Dual-Specificity Phosphatases in Neuroblastoma Cell Growth and Differentiation

 , , , and
, , , and 
Abstract
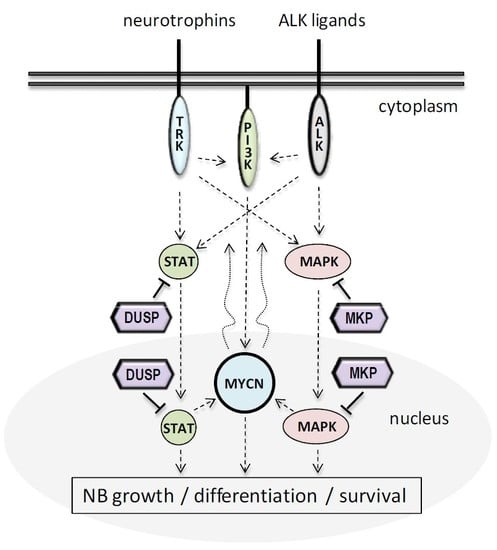
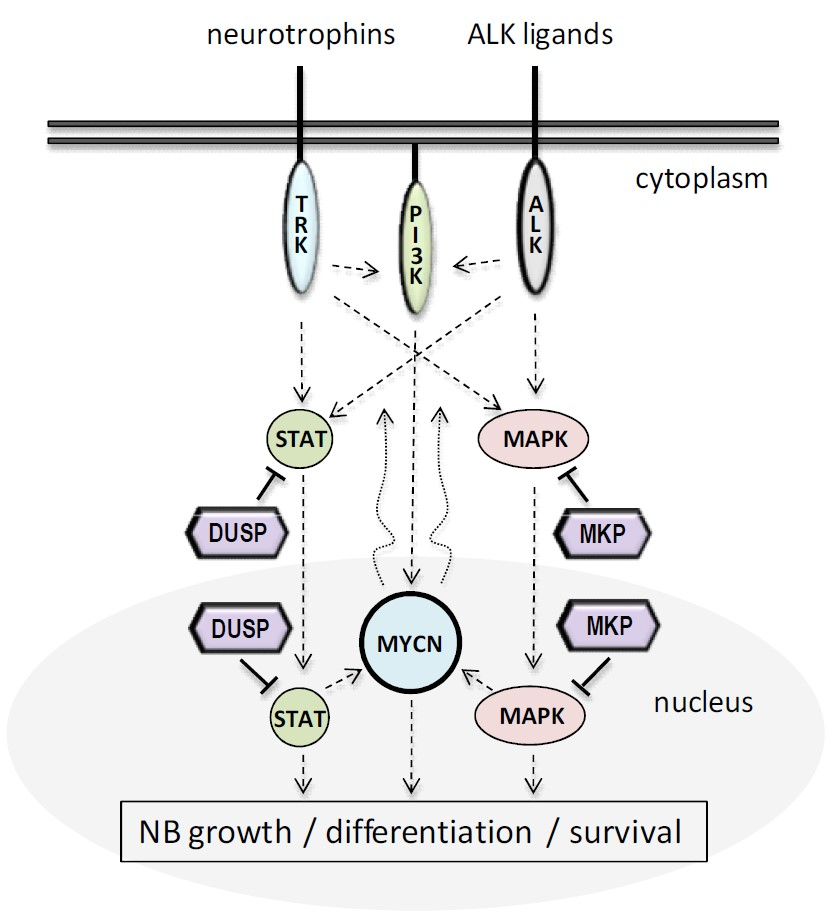
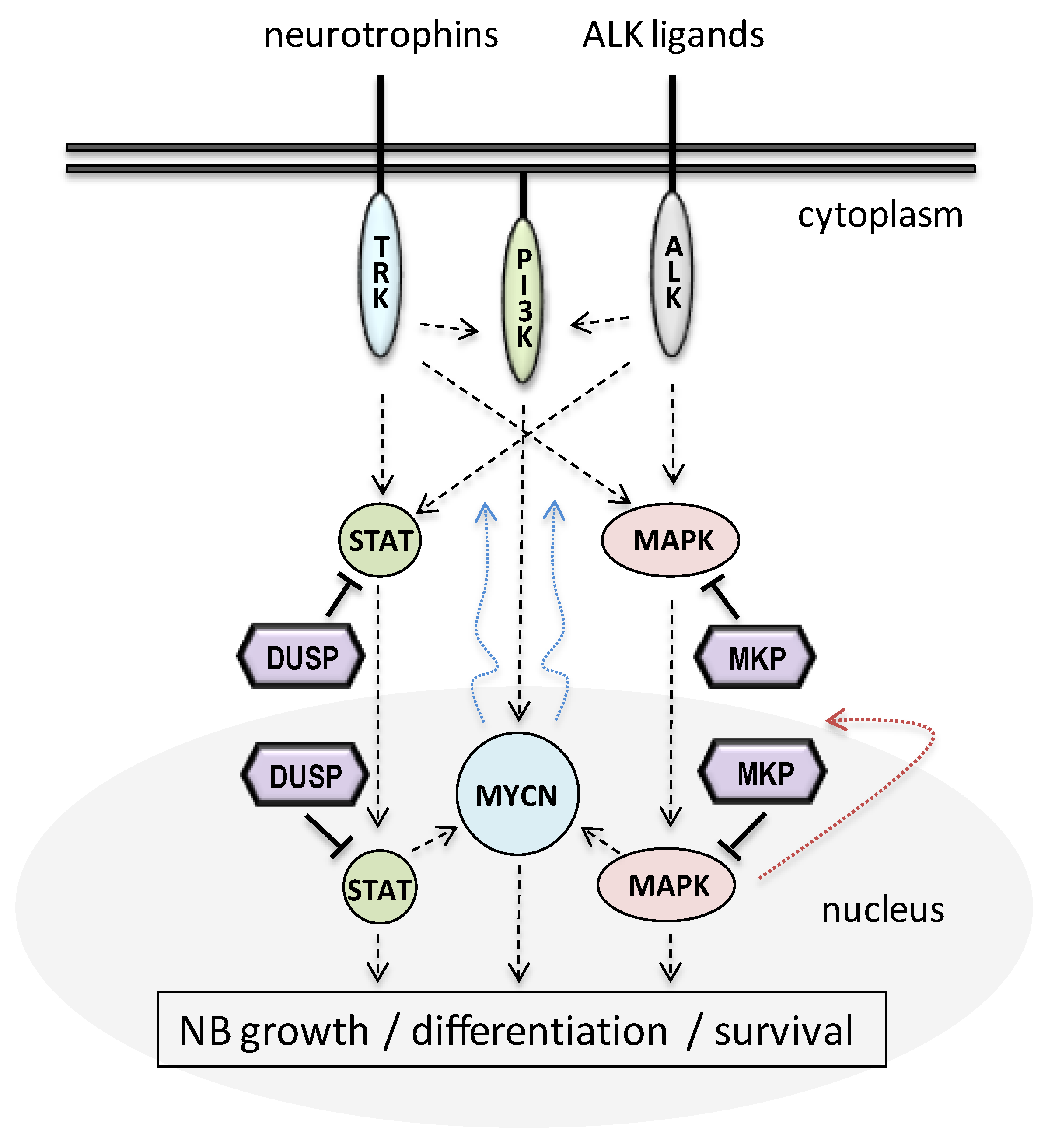
1. Introduction
2. Neuroblastoma Cell Growth and Differentiation
3. DUSPs in NB Cell Growth and Differentiation
3.1. MKPs in NB Cell Growth and Differentiation
3.1.1. DUSP1, DUSP4, and DUSP5
3.1.2. DUSP6, DUSP7, and DUSP9
3.1.3. DUSP8, DUSP10, and DUSP16
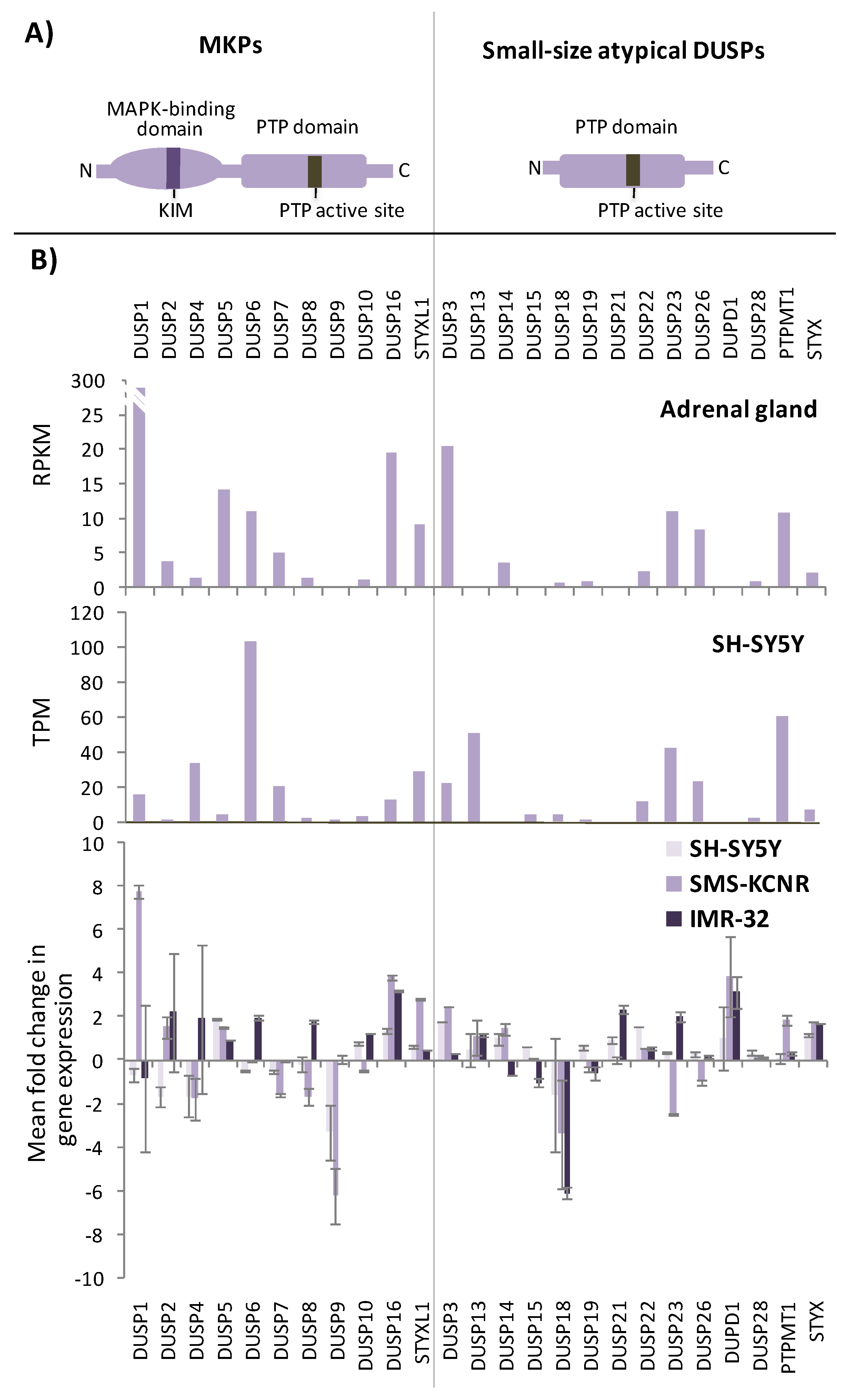
3.2. Small-Size Atypical DUSPs in NB Cell Growth and Differentiation
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALK | Anaplastic lymphoma kinase |
| BDNGF | Brain-derived neurotrophic factor |
| CCHS | Congenital central hypoventilation syndrome |
| DUSP | Dual-specificity phosphatase |
| EGFR | Epidermal growth factor receptor |
| ESC | Embryonic stem cell |
| FGFR | Fibroblast growth factor receptor |
| GWA | Genome-wide association |
| IFN | Interferon |
| KIM | Kinase interaction motif |
| MAPK | Mitogen-activated protein kinase |
| MKP | MAPK phosphatase |
| NB | Neuroblastoma |
| NGF | Nerve growth factor |
| PMA | Phorbol 12-myristate 13-acetate |
| PTP | Protein tyrosine phosphatase |
| RA | Retinoic acid |
| RTK | Receptor tyrosine kinase |
| TRH | Thyrotropin-releasing hormone |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Tsubota, S.; Kadomatsu, K. Origin and initiation mechanisms of neuroblastoma. Cell Tissue Res. 2018, 372, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Vo, K.T.; Matthay, K.K.; Neuhaus, J.; London, W.B.; Hero, B.; Ambros, P.F.; Nakagawara, A.; Miniati, D.; Wheeler, K.; Pearson, A.D.; et al. Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: A report from the international neuroblastoma risk group project. J. Clin. Oncol. 2014, 32, 3169–3176. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Zhang, L.; Reddivalla, N.; Hetherington, M. Neuroblastoma in children: Update on clinicopathologic and genetic prognostic factors. Pediatr. Hematol. Oncol. 2017, 34, 165–185. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Bagatell, R.; London, W.B.; Maris, J.M.; Cohn, S.L.; Mattay, K.K.; Hogarty, M.; COG Neuroblastoma Committee. Children’s Oncology Group’s 2013 blueprint for research: Neuroblastoma. Pediatr. Blood Cancer 2013, 60, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, V.P.; Matthay, K.K. Neuroblastoma: Clinical and biological approach to risk stratification and treatment. Cell Tissue Res. 2018, 372, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, L.; Haupt, R.; Garaventa, A.; Ponzoni, M. Investigational drugs in phase II clinical trials for the treatment of neuroblastoma. Expert Opin. Investig. Drugs 2017, 26, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, P.; Canete, A.; Castel, V. Advances in emerging drugs for the treatment of neuroblastoma. Expert Opin. Emerg. Drugs 2017, 22, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Greengard, E.G. Molecularly Targeted Therapy for Neuroblastoma. Children 2018, 5, 142. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Dyberg, C.; Fransson, S.; Wickstrom, M. Molecular mechanisms and therapeutic targets in neuroblastoma. Pharmacol. Res. 2018, 131, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Zage, P.E. Novel Therapies for Relapsed and Refractory Neuroblastoma. Children 2018, 5, 148. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lopez-Delisle, L.; Delattre, O.; Rohrer, H. The ALK receptor in sympathetic neuron development and neuroblastoma. Cell Tissue Res. 2018, 372, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Trigg, R.M.; Turner, S.D. ALK in Neuroblastoma: Biological and Therapeutic Implications. Cancers 2018, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Maloney, M.A.; Kun, S.S.; Keens, T.G.; Perez, I.A. Congenital central hypoventilation syndrome: Diagnosis and management. Expert Rev. Respir. Med. 2018, 12, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Barr, E.K.; Applebaum, M.A. Genetic Predisposition to Neuroblastoma. Children 2018, 5, 119. [Google Scholar] [CrossRef] [PubMed]
