prevents T-cell activation and inflammation in a rat model of inflammatory bowel disease. Br. J. Pharmacol. 2013, 168, 432–444. [Google Scholar] [CrossRef]
- Grgic, I.; Kiss, E.; Kaistha, B.P.; Busch, C.; Kloss, M.; Sautter, J.; Muller, A.; Kaistha, A.; Schmidt, C.; Raman, G.; et al. Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc. Natl. Acad. Sci. USA 2009, 106, 14518–14523. [Google Scholar] [CrossRef]
- Huang, C.; Shen, S.; Ma, Q.; Gill, A.; Pollock, C.A.; Chen, X.-M. KCa3.1 mediates activation of fibroblasts in diabetic renal interstitial fibrosis. Nephrol. Dial. Transplant. 2014, 29, 313–324. [Google Scholar] [CrossRef]
- Zhao, L.-M.; Wang, L.-P.; Wang, H.-F.; Ma, X.-Z.; Zhou, D.-X.; Deng, X.-L. The role of KCa3.1 channels in cardiac fibrosis induced by pressure overload in rats. Pflügers Arch. Eur. J. Physiol. 2015, 467, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Roach, K.M.; Wulff, H.; Feghali-Bostwick, C.; Amrani, Y.; Bradding, P. Increased constitutive αSMA and Smad2/3 expression in idiopathic pulmonary fibrosis myofibroblasts is KCa3.1-dependent. Respir. Res. 2014, 15, 155. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Deuse, T.; Chen, Y.-J.; Wulff, H.; Stubbendorff, M.; Köhler, R.; Miura, H.; Länger, F.; Reichenspurner, H.; Robbins, R.C.; et al. The potassium channel KCa3.1 as new therapeutic target for the prevention of obliterative airway disease. Transplantation 2013, 95, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Møller, L.S.; Fialla, A.D.; Schierwagen, R.; Biagini, M.; Reul, W.; Klein, S.; Rabjerg, M.; Hansen, L.K.; Schaffalitzky de Muckadell, O.; Koehler, R.; et al. KCa3.1 channels are upregulated in hepatocytes of cirrhotic patients. J. Hepatol. 2015, 62, S481. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Catalano, M.; Sciaccaluga, M.; Chece, G.; Cipriani, R.; Rosito, M.; Grimaldi, A.; Lauro, C.; Cantore, G.; Santoro, A.; et al. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis. 2013, 4, e773. [Google Scholar] [CrossRef] [PubMed]
