Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer


Abstract
:1. Introduction
2. The Role of ERK Cascade in Cancer and as a Therapeutic Target
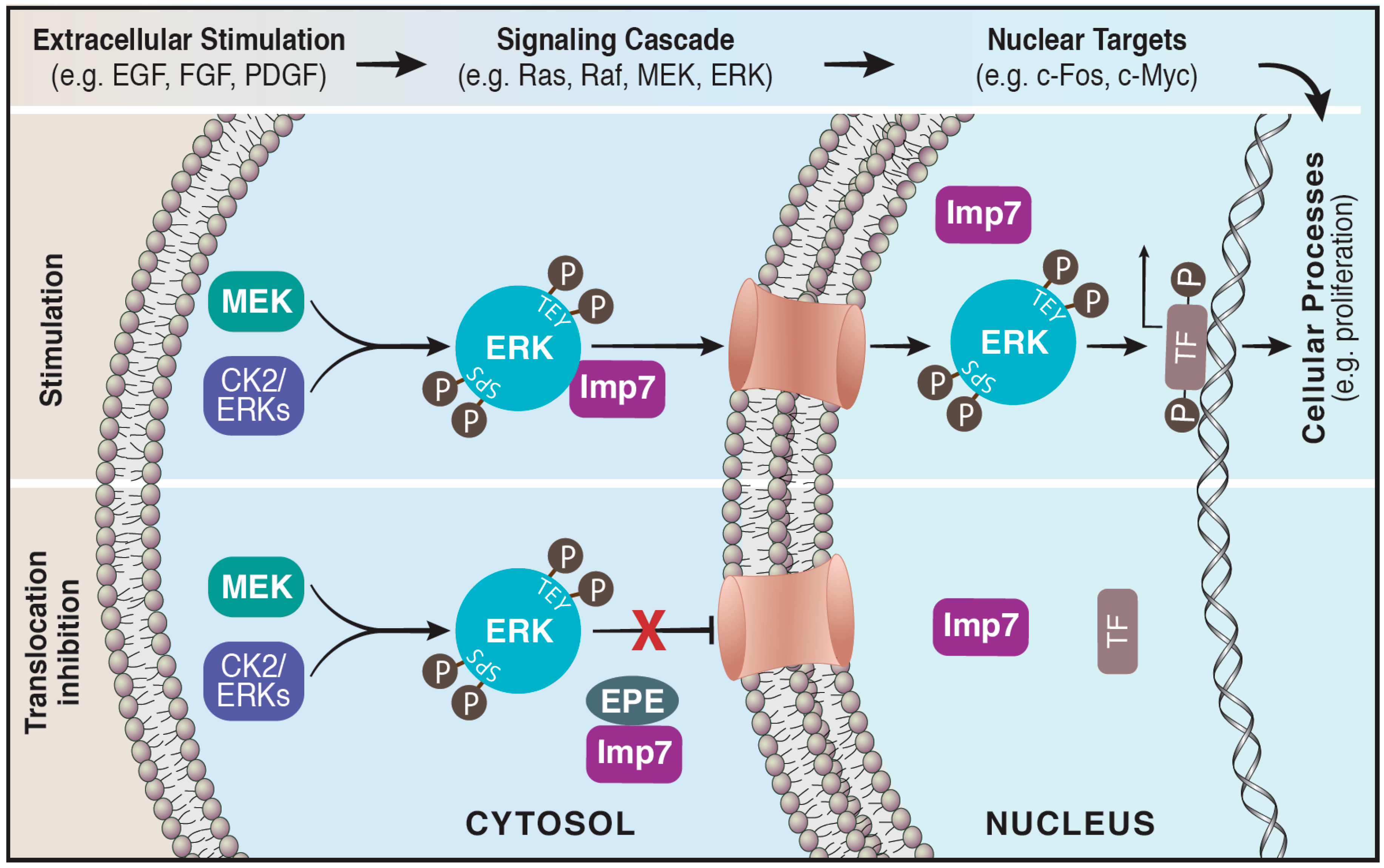
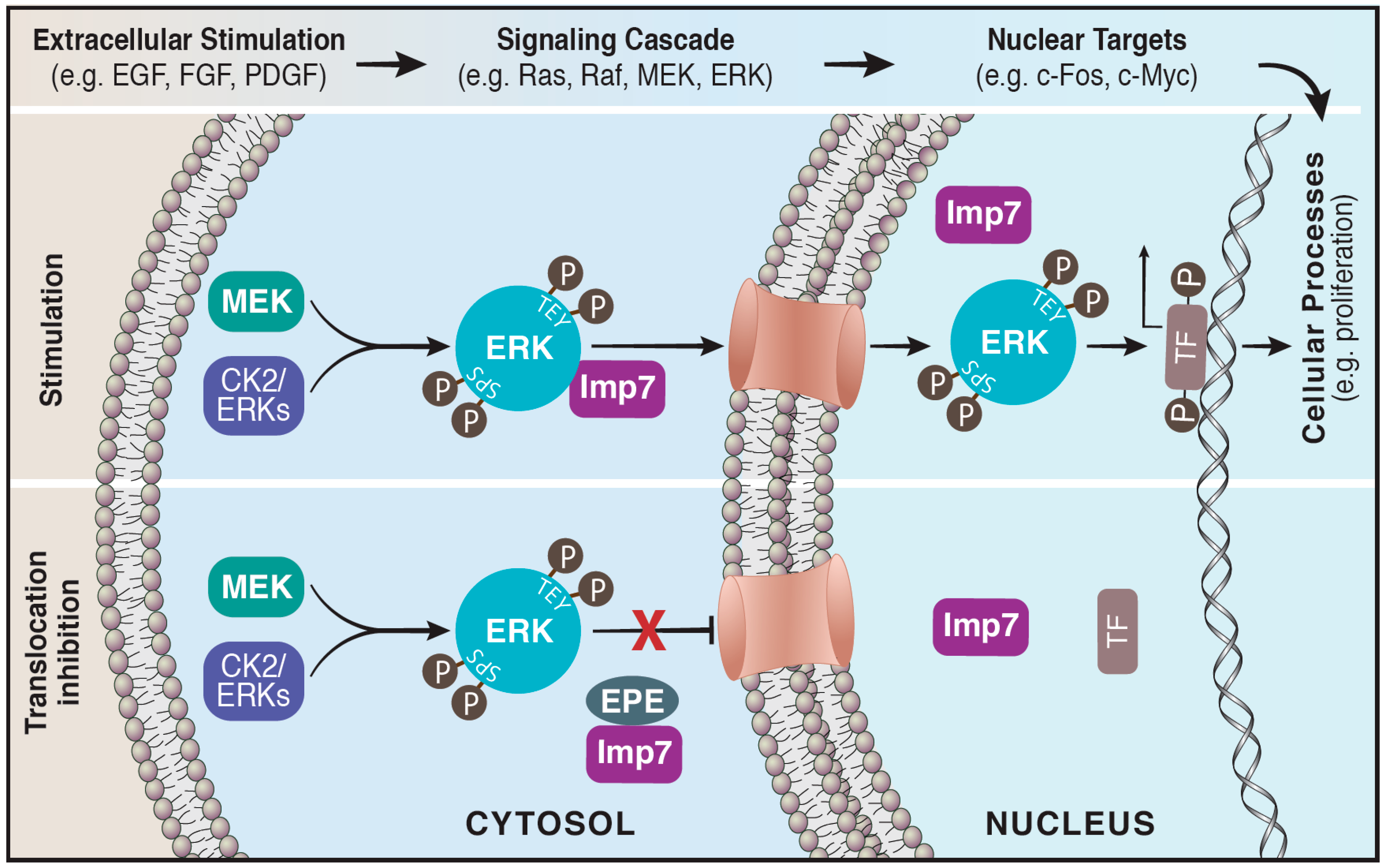
3. Translocation of ERK to the Nucleus
4. Nuclear Functions of ERK
5. Nuclear Substrates of ERK in Cancer Development and Maintenance
6. Nuclear Translocation of Other Signaling Proteins
7. Targeting ERK Nuclear Translocation for Cancer Treatment
8. Summary
Funding
Acknowledgments
Conflicts of Interest
References
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef]
- Eblen, S.T. Extracellular-Regulated Kinases: Signaling from Ras to ERK Substrates to Control Biological Outcomes. Adv. Cancer Res. 2018, 138, 99–142. [Google Scholar]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Patterson, K.I.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar]
- Zhou, B.; Wang, Z.X.; Zhao, Y.; Brautigan, D.L.; Zhang, Z.Y. The specificity of extracellular signal-regulated kinase 2 dephosphorylation by protein phosphatases. J. Biol. Chem. 2002, 277, 31818–31825. [Google Scholar] [CrossRef]
- Yao, Z.; Seger, R. The molecular Mechanism of MAPK/ERK inactivation. Curr. Genom. 2004, 5, 385–393. [Google Scholar] [CrossRef]
- Seternes, O.M.; Kidger, A.M.; Keyse, S.M. Dual-specificity MAP kinase phosphatases in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 124–143. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef]
- Chuderland, D.; Seger, R. Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Mol. Biotechnol. 2005, 29, 57–74. [Google Scholar] [CrossRef]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef]
- Morrison, D.K.; Davis, R.J. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 2003, 19, 91–118. [Google Scholar] [CrossRef]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Wainstein, E.; Seger, R. The dynamic subcellular localization of ERK: Mechanisms of translocation and role in various organelles. Curr. Opin. Cell Biol. 2016, 39, 15–20. [Google Scholar] [CrossRef]
- Yao, Z.; Seger, R. The ERK signaling cascade—Views from different subcellular compartments. Biofactors 2009, 35, 407–416. [Google Scholar] [CrossRef]
- Lawrence, M.C.; Jivan, A.; Shao, C.; Duan, L.; Goad, D.; Zaganjor, E.; Osborne, J.; McGlynn, K.; Stippec, S.; Earnest, S.; et al. The roles of MAPKs in disease. Cell Res. 2008, 18, 436–442. [Google Scholar] [CrossRef]
- Maik-Rachline, G.; Seger, R. The ERK cascade inhibitors: Towards overcoming resistance. Drug Resist. Updates 2016, 25, 1–12. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Gupta, J.; Nebreda, A.R. Roles of p38alpha mitogen-activated protein kinase in mouse models of inflammatory diseases and cancer. FEBS J. 2015, 282, 1841–1857. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Ngoei, K.R.; Zhao, T.T.; Yeap, Y.Y.; Ng, D.C. c-Jun N-terminal kinase (JNK) signaling: Recent advances and challenges. Biochim. Biophys. Acta 2010, 1804, 463–475. [Google Scholar] [CrossRef]
- Yoshizumi, M.; Kyotani, Y.; Zhao, J.; Nagayama, K.; Ito, S.; Tsuji, Y.; Ozawa, K. Role of big mitogen-activated protein kinase 1 (BMK1)/extracellular signal-regulated kinase 5 (ERK5) in the pathogenesis and progression of atherosclerosis. J. Pharmacol. Sci. 2012, 120, 259–263. [Google Scholar] [CrossRef]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef]
- Khan, A.Q.; Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Dermime, S.; Uddin, S. RAS-mediated oncogenic signaling pathways in human malignancies. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in the Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef]
- Wong, D.J.; Robert, L.; Atefi, M.S.; Lassen, A.; Avarappatt, G.; Cerniglia, M.; Avramis, E.; Tsoi, J.; Foulad, D.; Graeber, T.G.; et al. Antitumor activity of the ERK inhibitor SCH772984 against BRAF mutant, NRAS mutant and wild-type melanoma. Mol. Cancer 2014, 13, 194. [Google Scholar] [CrossRef]