- Ritenour, L.E.; Randall, M.P.; Bosse, K.R.; Diskin, S.J. Genetic susceptibility to neuroblastoma: Current knowledge and future directions. Cell Tissue Res. 2018, 372, 287–307. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, V.P.; Coggins, G.E.; Maris, J.M. Genetic susceptibility to neuroblastoma. Curr. Opin. Genet. Dev. 2017, 42, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.S.L.; Han, J.; James, S.J.; Png, C.W.; Weerasooriya, M.; Alonso, S.; Zhang, Y. Dual-Specificity Phosphatase 12 Targets p38 MAP Kinase to Regulate Macrophage Response to Intracellular Bacterial Infection. Front. Immunol. 2017, 8, 1259. [Google Scholar] [CrossRef] [PubMed]
- Nguyen le, B.; Diskin, S.J.; Capasso, M.; Wang, K.; Diamond, M.A.; Glessner, J.; Kim, C.; Attiyeh, E.F.; Mosse, Y.P.; Cole, K.; et al. Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility Loci. PLoS Genet. 2011, 7, e1002026. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Spontaneous regression of neuroblastoma. Cell Tissue Res. 2018, 372, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Mohlin, S.A.; Wigerup, C.; Pahlman, S. Neuroblastoma aggressiveness in relation to sympathetic neuronal differentiation stage. Semin. Cancer Biol. 2011, 21, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Tomolonis, J.A.; Agarwal, S.; Shohet, J.M. Neuroblastoma pathogenesis: Deregulation of embryonic neural crest development. Cell Tissue Res. 2018, 372, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, S.; Valli, E.; Erriquez, D.; Perini, G. MYCN-mediated transcriptional repression in neuroblastoma: The other side of the coin. Front. Oncol. 2013, 3, 42. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Westermark, U.K.; Wilhelm, M.; Frenzel, A.; Henriksson, M.A. The MYCN oncogene and differentiation in neuroblastoma. Semin. Cancer Biol. 2011, 21, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.; Ribeiro, D.; Arsenian-Henriksson, M.; Deller, T.; Rohrer, H. Proliferation and Survival of Embryonic Sympathetic Neuroblasts by MYCN and Activated ALK Signaling. J. Neurosci. 2016, 36, 10425–10439. [Google Scholar] [CrossRef] [PubMed]
- Reiff, T.; Huber, L.; Kramer, M.; Delattre, O.; Janoueix-Lerosey, I.; Rohrer, H. Midkine and Alk signaling in sympathetic neuron proliferation and neuroblastoma predisposition. Development 2011, 138, 4699–4708. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Iyer, R.; Varela, C.R.; Light, J.E.; Kolla, V.; Evans, A.E. Trk receptor expression and inhibition in neuroblastomas. Clin. Cancer Res. 2009, 15, 3244–3250. [Google Scholar] [CrossRef] [PubMed]
- Harel, L.; Costa, B.; Fainzilber, M. On the death Trk. Dev. Neurobiol. 2010, 70, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Thiele, C.J.; Li, Z.; McKee, A.E. On Trk—The TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin. Cancer Res. 2009, 15, 5962–5967. [Google Scholar] [CrossRef] [PubMed]
- Casey, M.J.; Stewart, R.A. Zebrafish as a model to study neuroblastoma development. Cell Tissue Res. 2018, 372, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kiyonari, S.; Kadomatsu, K. Neuroblastoma models for insights into tumorigenesis and new therapies. Expert Opin. Drug Discov. 2015, 10, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Edsjo, A.; Holmquist, L.; Pahlman, S. Neuroblastoma as an experimental model for neuronal differentiation and hypoxia-induced tumor cell dedifferentiation. Semin. Cancer Biol. 2007, 17, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic lymphoma kinase: Signalling in development and disease. Biochem. J. 2009, 420, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Megison, M.L.; Gillory, L.A.; Beierle, E.A. Cell survival signaling in neuroblastoma. Anti-Cancer Agents Med. Chem. 2013, 13, 563–575. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Anaplastic lymphoma kinase (ALK): Structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 2013, 68, 68–94. [Google Scholar] [CrossRef] [PubMed]