- Bulk, E.; Ay, A.-S.; Hammadi, M.; Ouadid-Ahidouch, H.; Schelhaas, S.; Hascher, A.; Rohde, C.; Thoennissen, N.H.; Wiewrodt, R.; Schmidt, E.; et al. Epigenetic dysregulation of K Ca 3.1 channels induces poor prognosis in lung cancer. Int. J. Cancer 2015, 137, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Rabjerg, M.; Oliván-Viguera, A.; Koch Hansen, L.; Jensen, L.; Sevelsted-Møller, L.; Walter, S.; Jensen, B.L.; Marcussen, N.; Köhler, R. High expression of KCa3.1 in patients with clear cell renal carcinoma predicts high metastatic risk and poor survival. PLoS ONE 2015, 10, e0122992. [Google Scholar] [CrossRef]
- Ramachandran, A.; Madesh, M.; Balasubramanian, K.A. Apoptosis in the intestinal epithelium: Its relevance in normal and pathophysiological conditions. J. Gastroenterol. Hepatol. 2000, 15, 109–120. [Google Scholar] [CrossRef]
- Wulff, H.; Miller, M.J.; Hansel, W.; Grissmer, S.; Cahalan, M.D.; Chandy, K.G. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: A potential immunosuppressant. Proc. Natl. Acad. Sci. USA 2000, 97, 8151–8156. [Google Scholar] [CrossRef]
- Sevelsted Møller, L.; Fialla, A.D.; Schierwagen, R.; Biagini, M.; Liedtke, C.; Laleman, W.; Klein, S.; Reul, W.; Koch Hansen, L.; Rabjerg, M.; et al. The calcium-activated potassium channel KCa3.1 is an important modulator of hepatic injury. Sci. Rep. 2016, 6, 28770. [Google Scholar] [CrossRef]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef] [PubMed]
- Köhler, R.; Oliván-Viguera, A.; Wulff, H. Endothelial small- and intermediate-conductance K channels and endothelium-dependent hyperpolarization as drug targets in cardiovascular disease. Adv. Pharmacol. 2016, 77, 65–104. [Google Scholar] [CrossRef] [PubMed]