- Moschos, S.J.; Sullivan, R.J.; Hwu, W.J.; Ramanathan, R.K.; Adjei, A.A.; Fong, P.C.; Shapira-Frommer, R.; Tawbi, H.A.; Rubino, J.; Rush, T.S., III; et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 2018, 3, 92352. [Google Scholar] [CrossRef]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol. Ther. 2018, 187, 45–60. [Google Scholar] [CrossRef]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef]
- Maik-Rachline, G.; Cohen, I.; Seger, R. RAF, MEK and ERK inhibitors as anti-cancer drugs: Intrinsic and acquired resistance as a major therapeutic challenge. In Resistance to Anti-Cancer Therapeutics Targeting Receptor Tyrosine Kinases and Downstream Pathways; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Unal, E.B.; Uhlitz, F.; Bluthgen, N. A compendium of ERK targets. FEBS Lett. 2017, 591, 2607–2615. [Google Scholar] [CrossRef]
- Ajenjo, N.; Canon, E.; Sanchez-Perez, I.; Matallanas, D.; Leon, J.; Perona, R.; Crespo, P. Subcellular localization determines the protective effects of activated ERK2 against distinct apoptogenic stimuli in myeloid leukemia cells. J. Biol. Chem. 2004, 279, 32813–32823. [Google Scholar] [CrossRef]
- Michailovici, I.; Harrington, H.A.; Azogui, H.H.; Yahalom-Ronen, Y.; Plotnikov, A.; Ching, S.; Stumpf, M.P.; Klein, O.D.; Seger, R.; Tzahor, E. Nuclear to cytoplasmic shuttling of ERK promotes differentiation of muscle stem/progenitor cells. Development 2014, 141, 2611–2620. [Google Scholar] [CrossRef]
- Formstecher, E.; Ramos, J.W.; Fauquet, M.; Calderwood, D.A.; Hsieh, J.C.; Canton, B.; Nguyen, X.T.; Barnier, J.V.; Camonis, J.; Ginsberg, M.H.; et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 2001, 1, 239–250. [Google Scholar] [CrossRef]
- Chen, R.H.; Sarnecki, C.; Blenis, J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol. Cell. Biol. 1992, 12, 915–927. [Google Scholar] [CrossRef]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef]
- Marfori, M.; Mynott, A.; Ellis, J.J.; Mehdi, A.M.; Saunders, N.F.; Curmi, P.M.; Forwood, J.K.; Boden, M.; Kobe, B. Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim. Biophys. Acta 2011, 1813, 1562–1577. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, H.; Hanoch, T.; Seger, R. Identification of a cytoplasmic-retention sequence in ERK2. J. Biol. Chem. 1999, 274, 30349–30352. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.M.; Rossomando, A.J.; Martino, P.; Erickson, A.K.; Her, J.-H.; Shabanowitz, J.; Hunt, D.F.; Weber, M.J.; Sturgill, T.W. Identification of the regulatory phosphorylation sites in pp42/mitogen activated protein kinase (MAP kinase). EMBO J. 1991, 10, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Wolf, I.; Rubinfeld, H.; Yoon, S.; Marmor, G.; Hanoch, T.; Seger, R. Involvement of the activation loop of ERK in the detachment from cytosolic anchoring (withdrawn but found online). J. Biol. Chem. 2001, 276, 24490–24497. [Google Scholar] [CrossRef] [PubMed]
- Chuderland, D.; Konson, A.; Seger, R. Identification and characterization of a general nuclear translocation signal in signaling proteins. Mol. Cell 2008, 31, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Chuderland, D.; Karamansha, Y.; Livnah, O.; Seger, R. Nuclear extracellular signal-regulated kinase 1 and 2 translocation is mediated by casein kinase 2 and accelerated by autophosphorylation. Mol. Cell. Biol. 2011, 31, 3515–3530. [Google Scholar] [CrossRef] [PubMed]
- Zeke, A.; Misheva, M.; Remenyi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef]
- Zehorai, E.; Yao, Z.; Plotnikov, A.; Seger, R. The subcellular localization of MEK and ERK—A novel nuclear translocation signal (NTS) paves a way to the nucleus. Mol. Cell. Endocrinol. 2010, 314, 213–220. [Google Scholar] [CrossRef]
- Flores, K.; Seger, R. Stimulated nuclear import by β-like importins. F1000Prime Rep. 2013, 5. [Google Scholar] [CrossRef]
- Flores, K.; Katz, A.A.; Yadav, S.S.; Seger, R. The nuclear translocation of mitogen-activated protein kinases: Molecular mechanisms and use as novel therapeutic target. Neuroendocrinology 2019, 108, 121–131. [Google Scholar] [CrossRef]