- Stafman, L.L.; Beierle, E.A. Cell Proliferation in Neuroblastoma. Cancers 2016, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Padovan-Merhar, O.M.; Raman, P.; Ostrovnaya, I.; Kalletla, K.; Rubnitz, K.R.; Sanford, E.M.; Ali, S.M.; Miller, V.A.; Mosse, Y.P.; Granger, M.P.; et al. Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet. 2016, 12, e1006501. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Eppstein, A.C.; Sandoval, J.A.; Klein, P.J.; Woodruff, H.A.; Grosfeld, J.L.; Hickey, R.J.; Malkas, L.H.; Schmidt, C.M. Differential sensitivity of chemoresistant neuroblastoma subtypes to MAPK-targeted treatment correlates with ERK, p53 expression, and signaling response to U0126. J. Pediatr. Surg. 2006, 41, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.S.; Rader, J.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Higashi, M.; Kimura, K.; Wakao, J.; Fumino, S.; Iehara, T.; Hosoi, H.; Sakai, T.; Tajiri, T. MEK inhibitors as a novel therapy for neuroblastoma: Their in vitro effects and predicting their efficacy. J. Pediatr. Surg. 2016, 51, 2074–2079. [Google Scholar] [CrossRef] [PubMed]
- Woodfield, S.E.; Zhang, L.; Scorsone, K.A.; Liu, Y.; Zage, P.E. Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer 2016, 16, 172. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Ruan, Y.; Tippett, T.; Narendran, A. Targeted inhibition of MEK1 by cobimetinib leads to differentiation and apoptosis in neuroblastoma cells. J. Exp. Clin. Cancer Res. CR 2015, 34, 104. [Google Scholar] [CrossRef] [PubMed]
- Umapathy, G.; El Wakil, A.; Witek, B.; Chesler, L.; Danielson, L.; Deng, X.; Gray, N.S.; Johansson, M.; Kvarnbrink, S.; Ruuth, K.; et al. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci. Signal. 2014, 7, ra102. [Google Scholar] [CrossRef] [PubMed]
- Umapathy, G.; Guan, J.; Gustafsson, D.E.; Javanmardi, N.; Cervantes-Madrid, D.; Djos, A.; Martinsson, T.; Palmer, R.H.; Hallberg, B. MEK inhibitor trametinib does not prevent the growth of anaplastic lymphoma kinase (ALK)-addicted neuroblastomas. Sci. Signal. 2017, 10, eaam755. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lv, F.; Xu, G.; Zhang, M.; Wu, Y.; Wu, Z. Phosphoproteomics reveals ALK promote cell progress via RAS/ JNK pathway in neuroblastoma. Oncotarget 2016, 7, 75968–75980. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Fan, Y.H.; Xu, X.; Zhang, H.; Dou, J.; Tang, Y.; Zhong, X.; Rojas, Y.; Yu, Y.; Zhao, Y.; et al. A small-molecule inhibitor of UBE2N induces neuroblastoma cell death via activation of p53 and JNK pathways. Cell Death Dis. 2014, 5, e1079. [Google Scholar] [CrossRef] [PubMed]
- Dedoni, S.; Olianas, M.C.; Onali, P. Interferon-beta counter-regulates its own pro-apoptotic action by activating p38 MAPK signalling in human SH-SY5Y neuroblastoma cells. Apoptosis 2014, 19, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Marengo, B.; De Ciucis, C.G.; Ricciarelli, R.; Furfaro, A.L.; Colla, R.; Canepa, E.; Traverso, N.; Marinari, U.M.; Pronzato, M.A.; Domenicotti, C. p38MAPK inhibition: A new combined approach to reduce neuroblastoma resistance under etoposide treatment. Cell Death Dis. 2013, 4, e589. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Li, Y.; Chi, Y. Role of p38 MAPK activation and mitochondrial cytochrome-c release in allicin-induced apoptosis in SK-N-SH cells. Anti-Cancer Drugs 2016, 27, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.S.; Choi, Y.K.; Song, S.S.; Kim, W.K.; Han, B.H. MKP-1 contributes to oxidative stress-induced apoptosis via inactivation of ERK1/2 in SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2005, 338, 1732–1738. [Google Scholar] [CrossRef] [PubMed]
- Dayem, A.A.; Kim, B.; Gurunathan, S.; Choi, H.Y.; Yang, G.; Saha, S.K.; Han, D.; Han, J.; Kim, K.; Kim, J.H.; et al. Biologically synthesized silver nanoparticles induce neuronal differentiation of SH-SY5Y cells via modulation of reactive oxygen species, phosphatases, and kinase signaling pathways. Biotechnol. J. 2014, 9, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Rossler, O.G.; Henss, I.; Thiel, G. Transcriptional response to muscarinic acetylcholine receptor stimulation: Regulation of Egr-1 biosynthesis by ERK, Elk-1, MKP-1, and calcineurin in carbachol-stimulated human neuroblastoma cells. Arch. Biochem. Biophys. 2008, 470, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Vangipuram, S.D.; Wang, Z.J.; Lyman, W.D. Resistance of stem-like cells from neuroblastoma cell lines to commonly used chemotherapeutic agents. Pediatr. Blood Cancer 2010, 54, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Reffas, S.; Schlegel, W. Compartment-specific regulation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinases (MAPKs) by ERK-dependent and non-ERK-dependent inductions of MAPK phosphatase (MKP)-3 and MKP-1 in differentiating P19 cells. Biochem. J. 2000, 352 Pt 3, 701–708. [Google Scholar]