- Hochedlinger, K.; Yamada, Y.; Beard, C.; Jaenisch, R. Ectopic Expression of Oct-4 Blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 2005, 121, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Grgic, I.; Kaistha, B.P.; Paschen, S.; Kaistha, A.; Busch, C.; Si, H.; Köhler, K.; Elsässer, H.-P.; Hoyer, J.; Köhler, R. Disruption of the Gardos channel (KCa3.1) in mice causes subtle erythrocyte macrocytosis and progressive splenomegaly. Pflügers Arch. Eur. J. Physiol. 2009, 458, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Heyken, W.-T.; Wolfle, S.E.; Tysiac, M.; Schubert, R.; Grgic, I.; Vilianovich, L.; Giebing, G.; Maier, T.; Gross, V.; et al. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ. Res. 2006, 99, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Gramsbergen, J.B.; Sivasaravanaparan, M.; Ditzel, N.; Sevelsted-Møller, L.M.; Oliván-Viguera, A.; Rabjerg, M.; Wulff, H.; Köhler, R. Genetic KCa3.1-deficiency produces locomotor hyperactivity and alterations in cerebral monoamine levels. PLoS ONE 2012, 7, e47744. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Nguyen, H.M.; Maezawa, I.; Grössinger, E.M.; Garing, A.L.; Köhler, R.; Jin, L.-W.; Wulff, H. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J. Cereb. Blood Flow Metab. 2016, 36, 2146–2161. [Google Scholar] [CrossRef] [PubMed]
- Tappenden, K.A. Intestinal adaptation following resection. J. Parenter. Enter. Nutr. 2014, 38, 23S–31S. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| −DOX | +DOX | p vs. − DOX | |||
|---|---|---|---|---|---|
| Mean + SEM | n (mice) | Mean + SEM | n (mice) | ||
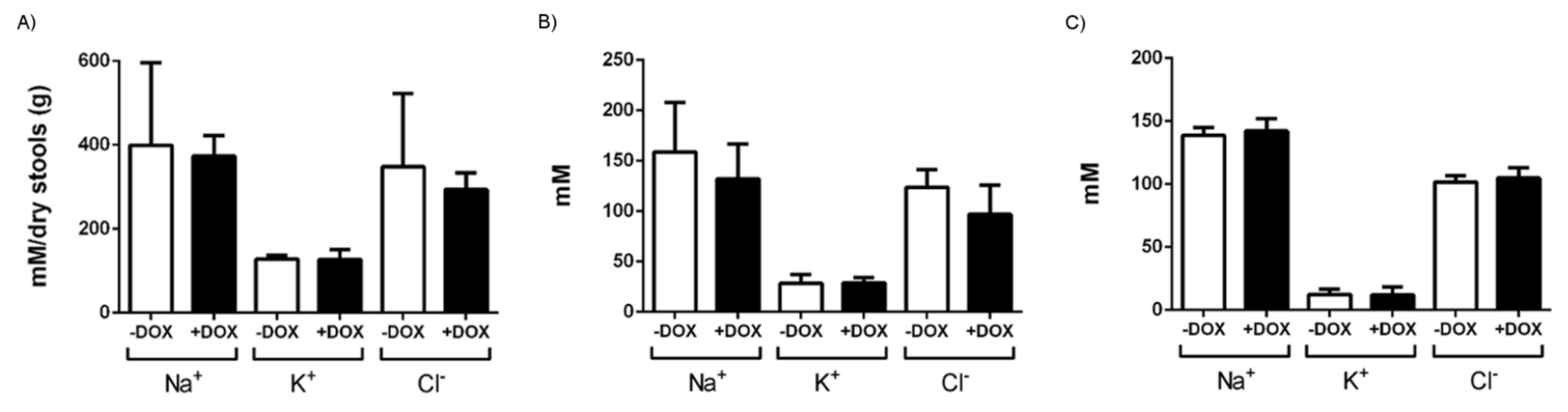
| Water intake 1st week (mL/day) | 4.7 ± 0.3 | 33 | 4.1 ± 0.4 | 34 | ns |
| Water intake 2nd week (mL/day) | 4.2 ± 0.3 | 33 | 6.8 ± 0.6 | 34 | 0.0005 |
| Food intake (g) | 1.4 ± 0.2 | 11 | 1.7 ± 0.2 | 9 | ns |
| Stool pellets: | |||||
| Wet weight (mg) | 97 ± 10 | 11 | 140 ± 11 | 14 | 0.009 |
| Dry weight (mg) | 27 ± 3 | 11 | 40 ± 3 | 14 | 0.009 |
| Water content (%) | 72 ± 1 | 11 | 71 ± 1 | 14 | ns |
| − DOX | + DOX | p vs. − DOX | |||
|---|---|---|---|---|---|
| Mean ± SEM (g organ/g BW) × 100 | n (mice) | Mean ± SEM | n (mice) | ||
| Organography | |||||
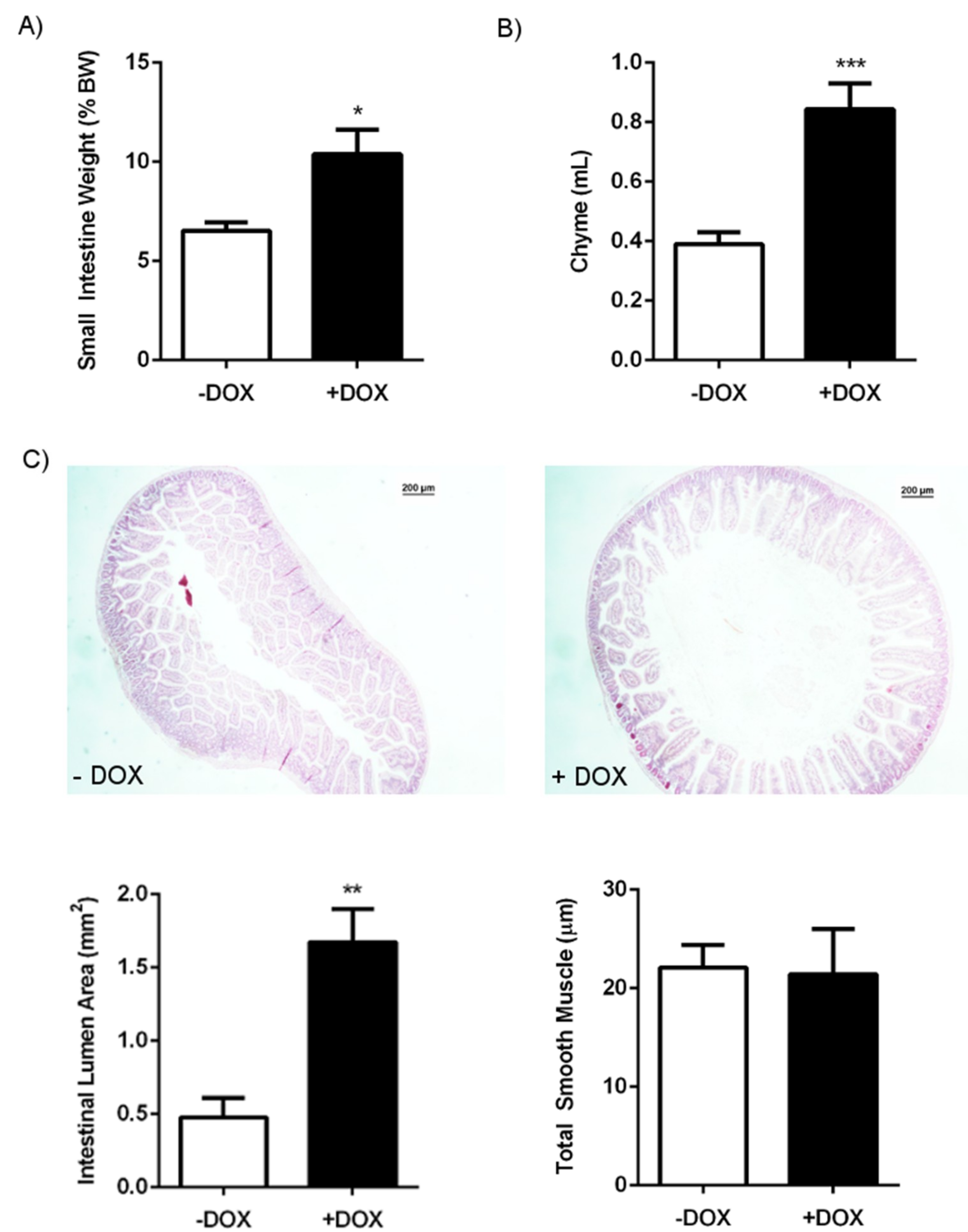
| GI package | 12.7 ± 0.6 | 12 | 17.5 ± 1.8 | 8 | 0.0328 |