- Zehorai, E.; Seger, R. Beta-like importins mediate the nuclear translocation of mitogen-activated protein kinases. Mol. Cell. Biol. 2014, 34, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Flores, K.; Maik-Rachline, G.; Zehorai, E.; Kapri-Pardes, E.; Berti, D.A.; Hanoch, T.; Besser, M.J.; Seger, R. The nuclear translocation of ERK1/2 as an anticancer target. Nat. Commun. 2015, 6, 6685. [Google Scholar] [CrossRef]
- Hu, X.; Kan, H.; Boye, A.; Jiang, Y.; Wu, C.; Yang, Y. Mitogen-activated protein kinase inhibitors reduce the nuclear accumulation of phosphorylated Smads by inhibiting Imp 7 or Imp 8 in HepG2 cells. Oncol. Lett. 2018, 15, 4867–4872. [Google Scholar] [CrossRef]
- Whitehurst, A.W.; Robinson, F.L.; Moore, M.S.; Cobb, M.H. The death effector domain protein PEA-15 prevents nuclear entry of ERK2 by inhibiting required interactions. J. Biol. Chem. 2004, 279, 12840–12847. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Crespo, P. Working without kinase activity: Phosphotransfer-independent functions of extracellular signal-regulated kinases. Sci. Signal. 2011, 4, re3. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.Y.; Lee, C.; Kwon, K.S. Extracellular signal-regulated kinase 2-dependent phosphorylation induces cytoplasmic localization and degradation of p21Cip1. Mol. Cell. Biol. 2009, 29, 3379–3389. [Google Scholar] [CrossRef] [PubMed]
- Pulverer, B.J.; Fisher, C.; Vousden, K.; Littlewood, T.; Evan, G.; Woodgett, J.R. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene 1994, 9, 59–70. [Google Scholar] [PubMed]
- Chuang, C.F.; Ng, S.Y. Functional divergence of the MAP kinase pathway. ERK1 and ERK2 activate specific transcription factors. FEBS Lett. 1994, 346, 229–234. [Google Scholar]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Junttila, M.R.; Westermarck, J. Mechanisms of MYC stabilization in human malignancies. Cell Cycle 2008, 7, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, K.; Huppi, K.; Spangler, G.; Siwarski, D.; Iyer, R.; Magrath, I. Point mutations in the c-Myc transactivation domain are common in Burkitt’s lymphoma and mouse plasmacytomas. Nat. Genet. 1993, 5, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Gille, H.; Sharrocks, A.D.; Shaw, P.E. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature 1992, 358, 414–417. [Google Scholar] [CrossRef]
- Okazaki, K.; Sagata, N. The Mos/MAP kinase pathway stabilizes c-Fos by phosphorylation and augments its transforming activity in NIH 3T3 cells. EMBO J. 1995, 14, 5048–5059. [Google Scholar] [CrossRef]
- Chen, R.H.; Abate, C.; Blenis, J. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 1993, 90, 10952–10956. [Google Scholar] [CrossRef]
- Chen, R.H.; Juo, P.C.; Curran, T.; Blenis, J. Phosphorylation of c-Fos at the C-terminus enhances its transforming activity. Oncogene 1996, 12, 1493–1502. [Google Scholar]
- Murphy, L.O.; Smith, S.; Chen, R.H.; Fingar, D.C.; Blenis, J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002, 4, 556–564. [Google Scholar] [CrossRef]
- Healy, S.; Khan, P.; Davie, J.R. Immediate early response genes and cell transformation. Pharmacol. Ther. 2013, 137, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Iyer, S.V.; Ward, C.; Link, T.; Diaz, F.J.; Dhar, A.; Tawfik, O.W.; Weinman, S.A.; Azuma, Y.; Izumi, T.; et al. MTBP inhibits the Erk1/2-Elk-1 signaling in hepatocellular carcinoma. Oncotarget 2018, 9, 21429–21443. [Google Scholar] [CrossRef] [PubMed]
- Stefanovsky, V.; Langlois, F.; Gagnon-Kugler, T.; Rothblum, L.I.; Moss, T. Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol. Cell 2006, 21, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Stefanovsky, V.Y.; Pelletier, G.; Hannan, R.; Gagnon-Kugler, T.; Rothblum, L.I.; Moss, T. An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol. Cell 2001, 8, 1063–1073. [Google Scholar] [CrossRef]