- Ryser, S.; Tortola, S.; Schlegel, W. Map kinase phosphatase-1 gene expression and regulation in neuroendocrine cells. J. Recept. Signal Transduct. Res. 2002, 22, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Ryser, S.; Tortola, S.; van Haasteren, G.; Muda, M.; Li, S.; Schlegel, W. MAP kinase phosphatase-1 gene transcription in rat neuroendocrine cells is modulated by a calcium-sensitive block to elongation in the first exon. J. Biol. Chem. 2001, 276, 33319–33327. [Google Scholar] [CrossRef] [PubMed]
- Aurtenetxe, O.; Zaldumbide, L.; Erramuzpe, A.; Lopez, R.; Lopez, J.I.; Cortes, J.M.; Pulido, R.; Nunes-Xavier, C.E. DUSP5 expression associates with poor prognosis in human neuroblastoma. Exp. Mol. Pathol. 2018, 105, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Kojima, S.; Kishimoto, T.; Kuwabara, S.; Yamaguchi, A. Over-expression of map kinase phosphatase-1 (MKP-1) suppresses neuronal death through regulating JNK signaling in hypoxia/re-oxygenation. Brain Res. 2012, 1436, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Han, Y.M.; Oh, M.; Kim, W.K.; Oh, K.J.; Lee, S.C.; Bae, K.H.; Han, B.S. DUSP4 regulates neuronal differentiation and calcium homeostasis by modulating ERK1/2 phosphorylation. Stem Cells Dev. 2015, 24, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, I.; Kumps, C.; Claeys, S.; Lindner, S.; Beckers, A.; Janssens, E.; Carter, D.R.; Cazes, A.; Cheung, B.B.; De Mariano, M.; et al. Upregulation of MAPK Negative Feedback Regulators and RET in Mutant ALK Neuroblastoma: Implications for Targeted Treatment. Clin. Cancer Res. 2015, 21, 3327–3339. [Google Scholar] [CrossRef] [PubMed]
- Staege, M.S.; Muller, K.; Kewitz, S.; Volkmer, I.; Mauz-Korholz, C.; Bernig, T.; Korholz, D. Expression of dual-specificity phosphatase 5 pseudogene 1 (DUSP5P1) in tumor cells. PLoS ONE 2014, 9, e89577. [Google Scholar] [CrossRef] [PubMed]
- Mourey, R.J.; Vega, Q.C.; Campbell, J.S.; Wenderoth, M.P.; Hauschka, S.D.; Krebs, E.G.; Dixon, J.E. A novel cytoplasmic dual specificity protein tyrosine phosphatase implicated in muscle and neuronal differentiation. J. Biol. Chem. 1996, 271, 3795–3802. [Google Scholar] [CrossRef] [PubMed]
- Muda, M.; Boschert, U.; Dickinson, R.; Martinou, J.C.; Martinou, I.; Camps, M.; Schlegel, W.; Arkinstall, S. MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J. Biol. Chem. 1996, 271, 4319–4326. [Google Scholar] [CrossRef] [PubMed]
- Vician, L.; Basconcillo, R.; Herschman, H.R. Identification of genes preferentially induced by nerve growth factor versus epidermal growth factor in PC12 pheochromocytoma cells by means of representational difference analysis. J. Neurosci. Res. 1997, 50, 32–43. [Google Scholar] [CrossRef]
- Liu, T.; Bohlken, A.; Kuljaca, S.; Lee, M.; Nguyen, T.; Smith, S.; Cheung, B.; Norris, M.D.; Haber, M.; Holloway, A.J.; et al. The retinoid anticancer signal: Mechanisms of target gene regulation. Br. J. Cancer 2005, 93, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Mendell, A.L.; MacLusky, N.J. The testosterone metabolite 3alpha-androstanediol inhibits oxidative stress-induced ERK phosphorylation and neurotoxicity in SH-SY5Y cells through an MKP3/DUSP6-dependent mechanism. Neurosci. Lett. 2018, 696, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Marshall, G.M.; Liu, P.Y.; Gherardi, S.; Scarlett, C.J.; Bedalov, A.; Xu, N.; Iraci, N.; Valli, E.; Ling, D.; Thomas, W.; et al. SIRT1 promotes N-Myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N-Myc protein stability. PLoS Genet. 2011, 7, e1002135. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Oh, M.; Lee, K.S.; Kim, W.K.; Oh, K.J.; Lee, S.C.; Bae, K.H.; Han, B.S. Profiling analysis of protein tyrosine phosphatases during neuronal differentiation. Neurosci. Lett. 2016, 612, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Zega, K.; Jovanovic, V.M.; Vitic, Z.; Niedzielska, M.; Knaapi, L.; Jukic, M.M.; Partanen, J.; Friedel, R.H.; Lang, R.; Brodski, C. Dusp16 Deficiency Causes Congenital Obstructive Hydrocephalus and Brain Overgrowth by Expansion of the Neural Progenitor Pool. Front. Mol. Neurosci. 2017, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Park, B.C.; Kim, H.A.; Song, M.; Park, S.G.; Lee, D.H.; Kim, H.J.; Choi, H.K.; Kim, J.T.; Cho, S. Positive regulation of apoptosis signal-regulating kinase 1 by dual-specificity phosphatase 13A. Cell. Mol. Life Sci. CMLS 2010, 67, 2619–2629. [Google Scholar] [CrossRef] [PubMed]
- Caren, H.; Djos, A.; Nethander, M.; Sjoberg, R.M.; Kogner, P.; Enstrom, C.; Nilsson, S.; Martinsson, T. Identification of epigenetically regulated genes that predict patient outcome in neuroblastoma. BMC Cancer 2011, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Lin, C.H.; Yang, C.H.; Tan, T.H.; Chen, Y.R. Biochemical and biological characterization of a neuroendocrine-associated phosphatase. J. Neurochem. 2006, 98, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Yang, C.H.; Yeh, C.L.; Lin, C.H.; Chen, Y.R. NEAP causes down-regulation of EGFR, subsequently induces the suppression of NGF-induced differentiation in PC12 cells. J. Neurochem. 2008, 107, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.I.; Brummer, T.; Daly, R.J.; O’Brien, P.M. DUSP26 negatively affects the proliferation of epithelial cells, an effect not mediated by dephosphorylation of MAPKs. Biochim. Biophys. Acta 2010, 1803, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Vasudevan, S.A.; Yu, Y.; Ge, N.; Ludwig, A.D.; Wesson, C.L.; Wang, K.; Burlingame, S.M.; Zhao, Y.J.; Rao, P.H.; et al. Dual-specificity phosphatase 26 is a novel p53 phosphatase and inhibits p53 tumor suppressor functions in human neuroblastoma. Oncogene 2010, 29, 4938–4946. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ma, I.T.; Patel, R.H.; Shang, X.; Chen, Z.; Zhao, Y.; Cheng, J.; Fan, Y.; Rojas, Y.; Barbieri, E.; et al. NSC-87877 inhibits DUSP26 function in neuroblastoma resulting in p53-mediated apoptosis. Cell Death Dis. 2015, 6, e1841. [Google Scholar] [CrossRef] [PubMed]
- Dedoni, S.; Olianas, M.C.; Onali, P. Interferon-beta induces apoptosis in human SH-SY5Y neuroblastoma cells through activation of JAK-STAT signaling and down-regulation of PI3K/Akt pathway. J. Neurochem. 2010, 115, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- King, D.; Yeomanson, D.; Bryant, H.E. PI3King the lock: Targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J. Pediatr. Hematol./Oncol. 2015, 37, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Sattu, K.; Hochgrafe, F.; Wu, J.; Umapathy, G.; Schonherr, C.; Ruuth, K.; Chand, D.; Witek, B.; Fuchs, J.; Li, P.K.; et al. Phosphoproteomic analysis of anaplastic lymphoma kinase (ALK) downstream signaling pathways identifies signal transducer and activator of transcription 3 as a functional target of activated ALK in neuroblastoma cells. FEBS J. 2013, 280, 5269–5282. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Li, Z.; Thiele, C.J. Inhibition of STAT3 with orally active JAK inhibitor, AZD1480, decreases tumor growth in Neuroblastoma and Pediatric Sarcomas In vitro and In vivo. Oncotarget 2013, 4, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Nafady, A.; Takatori, A.; Kishida, S.; Ohira, M.; Suenaga, Y.; Hossain, S.; Akter, J.; Ogura, A.; Nakamura, Y.; et al. ALK is a MYCN target gene and regulates cell migration and invasion in neuroblastoma. Sci. Rep. 2013, 3, 3450. [Google Scholar] [CrossRef] [PubMed]