| Small intestine | 6.5 ± 0.4 | 12 | 10.4 ± 1.2 | 8 | 0.0120 |
| Stomach | 1.7 ± 0.2 | 10 | 2.1 ± 0.3 | 8 | ns |
| Caecum | 1.6 ± 0.1 | 12 | 4.9 ± 0.6 | 8 | <0.0001 |
| Colon | 1.6 ± 0.2 | 12 | 1.5 ± 0.1 | 8 | ns |
| + DOX + Senicapoc | n (mice) | p vs. − DOX | |
|---|---|---|---|
| (% of –DOX) | |||
| Water intake 1st week | 79 ± 27 | 4 | ns |
| Water intake 2nd week | 98 ± 17 | 5 | ns |
| Food intake | 147 ± 22 | 5 | ns |
| Organography | |||
| GI package | 121 ± 9 | 6 | ns |
| Small intestine | 123 ± 17 | 6 | ns |
| Stomach | 120 ± 17 | 6 | ns |
| Caecum | 125 ± 12 | 6 | ns |
| Colon | 100 ± 20 | 6 | ns |
| Chyme | 74 ± 9 | 6 | ns |
| ILA | 105 ± 30 | 6 | ns |
| Duodenum contractility | |||
| Frequency | 102 ± 23 | 6 | ns |
| Amplitude | 90 ± 13 | 6 | ns |
| Colon contractility | |||
| Frequency | 96 ± 4 | 6 | ns |
| Amplitude | 105 ± 3 | 6 | ns |
| − DOX | + DOX | p vs. − DOX | |||
|---|---|---|---|---|---|
| Mean ± SEM | n (mice) | Mean ± SEM | n (mice) | ||
| Water intake 1st week (mL/d) Water intake 2nd week (mL/d) Food intake (g/d) Stool pellets: | 1.8 ± 0.1 0.9 ± 0.1 1.4 ± 0.2 | 9 9 9 | 1.9 ± 1.0 0.9 ± 0.1 1.7 ± 0.2 | 12 12 12 | ns ns ns |
| Wet weight (mg) | 236 ± 31 | 9 | 213 ± 29 | 15 | ns |
| Dry weight (mg) | 40.7 ± 6.3 | 9 | 36.0 ± 5.2 | 15 | ns |
| Water content (%) | 81.3 ± 0.5 | 9 | 81.5 ± 1.3 | 15 | ns |
| Organography | |||||
| GI package | 19.8 ± 1.0 | 9 | 21.8 ± 0.3 | 15 | ns |
| Small intestine | 6.3 ± 0.3 | 9 | 7.2 ± 0.4 | 15 | ns |
| Stomach | 1.8 ± 0.1 | 9 | 2.1 ± 0.1 | 15 | ns |
| Caecum | 2.6 ± 0.1 | 9 | 3.1 ± 0.2 | 15 | ns |
| Colon | 1.9 ± 0.1 | 9 | 2.0 ± 0.1 | 15 | ns |
| Chyme (mL) | 0.3 ± 0.0 | 9 | 0.4 ± 0.0 | 15 | ns |
| ILA (mm2) | 0.1 ± 0.0 | 9 | 0.2 ± 0.0 | 15 | ns |
| Duodenum contractility | |||||
| Frequency (% of − DOX) | 100 ± 2 | 8 | 99 ± 2 | 8 | ns |
| Amplitude (% of − DOX) | 100 ± 16 | 8 | 99 ± 13 | 8 | ns |
| Frequency (% change-TRAM-34) | 103 ± 1 | 8 | 100 ± 3 | 8 | ns |
| Amplitude (% change-TRAM-34) | 99 ± 18 | 8 | 98 ± 24 | 8 | ns |