- Ayrault, O.; Andrique, L.; Fauvin, D.; Eymin, B.; Gazzeri, S.; Seite, P. Human tumor suppressor p14ARF negatively regulates rRNA transcription and inhibits UBF1 transcription factor phosphorylation. Oncogene 2006, 25, 7577–7586. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, K.; Sueoka, N.; Sato, A.; Sakuragi, T.; Sakao, Y.; Tominaga, M.; Suzuki, T.; Yoshida, Y.; Junko, K.; Yamamoto, T.; et al. Alteration of expression or phosphorylation status of tob, a novel tumor suppressor gene product, is an early event in lung cancer. Cancer Lett. 2003, 202, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Kawamura-Tsuzuku, J.; Ohsugi, M.; Yoshida, M.; Emi, M.; Nakamura, Y.; Onda, M.; Yoshida, Y.; Nishiyama, A.; Yamamoto, T. Tob, a novel protein that interacts with p185erbB2, is associated with anti-proliferative activity. Oncogene 1996, 12, 705–713. [Google Scholar]
- Guan, R.; Peng, L.; Wang, D.; He, H.; Wang, D.; Zhang, R.; Wang, H.; Hao, H.; Zhang, J.; Song, H.; et al. Decreased TOB1 expression and increased phosphorylation of nuclear TOB1 promotes gastric cancer. Oncotarget 2017, 8, 75243–75253. [Google Scholar] [CrossRef]
- Suzuki, T.; Junko, K.; Ajima, R.; Nakamura, T.; Yoshida, Y.; Yamamoto, T. Phosphorylation of three regulatory serines of Tob by Erk1 and Erk2 is required for Ras-mediated cell proliferation and transformation. Genes Dev. 2002, 16, 1356–1370. [Google Scholar] [CrossRef]
- Yang, W.; Dolloff, N.G.; El-Deiry, W.S. ERK and MDM2 prey on FOXO3a. Nat. Cell Biol. 2008, 10, 125–126. [Google Scholar] [CrossRef]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Procaccia, S.; Ordan, M.; Cohen, I.; Bendetz-Nezer, S.; Seger, R. Direct binding of MEK1 and MEK2 to AKT induces Foxo1 phosphorylation, cellular migration and metastasis. Sci. Rep. 2017, 7, 43078. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Krigsfeld, G.; Mayes, P.A.; Patel, L.; Dicker, D.T.; Patel, A.S.; Dolloff, N.G.; Messaris, E.; Scata, K.A.; Wang, W.; et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci. Transl. Med. 2013, 5, 171ra17. [Google Scholar] [CrossRef] [PubMed]
- Leder, S.; Weber, Y.; Altafaj, X.; Estivill, X.; Joost, H.G.; Becker, W. Cloning and characterization of DYRK1B, a novel member of the DYRK family of protein kinases. Biochem. Biophys. Res. Commun. 1999, 254, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. A wake-up call to quiescent cancer cells—Potential use of DYRK1B inhibitors in cancer therapy. FEBS J. 2018, 285, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Ashford, A.L.; Dunkley, T.P.; Cockerill, M.; Rowlinson, R.A.; Baak, L.M.; Gallo, R.; Balmanno, K.; Goodwin, L.M.; Ward, R.A.; Lochhead, P.A.; et al. Identification of DYRK1B as a substrate of ERK1/2 and characterisation of the kinase activity of DYRK1B mutants from cancer and metabolic syndrome. Cell. Mol. Life Sci. 2016, 73, 883–900. [Google Scholar] [CrossRef] [PubMed]
- Jaaro, H.; Rubinfeld, H.; Hanoch, T.; Seger, R. Nuclear translocation of mitogen-activated protein kinase kinase (MEK1) in response to mitogenic stimulation. Proc. Natl. Acad. Sci. USA 1997, 94, 3742–3747. [Google Scholar] [CrossRef] [PubMed]
- Neri, L.M.; Billi, A.M.; Manzoli, L.; Rubbini, S.; Gilmour, R.S.; Cocco, L.; Martelli, A.M. Selective nuclear translocation of protein kinase C alpha in Swiss 3T3 cells treated with IGF-I, PDGF and EGF. FEBS Lett. 1994, 347, 63–68. [Google Scholar] [CrossRef]
- Trubiani, O.; Rana, R.A.; Stuppia, L.; Di Primio, R. Nuclear translocation of beta II PKC isoenzyme in phorbol ester-stimulated KM-3 pre-B human leukemic cells. Exp. Cell Res. 1995, 221, 172–178. [Google Scholar] [CrossRef]
- Haller, H.; Ziegler, W.; Lindschau, C.; Luft, F.C. Endothelial cell tyrosine kinase receptor and G protein-coupled receptor activation involves distinct protein kinase C isoforms. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 678–686. [Google Scholar] [CrossRef]
- Chen, C.C.; Wang, J.K.; Chen, W.C. TPA induces translocation but not down-regulation of new PKC isoform eta in macrophages, MDCK cells and astrocytes. FEBS Lett. 1997, 412, 30–34. [Google Scholar] [CrossRef]
- Jones, T.; Courage, C.; Hubbard, A.; Gescher, A. Cellular relocalisation of protein kinase C-theta caused by staurosporine and some of its analogues. Biochem. Pharmacol. 1997, 53, 1413–1418. [Google Scholar] [CrossRef]