- Schonherr, C.; Ruuth, K.; Kamaraj, S.; Wang, C.L.; Yang, H.L.; Combaret, V.; Djos, A.; Martinsson, T.; Christensen, J.G.; Palmer, R.H.; et al. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene 2012, 31, 5193–5200. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Pulido, R. The extended human PTPome: A growing tyrosine phosphatase family. FEBS J. 2016, 283, 1404–1429. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R.; Hooft van Huijsduijnen, R. Protein tyrosine phosphatases: Dual-specificity phosphatases in health and disease. FEBS J. 2008, 275, 848–866. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R.; Stoker, A.W.; Hendriks, W.J. PTPs emerge as PIPs: Protein tyrosine phosphatases with lipid-phosphatase activities in human disease. Hum. Mol. Genet. 2013, 22, R66–R76. [Google Scholar] [CrossRef] [PubMed]
- Rios, P.; Nunes-Xavier, C.E.; Tabernero, L.; Kohn, M.; Pulido, R. Dual-specificity phosphatases as molecular targets for inhibition in human disease. Antioxid. Redox Signal. 2014, 20, 2251–2273. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Tan, T.H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Xavier, C.; Roma-Mateo, C.; Rios, P.; Tarrega, C.; Cejudo-Marin, R.; Tabernero, L.; Pulido, R. Dual-specificity MAP kinase phosphatases as targets of cancer treatment. Anti-Cancer Agents Med. Chem. 2011, 11, 109–132. [Google Scholar] [CrossRef]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Cejudo-Marín, R.; Tárrega, C.; Nunes-Xavier, C.E.; Pulido, R. Caspase-3 cleavage of DUSP6/MKP3 at the interdomain region generates active MKP3 fragments that regulate ERK1/2 subcellular localization and function. J. Mol. Biol. 2012, 420, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Kidger, A.M.; Rushworth, L.K.; Stellzig, J.; Davidson, J.; Bryant, C.J.; Bayley, C.; Caddye, E.; Rogers, T.; Keyse, S.M.; Caunt, C.J. Dual-specificity phosphatase 5 controls the localized inhibition, propagation, and transforming potential of ERK signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E317–E326. [Google Scholar] [CrossRef] [PubMed]
- Tarrega, C.; Rios, P.; Cejudo-Marin, R.; Blanco-Aparicio, C.; van den Berk, L.; Schepens, J.; Hendriks, W.; Tabernero, L.; Pulido, R. ERK2 shows a restrictive and locally selective mechanism of recognition by its tyrosine phosphatase inactivators not shared by its activator MEK1. J. Biol. Chem. 2005, 280, 37885–37894. [Google Scholar] [CrossRef] [PubMed]
- Kidger, A.M.; Keyse, S.M. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 2016, 50, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Low, H.B.; Zhang, Y. Regulatory Roles of MAPK Phosphatases in Cancer. Immune Netw. 2016, 16, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Seternes, O.M.; Kidger, A.M.; Keyse, S.M. Dual-specificity MAP kinase phosphatases in health and disease. Biochim. Biophys. Acta. Mol. Cell Res. 2019, 1866, 124–143. [Google Scholar] [CrossRef] [PubMed]
- Pavic, K.; Duan, G.; Kohn, M. VHR/DUSP3 phosphatase: Structure, function and regulation. FEBS J. 2015, 282, 1871–1890. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Nunes-Xavier, C.E.; Bayon, Y.; Pulido, R. The Extended Family of Protein Tyrosine Phosphatases. Methods Mol. Biol. 2016, 1447, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.I.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.J.; Keyse, S.M. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J. Cell Sci. 2006, 119, 4607–4615. [Google Scholar] [CrossRef] [PubMed]
- Kutty, R.G.; Talipov, M.R.; Bongard, R.D.; Lipinski, R.A.J.; Sweeney, N.L.; Sem, D.S.; Rathore, R.; Ramchandran, R. Dual Specificity Phosphatase 5-Substrate Interaction: A Mechanistic Perspective. Compr. Physiol. 2017, 7, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Shi, H.; Gatzke, F.; Bennett, A.M. Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cell. Mol. Life Sci. CMLS 2013, 70, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Torrance, E.; Al-Harthi, S.; Shweash, M.; Alnasser, S.; Neamatallah, T.; Schroeder, J.; Plevin, R. MKP-2: Out of the DUSP-bin and back into the limelight. Biochem. Soc. Trans. 2012, 40, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.M.; Downer, E.J.; Toulouse, A.; Nolan, Y.M. Mitogen-Activated Protein Kinase Phosphatase (MKP)-1 in Nervous System Development and Disease. Mol. Neurobiol. 2015, 51, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Yang, L.C.; Guo, K.; Wang, Y.P.; Li, Y.G. Mitogen-activated protein kinase phosphatase-1: A critical phosphatase manipulating mitogen-activated protein kinase signaling in cardiovascular disease (review). Int. J. Mol. Med. 2015, 35, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, S.M.; Prabhala, P.; Ammit, A.J. Role and regulation of MKP-1 in airway inflammation. Respir. Res. 2017, 18, 154. [Google Scholar] [CrossRef] [PubMed]