| Colon contractility | |||||
| Frequency (% of − DOX) | 100 ± 6 | 8 | 116 ± 7 | 8 | ns |
| Amplitude (% of − DOX) | 100 ± 17 | 8 | 96 ± 19 | 8 | ns |
| Frequency (% change-TRAM-34) | 99 ± 4 | 8 | 98 ± 4 | 8 | ns |
| Amplitude (% change-TRAM-34) | 105 ± 20 | 8 | 96 ± 12 | 8 | ns |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valero, M.S.; Ramón-Gimenez, M.; Lozano-Gerona, J.; Delgado-Wicke, P.; Calmarza, P.; Oliván-Viguera, A.; López, V.; Garcia-Otín, Á.-L.; Valero, S.; Pueyo, E.; et al. KCa3.1 Transgene Induction in Murine Intestinal Epithelium Causes Duodenal Chyme Accumulation and Impairs Duodenal Contractility. Int. J. Mol. Sci. 2019, 20, 1193. https://doi.org/10.3390/ijms20051193
Valero MS, Ramón-Gimenez M, Lozano-Gerona J, Delgado-Wicke P, Calmarza P, Oliván-Viguera A, López V, Garcia-Otín Á-L, Valero S, Pueyo E, et al. KCa3.1 Transgene Induction in Murine Intestinal Epithelium Causes Duodenal Chyme Accumulation and Impairs Duodenal Contractility. International Journal of Molecular Sciences. 2019; 20(5):1193. https://doi.org/10.3390/ijms20051193
Chicago/Turabian StyleValero, Marta Sofía, Mariano Ramón-Gimenez, Javier Lozano-Gerona, Pablo Delgado-Wicke, Pilar Calmarza, Aida Oliván-Viguera, Víctor López, Ángel-Luis Garcia-Otín, Salvador Valero, Esther Pueyo, and et al. 2019. "KCa3.1 Transgene Induction in Murine Intestinal Epithelium Causes Duodenal Chyme Accumulation and Impairs Duodenal Contractility" International Journal of Molecular Sciences 20, no. 5: 1193. https://doi.org/10.3390/ijms20051193
APA StyleValero, M. S., Ramón-Gimenez, M., Lozano-Gerona, J., Delgado-Wicke, P., Calmarza, P., Oliván-Viguera, A., López, V., Garcia-Otín, Á.-L., Valero, S., Pueyo, E., Hamilton, K. L., Miura, H., & Köhler, R. (2019). KCa3.1 Transgene Induction in Murine Intestinal Epithelium Causes Duodenal Chyme Accumulation and Impairs Duodenal Contractility. International Journal of Molecular Sciences, 20(5), 1193. https://doi.org/10.3390/ijms20051193