- Holt, S.J.; Alexander, P.; Inman, C.B.; Davies, D.E. Epidermal growth factor induced tyrosine phosphorylation of nuclear proteins associated with translocation of epidermal growth factor receptor into the nucleus. Biochem. Pharmacol. 1994, 47, 117–126. [Google Scholar] [CrossRef]
- Chen, J.; Liu, M.Y.; Parish, C.R.; Chong, B.H.; Khachigian, L. Nuclear import of early growth response-1 involves importin-7 and the novel nuclear localization signal serine-proline-serine. Int. J. Biochem. Cell Biol. 2011, 43, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Maik-Rachline, G.; Zehorai, E.; Hanoch, T.; Blenis, J.; Seger, R. The nuclear translocation of the kinases p38 and JNK promotes inflammation-induced cancer. Sci. Signal. 2018, 11, eaao3428. [Google Scholar] [CrossRef] [PubMed]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Muller, J.; Cross, M.J. The role of ERK5 in endothelial cell function. Biochem. Soc. Trans. 2014, 42, 1584–1589. [Google Scholar] [CrossRef]
- Gomez, N.; Erazo, T.; Lizcano, J.M. ERK5 and Cell Proliferation: Nuclear Localization Is What Matters. Front. Cell Dev. Biol. 2016, 4, 105. [Google Scholar] [CrossRef]
- Kasler, H.G.; Victoria, J.; Duramad, O.; Winoto, A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 2000, 20, 8382–8389. [Google Scholar] [CrossRef]
- Raviv, Z.; Kalie, E.; Seger, R. MEK5 and ERK5 are localized in the nuclei of resting as well as stimulated cells, while MEKK2 translocates from the cytosol to the nucleus upon stimulation. J. Cell Sci. 2004, 117, 1773–1784. [Google Scholar] [CrossRef]
- Yao, Z.; Yoon, S.; Kalie, E.; Raviv, Z.; Seger, R. Calcium regulation of EGF-induced ERK5 activation: Role of Lad1-MEKK2 interaction. PLoS ONE 2010, 5, e12627. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, S.; Nishida, E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 2006, 7, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Barros, J.C.; Marshall, C.J. Activation of either ERK1/2 or ERK5 MAP kinase pathways can lead to disruption of the actin cytoskeleton. J. Cell Sci. 2005, 118, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, K.; Terasawa, K.; Morimoto, H.; Nishida, E. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol. Cell. Biol. 2006, 26, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Erazo, T.; Moreno, A.; Ruiz-Babot, G.; Rodriguez-Asiain, A.; Morrice, N.A.; Espadamala, J.; Bayascas, J.R.; Gomez, N.; Lizcano, J.M. Canonical and kinase activity-independent mechanisms for extracellular signal-regulated kinase 5 (ERK5) nuclear translocation require dissociation of Hsp90 from the ERK5-Cdc37 complex. Mol. Cell. Biol. 2013, 33, 1671–1686. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.K.; McCracken, S.R.; Soofi, M.; Fleming, J.; Yu, A.X.; Ahmad, I.; Morland, R.; Machesky, L.; Nixon, C.; Edwards, D.R.; et al. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br. J. Cancer 2011, 104, 664–672. [Google Scholar] [CrossRef] [PubMed]
- McCracken, S.R.; Ramsay, A.; Heer, R.; Mathers, M.E.; Jenkins, B.L.; Edwards, J.; Robson, C.N.; Marquez, R.; Cohen, P.; Leung, H.Y. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene 2008, 27, 2978–2988. [Google Scholar] [CrossRef]
- Simoes, A.E.; Pereira, D.M.; Gomes, S.E.; Brito, H.; Carvalho, T.; French, A.; Castro, R.E.; Steer, C.J.; Thibodeau, S.N.; Rodrigues, C.M.; et al. Aberrant MEK5/ERK5 signalling contributes to human colon cancer progression via NF-kappaB activation. Cell Death Dis. 2015, 6, e1718. [Google Scholar] [CrossRef]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates, J.R., III; Gray, N.S.; Lee, J.D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010, 18, 258–267. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Yeung, Y.T.; Aziz, F.; Guerrero-Castilla, A.; Arguelles, S. Signaling Pathways in Inflammation and Anti-inflammatory Therapies. Curr. Pharm. Des. 2018, 24, 1449–1484. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Igea, A.; Nebreda, A.R. The Stress Kinase p38alpha as a Target for Cancer Therapy. Cancer Res. 2015, 75, 3997–4002. [Google Scholar] [CrossRef] [PubMed]