- Ralph, J.A.; Morand, E.F. MAPK phosphatases as novel targets for rheumatoid arthritis. Expert Opin. Ther. Targets 2008, 12, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, Y.; Yu, H.; Shen, B.; Liang, Y.; Jin, R.; Liu, X.; Shi, L.; Cai, X. Role of DUSP1/MKP1 in tumorigenesis, tumor progression and therapy. Cancer Med. 2016, 5, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Wancket, L.M.; Frazier, W.J.; Liu, Y. Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology, physiology, and disease. Life Sci. 2012, 90, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Abdal Dayem, A.; Lee, S.B.; Choi, H.Y.; Cho, S.G. Silver Nanoparticles: Two-Faced Neuronal Differentiation-Inducing Material in Neuroblastoma (SH-SY5Y) Cells. Int. J. Mol. Sci. 2018, 19, 1470. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Berger, A.H.; Diolombi, M.S.; Ng, C.; Fung, J.; Bronson, R.T.; Castillo-Martin, M.; Thin, T.H.; Cordon-Cardo, C.; et al. Compound haploinsufficiency of Dok2 and Dusp4 promotes lung tumorigenesis. J. Clin. Investig. 2018, 129, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Hijiya, N.; Tsukamoto, Y.; Nakada, C.; Tung Nguyen, L.; Kai, T.; Matsuura, K.; Shibata, K.; Inomata, M.; Uchida, T.; Tokunaga, A.; et al. Genomic Loss of DUSP4 Contributes to the Progression of Intraepithelial Neoplasm of Pancreas to Invasive Carcinoma. Cancer Res. 2016, 76, 2612–2625. [Google Scholar] [CrossRef] [PubMed]
- Ichimanda, M.; Hijiya, N.; Tsukamoto, Y.; Uchida, T.; Nakada, C.; Akagi, T.; Etoh, T.; Iha, H.; Inomata, M.; Takekawa, M.; et al. Downregulation of dual-specificity phosphatase 4 enhances cell proliferation and invasiveness in colorectal carcinomas. Cancer Sci. 2018, 109, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Li, M.; Zhu, H.; Lu, X.; Miao, J.; Du, S.; Xia, X.; Guan, W. DUSP4 promotes doxorubicin resistance in gastric cancer through epithelial-mesenchymal transition. Oncotarget 2017, 8, 94028–94039. [Google Scholar] [CrossRef] [PubMed]
- Menyhart, O.; Budczies, J.; Munkacsy, G.; Esteva, F.J.; Szabo, A.; Miquel, T.P.; Gyorffy, B. DUSP4 is associated with increased resistance against anti-HER2 therapy in breast cancer. Oncotarget 2017, 8, 77207–77218. [Google Scholar] [CrossRef] [PubMed]
- Buffet, C.; Hecale-Perlemoine, K.; Bricaire, L.; Dumont, F.; Baudry, C.; Tissier, F.; Bertherat, J.; Cochand-Priollet, B.; Raffin-Sanson, M.L.; Cormier, F.; et al. DUSP5 and DUSP6, two ERK specific phosphatases, are markers of a higher MAPK signaling activation in BRAF mutated thyroid cancers. PLoS ONE 2017, 12, e0184861. [Google Scholar] [CrossRef] [PubMed]
- Higa, T.; Takahashi, H.; Higa-Nakamine, S.; Suzuki, M.; Yamamoto, H. Up-regulation of DUSP5 and DUSP6 by gonadotropin-releasing hormone in cultured hypothalamic neurons, GT1-7 cells. Biomed. Res. 2018, 39, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Kucharska, A.; Rushworth, L.K.; Staples, C.; Morrice, N.A.; Keyse, S.M. Regulation of the inducible nuclear dual-specificity phosphatase DUSP5 by ERK MAPK. Cell. Signal. 2009, 21, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Xavier, C.E.; Tárrega, C.; Cejudo-Marín, R.; Frijhoff, J.; Sandin, A.; Ostman, A.; Pulido, R. Differential up-regulation of MAP kinase phosphatases MKP3/DUSP6 and DUSP5 by Ets2 and c-Jun converge in the control of the growth arrest versus proliferation response of MCF-7 breast cancer cells to phorbol ester. J. Biol. Chem. 2010, 285, 26417–26430. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.Y.; Yin, J.Y.; Tang, Q.; Zhai, L.L.; Zhang, T.J.; Wang, Y.X.; Yang, D.Q.; Qian, J.; Lin, J.; Deng, Z.Q. High expression of dual-specificity phosphatase 5 pseudogene 1 (DUSP5P1) is associated with poor prognosis in acute myeloid leukemia. Int. J. Clin. Exp. Pathol. 2015, 8, 16073–16080. [Google Scholar] [PubMed]