- Ben-Levy, R.; Hooper, S.; Wilson, R.; Paterson, H.F.; Marshall, C.J. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr. Biol. 1998, 8, 1049–1057. [Google Scholar] [CrossRef]
- Posen, Y.; Kalchenko, V.; Seger, R.; Brandis, A.; Scherz, A.; Salomon, Y. Manipulation of redox signaling in mammalian cells enabled by controlled photogeneration of reactive oxygen species. J. Cell Sci. 2005, 118, 1957–1969. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Ming, X.; Deng, P.; Jiang, Y. Mechanisms regulating the nuclear translocation of p38 MAP kinase. J. Cell. Biochem. 2010, 110, 1420–1429. [Google Scholar] [CrossRef]
- McCrea, P.D.; Gottardi, C.J. Beyond beta-catenin: Prospects for a larger catenin network in the nucleus. Nat. Rev. Mol. Cell Biol. 2016, 17, 55–64. [Google Scholar] [CrossRef]
- Jamieson, C.; Sharma, M.; Henderson, B.R. Targeting the beta-catenin nuclear transport pathway in cancer. Semin. Cancer Biol. 2014, 27, 20–29. [Google Scholar] [CrossRef]
- Kim, W.; Kim, M.; Jho, E.H. Wnt/beta-catenin signalling: From plasma membrane to nucleus. Biochem. J. 2013, 450, 9–21. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef]
- Fagotto, F.; Gluck, U.; Gumbiner, B.M. Nuclear localization signal-independent and importin/karyopherin-independent nuclear import of beta-catenin. Curr. Biol. 1998, 8, 181–190. [Google Scholar] [CrossRef]
- Jian, H.; Shen, X.; Liu, I.; Semenov, M.; He, X.; Wang, X.F. Smad3-dependent nuclear translocation of beta-catenin is required for TGF-beta1-induced proliferation of bone marrow-derived adult human mesenchymal stem cells. Genes Dev. 2006, 20, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.N.; Del Viso, F.; Duncan, A.R.; Robson, A.; Hwang, W.; Kulkarni, S.; Liu, K.J.; Khokha, M.K. RAPGEF5 Regulates Nuclear Translocation of beta-Catenin. Dev. Cell 2018, 44, 248–260.e4. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Tao, Q.; Kofron, M.; Chen, J.S.; Schloemer, A.; Davis, R.J.; Hsieh, J.C.; Wylie, C.; Heasman, J.; Kuan, C.Y. Jun NH2-terminal kinase (JNK) prevents nuclear beta-catenin accumulation and regulates axis formation in Xenopus embryos. Proc. Natl. Acad. Sci. USA 2006, 103, 16313–16318. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Rosen, N. Resistance to BRAF inhibition in melanomas. N. Engl. J. Med. 2011, 364, 772–774. [Google Scholar] [CrossRef] [PubMed]
- Arafeh, R.; Flores, K.; Keren-Paz, A.; Maik-Rachline, G.; Gutkind, N.; Rosenberg, S.; Seger, R.; Samuels, Y. Combined inhibition of MEK and nuclear ERK translocation has synergistic antitumor activity in melanoma cells. Sci. Rep. 2017, 7, 16345. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene | Gene Expression | p-Value | Fold Change | |
|---|---|---|---|---|
| Tumor | Normal | |||
| MYOCD | 3.53 | 6.16 | 1.4 × 10−119 | −6.19 |
| NR0B2 | 2.20 | 4.39 | 9.1 × 10−43 | −4.55 |
| PGR | 5.45 | 7.59 | 3.6 × 10−94 | −4.43 |
| FOS | 11.72 | 13.84 | 3.2 × 10−172 | −4.34 |
| EGR1 | 11.63 | 13.67 | 2.2 × 10−16 | −4.11 |
| DUSP1 | 11.58 | 13.56 | 2.2 × 10−230 | −3.95 |
| THRB | 7.62 | 9.40 | 2.9 × 10−214 | −3.44 |
| IRX2 | 5.20 | 6.81 | 1.3 × 10−31 | −3.06 |
| PPARG | 7.39 | 8.96 | 5.0 × 10−109 | −2.96 |
| TAL1 | 5.28 | 6.74 | 2.7 × 10−133 | −2.76 |
| NR4A2 | 7.74 | 9.19 | 3.5 × 10−94 | −2.72 |
| ESR1 | 6.84 | 8.16 | 3.6 × 10−41 | −2.50 |
| MITF | 7.83 | 9.05 | 2.8 × 10−93 | −2.33 |
| ARRB1 | 8.25 | 9.32 | 3.0 × 10−92 | −2.10 |
| RORA | 8.31 | 9.38 | 1.8 × 10−132 | −2.09 |
| JUN | 12.03 | 13.09 | 5.0 × 10−106 | −2.08 |
| RPS6KA5 | 6.83 | 7.65 | 8.3 × 10−69 | −1.77 |
| JUNB | 11.68 | 12.47 | 7.1 × 10−53 | −1.73 |
| NCOA2 | 8.61 | 9.37 | 4.9 × 10−60 | −1.70 |
| TOB1 | 10.46 | 11.11 | 5.3 × 10−77 | −1.56 |
| ETV3 | 7.01 | 7.61 | 8.2 × 10−45 | −1.53 |
| ETS1 | 10.65 | 11.23 | 1.8 × 10−44 | −1.50 |
| GATA1 | 0.88 | 1.46 | 1.7 × 10−31 | −1.49 |
| NFATC1 | 8.07 | 8.62 | 1.9 × 10−44 | −1.47 |
| PDCD4 | 11.09 | 11.60 | 7.2 × 10−32 | −1.43 |
| RXRA | 10.90 | 11.41 | 3.3 × 10−72 | −1.43 |
| MKL2 | 9.86 | 10.27 | 1.9 × 10−33 | −1.34 |
| CREM | 8.64 | 9.04 | 2.0 × 10−30 | −1.32 |
| ATF2 | 8.49 | 8.85 | 1.0 × 10−39 | −1.29 |
| DCP1A | 9.20 | 9.56 | 3.5 × 10−65 | −1.28 |
| SNAI2 | 7.85 | 8.19 | 9.8 × 10−12 | −1.27 |
| ETV1 | 8.04 | 8.37 | 2.0 × 10−8 | −1.26 |
| CEBPA | 9.19 | 9.50 | 1.2 × 10−5 | −1.24 |
| CDKN1A | 11.39 | 11.69 | 5.3 × 10−10 | −1.24 |
| ESR2 | 3.71 | 3.98 | 1.8 × 10−7 | −1.20 |
| EP300 | 10.94 | 11.19 | 2.7 × 10−27 | −1.19 |
| CIITA | 7.74 | 7.95 | 5.8 × 10−5 | −1.16 |
| PHF2 | 10.10 | 10.30 | 1.0 × 10−19 | −1.15 |
| HNRNPH2 | 11.13 | 11.30 | 2.1 × 10−50 | −1.13 |
| SSBP3 | 9.46 | 9.61 | 2.5 × 10−11 | −1.11 |
| FBXW7 | 8.59 | 8.73 | 3.8 × 10−7 | −1.10 |
| HDAC6 | 10.22 | 10.34 | 8.7 × 10−6 | −1.09 |
| RB1 | 10.07 | 10.19 | 5.1 × 10−8 | −1.08 |
| NUP98 | 11.03 | 11.14 | 2.0 × 10−14 | −1.08 |
| ALOX5 | 8.31 | 8.42 | 0.15 | −1.08 |
| GTF2I | 11.10 | 11.20 | 1.1 × 10−3 | −1.07 |
| THRAP3 | 11.58 | 11.67 | 1.1 × 10−13 | −1.07 |
| SP1 | 11.25 | 11.35 | 1.0 × 10−6 | −1.07 |
| POLR2A | 12.20 | 12.29 | 2.4 × 10−5 | −1.07 |
| LMNA | 12.92 | 13.00 | 0.02 | −1.06 |
| CDKN1B | 10.46 | 10.54 | 2.0 × 10−5 | −1.06 |
| MYC | 10.60 | 10.67 | 0.23 | −1.05 |
| GLI2 | 6.09 | 6.15 | 0.33 | −1.04 |
| TNKS1BP1 | 11.86 | 11.90 | 0.08 | −1.03 |
| NUMA1 | 12.89 | 12.93 | 0.07 | −1.03 |
| NUP214 | 10.49 | 10.53 | 0.01 | −1.03 |
| NUP153 | 10.21 | 10.24 | 0.08 | −1.02 |
| ELK4 | 7.07 | 7.10 | 0.43 | −1.02 |
| H3F3B | 13.57 | 13.59 | 0.48 | −1.01 |
| SP3 | 10.69 | 10.70 | 0.45 | −1.01 |
| CEBPB | 10.07 | 10.08 | 0.81 | −1.01 |
| HMG20A | 9.64 | 9.65 | 0.46 | −1.01 |
| AEBP1 | 11.97 | 11.95 | 0.76 | 1.01 |
| SRRM2 | 13.32 | 13.30 | 0.46 | 1.01 |
| NEUROD1 | 0.99 | 0.96 | 0.68 | 1.02 |
| MAFA | 0.68 | 0.65 | 0.44 | 1.02 |
| TERF2 | 9.25 | 9.22 | 3.5 × 10−3 | 1.02 |
| HIF1A | 11.66 | 11.62 | 0.17 | 1.03 |
| MED1 | 10.17 | 10.12 | 0.02 | 1.03 |
| MKNK1 | 9.01 | 8.96 | 0.02 | 1.03 |
| RPS6KB1 | 9.48 | 9.41 | 6.2 × 10−5 | 1.05 |
| XRN2 | 10.91 | 10.83 | 3.2 × 10−8 | 1.06 |
| PAPOLA | 11.77 | 11.69 | 4.2 × 10−13 | 1.06 |
| RRN3 | 10.17 | 10.08 | 4.2 × 10−9 | 1.06 |
| HNRNPK | 13.51 | 13.39 | 2.3 × 10−41 | 1.09 |
| MAPKAPK2 | 11.56 | 11.43 | 8.2 × 10−12 | 1.10 |
| SAFB2 | 10.57 | 10.43 | 2.4 × 10−11 | 1.10 |
| NUP50 | 10.26 | 10.12 | 1.5 × 10−15 | 1.10 |
| TPR | 11.44 | 11.29 | 1.2 × 10−13 | 1.11 |
| UBTF | 11.53 | 11.37 | 2.6 × 10−25 | 1.12 |
| MZF1 | 8.35 | 8.18 | 9.1 × 10−10 | 1.13 |
| TP53BP1 | 9.98 | 9.79 | 9.4 × 10−10 | 1.14 |
| KHDRBS1 | 11.81 | 11.61 | 2.6 × 10−48 | 1.15 |
| PHOX2A | 0.83 | 0.63 | 1.7 × 10−4 | 1.15 |
| SF3B2 | 12.30 | 12.08 | 1.2 × 10−70 | 1.16 |
| DYRK1B | 9.02 | 8.79 | 2.7 × 10−17 | 1.17 |
| TGIF1 | 10.36 | 10.13 | 1.9 × 10−16 | 1.17 |
| EWSR1 | 12.12 | 11.88 | 1.9 × 10−68 | 1.18 |
| HIST1H3A | 0.73 | 0.49 | 2.5 × 10−20 | 1.18 |
| DDX47 | 10.13 | 9.88 | 2.5 × 10−82 | 1.19 |
| NCOA6 | 10.61 | 10.36 | 2.3 × 10−37 | 1.20 |
| NCOR2 | 11.93 | 11.66 | 4.3 × 10−22 | 1.21 |
| HNRNPD | 11.96 | 11.68 | 1.9 × 10−74 | 1.22 |
| BAZ1B | 11.14 | 10.85 | 1.7 × 10−61 | 1.22 |
| ERF | 10.21 | 9.91 | 6.5 × 10−29 | 1.23 |
| FAM103A1 | 8.52 | 8.22 | 1.9 × 10−66 | 1.23 |
| REXO1 | 9.57 | 9.27 | 3.9 × 10−44 | 1.24 |
| SUPT5H | 11.72 | 11.40 | 1.1 × 10−105 | 1.25 |
| CSNK2A1 | 10.37 | 10.05 | 2.1 × 10−89 | 1.25 |
| RARG | 9.55 | 9.23 | 1.7 × 10−10 | 1.25 |
| TP53 | 10.34 | 10.00 | 7.2 × 10−37 | 1.26 |
| SAFB | 10.96 | 10.62 | 5.7 × 10−71 | 1.27 |
| WIZ | 10.34 | 9.98 | 2.4 × 10−80 | 1.28 |
| ZC3HC1 | 8.66 | 8.29 | 1.6 × 10−102 | 1.29 |
| PML | 10.91 | 10.54 | 4.4 × 10−45 | 1.29 |
| ELK1 | 9.77 | 9.35 | 6.8 × 10−109 | 1.35 |
| GATA4 | 2.77 | 2.25 | 6.3 × 10−5 | 1.44 |
| MYBBP1A | 10.22 | 9.56 | 1.4 × 10−111 | 1.57 |
| RUNX1 | 10.24 | 9.56 | 4.8 × 10−24 | 1.61 |
| RUNX2 | 7.40 | 6.70 | 1.9 × 10−39 | 1.62 |
| CCDC86 | 9.51 | 8.81 | 3.6 × 10−143 | 1.62 |
| NR5A1 | 1.28 | 0.47 | 4.6 × 10−45 | 1.74 |
| INCENP | 8.72 | 7.91 | 1.6 × 10−84 | 1.75 |
| TWIST1 | 5.73 | 4.91 | 9.8 × 10−19 | 1.77 |
| CAD | 10.36 | 9.52 | 5.4 × 10−150 | 1.80 |
| PDX1 | 2.03 | 1.14 | 5.2 × 10−24 | 1.84 |
| MAZ | 12.19 | 11.31 | 6.7 × 10−173 | 1.85 |
| TCF3 | 10.71 | 9.77 | 5.2 × 10−188 | 1.92 |
| LMNB1 | 9.62 | 7.90 | 3.2 × 10−153 | 3.28 |
| ESPL1 | 7.68 | 5.59 | 4.7 × 10−104 | 4.25 |
| TOP2A | 10.37 | 7.10 | 6.8 × 10−194 | 9.64 |
| *UBF | NA | NA | NA | NA |
| *NSMF | NA | NA | NA | NA |
| *RBFOX2 | NA | NA | NA | NA |
| *SRSF11 | NA | NA | NA | NA |
| Gene | Gene Expression | p-Value | Fold Change | Other Localizations | |
|---|---|---|---|---|---|
| Tumor | Normal | ||||
| NR4A1 | 10.47 | 12.37 | 3.4 × 10−118 | −3.73 | cytosol |
| CBX7 | 9.50 | 10.94 | 1.9 × 10−223 | −2.72 | cytosol |
| CRYAA | 0.99 | 2.24 | 4.1 × 10−17 | −2.38 | cytosol |
| ERG | 7.80 | 8.98 | 1.4 × 10−106 | −2.27 | cytosol |
| RBPMS | 9.75 | 10.88 | 1.3 × 10−124 | −2.19 | cytosol |
| FOXO1 | 9.25 | 10.36 | 2.7 × 10−189 | −2.17 | cytosol |
| CRY2 | 9.75 | 10.82 | 8.4 × 10−219 | −2.09 | cytosol |
| NR3C1 | 9.93 | 10.89 | 3.1 × 10−161 | −1.95 | cytosol + mitochondria |
| AIM1 | 9.64 | 10.57 | 2.4 × 10−52 | −1.91 | cytosol + microtubules |
| EGFR | 9.23 | 10.06 | 2.5 × 10−76 | −1.78 | plasma membrane |
| ETS2 | 11.23 | 12.01 | 1.6 × 10−69 | −1.72 | cytosol + plasma membrane |
| NCOA1 | 10.21 | 10.74 | 3.5 × 10−112 | −1.44 | cytosol + plasma membrane |
| FGFR1 | 10.27 | 10.77 | 2.3 × 10−24 | −1.42 | plasma membrane |
| RPS6KA3 | 10.33 | 10.82 | 1.2 × 10−85 | −1.40 | cytosol |
| SMAD4 | 10.43 | 10.92 | 2.5 × 10−158 | −1.40 | cytosol |
| STAT5A | 9.48 | 9.94 | 5.8 × 10−36 | −1.38 | cytosol |
| SORBS3 | 10.76 | 11.22 | 4.4 × 10−72 | −1.37 | focal adhesion sites |
| FOXO3 | 10.44 | 10.82 | 1.6 × 10−48 | −1.30 | cytosol |
| BCL6 | 9.96 | 10.29 | 1.4 × 10−14 | −1.25 | Golgi |
| CRY1 | 8.74 | 9.03 | 1.9 × 10−33 | −1.22 | microtubules |
| SREBF2 | 11.72 | 11.98 | 2.6 × 10−24 | −1.20 | mitochondria |
| WASL | 10.75 | 11.00 | 4.9 × 10−43 | −1.19 | cytosol |
| SMAD3 | 10.45 | 10.70 | 7.8 × 10−25 | −1.19 | cytosol |
| SMAD2 | 10.63 | 10.79 | 3.6 × 10−19 | −1.12 | cytosol |
| SMAD1 | 9.25 | 9.40 | 9.9 × 10−12 | −1.11 | cytosol + plasma membrane |
| RPS6KA1 | 10.22 | 10.25 | 0.59 | −1.02 | cytosol |
| PIP5K1C | 10.36 | 10.32 | 0.16 | 1.03 | cytosol |
| MKL1 | 9.98 | 9.90 | 1.2 × 10−4 | 1.06 | cytosol |
| RAI14 | 10.11 | 10.02 | 1.4 × 10−2 | 1.06 | cytosol |
| NFATC4 | 8.76 | 8.59 | 1.4 × 10−3 | 1.12 | cytosol |
| TGS1 | 9.07 | 8.91 | 3.1 × 10−18 | 1.12 | cytosol |
| HNRNPH1 | 12.00 | 11.83 | 2.4 × 10−21 | 1.13 | cytosol |
| ETV6 | 9.99 | 9.81 | 5.6 × 10−9 | 1.14 | cytosol |
| STAT1 | 12.23 | 11.80 | 3.1 × 10−43 | 1.35 | cytosol |
| PPP1R9B | 10.73 | 10.29 | 5.6 × 10−72 | 1.36 | cytoskeleton |
| DAZAP1 | 10.82 | 10.34 | 2.7 × 10−113 | 1.39 | cytosol |
| POU5F1 | 3.91 | 3.42 | 1.5 × 10−12 | 1.40 | cytosol |
| HSF1 | 10.98 | 10.48 | 3.3 × 10−148 | 1.41 | cytosol |
| MYB | 6.43 | 5.58 | 2.4 × 10−11 | 1.80 | plasma membrane |
| SMC4 | 10.42 | 9.42 | 5.6 × 10−157 | 2.00 | cytosol |
| MAGEA11 | 1.33 | 0.27 | 1.0 × 10−159 | 2.08 | cytosol |
| PTTG1 | 8.68 | 6.19 | 1.2 × 10−140 | 5.63 | cytosol |
| TPX2 | 9.75 | 6.59 | 5.7 × 10−216 | 8.95 | microtubules |
| *PRRC2A | NA | NA | NA | NA | cytosol + plasma membrane |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maik-Rachline, G.; Hacohen-Lev-Ran, A.; Seger, R. Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. Int. J. Mol. Sci. 2019, 20, 1194. https://doi.org/10.3390/ijms20051194
Maik-Rachline G, Hacohen-Lev-Ran A, Seger R. Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. International Journal of Molecular Sciences. 2019; 20(5):1194. https://doi.org/10.3390/ijms20051194
Chicago/Turabian StyleMaik-Rachline, Galia, Avital Hacohen-Lev-Ran, and Rony Seger. 2019. "Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer" International Journal of Molecular Sciences 20, no. 5: 1194. https://doi.org/10.3390/ijms20051194
APA StyleMaik-Rachline, G., Hacohen-Lev-Ran, A., & Seger, R. (2019). Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. International Journal of Molecular Sciences, 20(5), 1194. https://doi.org/10.3390/ijms20051194