- Ekerot, M.; Stavridis, M.P.; Delavaine, L.; Mitchell, M.P.; Staples, C.; Owens, D.M.; Keenan, I.D.; Dickinson, R.J.; Storey, K.G.; Keyse, S.M. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem. J. 2008, 412, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Scott, D.A.; Hatch, E.; Tian, X.; Mansour, S.L. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development 2007, 134, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.K.; Abdollah, N.A.; Shafie, N.H.; Yusof, N.M.; Razak, S.R.A. Dual-specificity phosphatase 6 (DUSP6): A review of its molecular characteristics and clinical relevance in cancer. Cancer Biol. Med. 2018, 15, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.F.; Ferruzo, P.Y.M.; Russo, L.C.; Farias, J.O.; Forti, F.L. DUSP3/VHR: A Druggable Dual Phosphatase for Human Diseases. Rev. Physiol. Biochem. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Chen, H.H.; Luche, R.; Wei, B.; Tonks, N.K. Characterization of two distinct dual specificity phosphatases encoded in alternative open reading frames of a single gene located on human chromosome 10q22.2. J. Biol. Chem. 2004, 279, 41404–41413. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, K.; Satoh, T.; Tanuma, N.; Masuda, K.; Takekawa, M.; Shima, H.; Kikuchi, K. Characterization of a novel low-molecular-mass dual-specificity phosphatase-3 (LDP-3) that enhances activation of JNK and p38. Biochem. J. 2004, 383, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, Y.; Gu, S.; Li, N.; Zheng, D.; Li, D.; Zheng, Z.; Ji, C.; Xie, Y.; Mao, Y. Molecular cloning and characterization of a novel dual-specificity phosphatase 23 gene from human fetal brain. Int. J. Biochem. Cell Biol. 2004, 36, 1542–1553. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, L.L.; Ng, M.R.; Sowa, M.E.; Selfors, L.M.; White, A.; Zervantonakis, I.K.; Singh, P.; Dhakal, S.; Harper, J.W.; Brugge, J.S. A protein interaction map for cell-cell adhesion regulators identifies DUSP23 as a novel phosphatase for beta-catenin. Sci. Rep. 2016, 6, 27114. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.Y.; Chang, C.W.; Cheong, M.L.; Chen, H.C.; Lee, D.Y.; Chang, G.D.; Chen, H. Dual-specificity phosphatase 23 mediates GCM1 dephosphorylation and activation. Nucleic Acids Res. 2011, 39, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.A.; Skoko, J.; Wang, K.; Burlingame, S.M.; Patel, P.N.; Lazo, J.S.; Nuchtern, J.G.; Yang, J. MKP-8, a novel MAPK phosphatase that inhibits p38 kinase. Biochem. Biophys. Res. Commun. 2005, 330, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Yeh, Y.J.; Wang, J.Y.; Liu, Y.W.; Chen, Y.L.; Cheng, H.W.; Cheng, C.M.; Chuang, Y.J.; Yuh, C.H.; Chen, Y.R. NEAP/DUSP26 suppresses receptor tyrosine kinases and regulates neuronal development in zebrafish. Sci. Rep. 2017, 7, 5241. [Google Scholar] [CrossRef] [PubMed]
- Tanuma, N.; Nomura, M.; Ikeda, M.; Kasugai, I.; Tsubaki, Y.; Takagaki, K.; Kawamura, T.; Yamashita, Y.; Sato, I.; Sato, M.; et al. Protein phosphatase Dusp26 associates with KIF3 motor and promotes N-cadherin-mediated cell-cell adhesion. Oncogene 2009, 28, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Lazo, J.S.; McQueeney, K.E.; Burnett, J.C.; Wipf, P.; Sharlow, E.R. Small molecule targeting of PTPs in cancer. Int. J. Biochem. Cell Biol. 2018, 96, 171–181. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
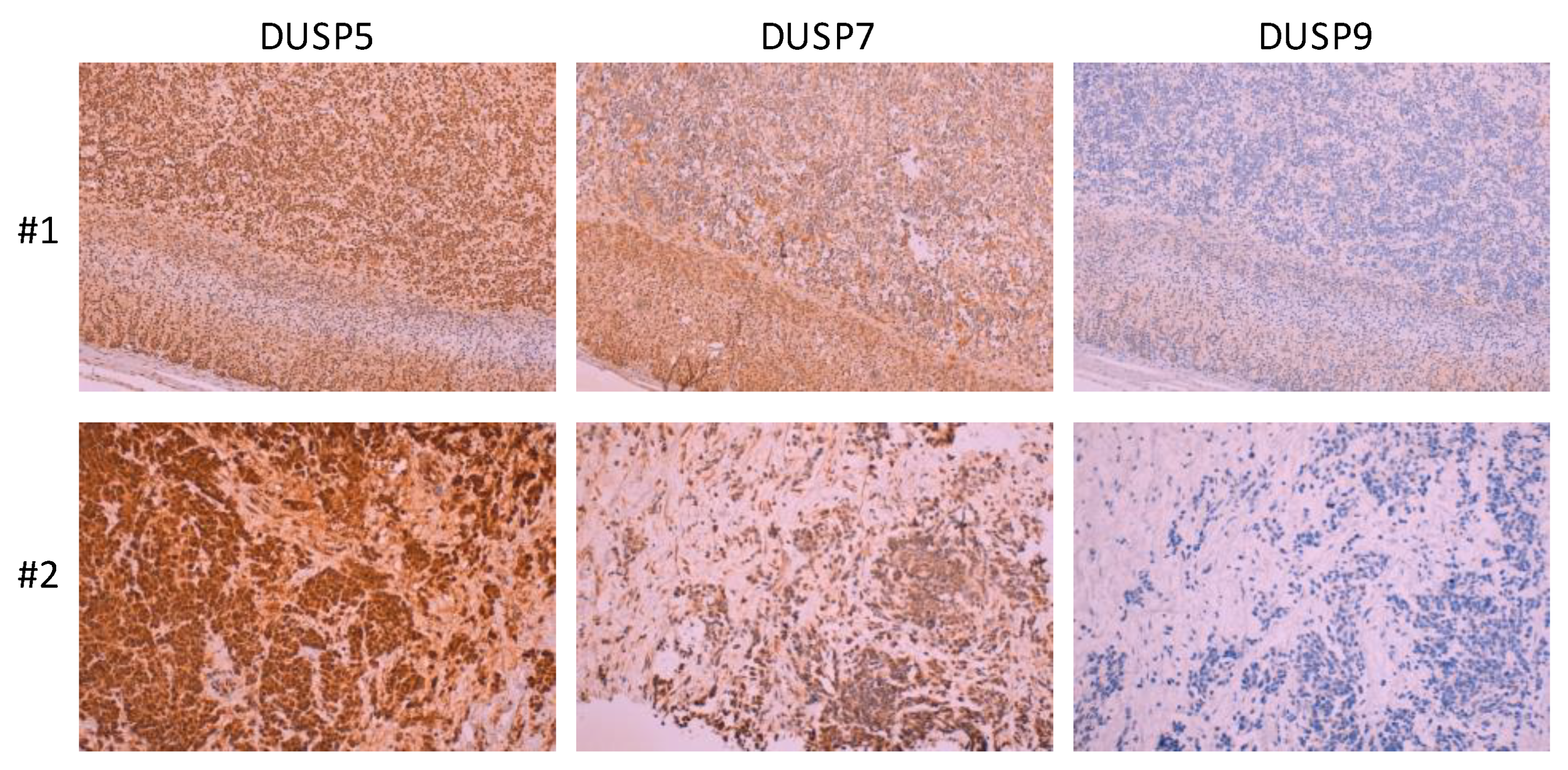{kind=link}
| Gene/Protein | MAPK Substrates 1 Localization | Alterations in NB Cell Lines and NB Tumors |
|---|---|---|
| DUSP1/MKP1 | JNK, p38 > ERK Nuclear | -SH-SY5Y cells: DUSP1 KD: ↓ H2O2-induced apoptosis, ↑ pERK1/2 [60]; ↓ DUSP1 mRNA upon AgNP treatment [61]; ↑ DUSP1 upon carbachol stimulation [62]. -SK-N-SH and SK-N-BE cells: ↓ DUSP1 protein in CD133+ cells resistant to chemotherapy [63]. -P19 cells: ↑ DUSP1 protein upon RA differentiation [64]. -GH4C1 neuroendocrine cells: ↑ DUSP1 upon TRH and EGF stimulation [65,66]. -SMS-KCNR cells: ↑ DUSP1 mRNA upon RA-induced differentiation [67]. -N1E-115 cells: ↑ DUSP1 upon hypoxia/re-oxygenation; DUSP1 KD: ↑ pJNK, ↑neuronal death; DUSP1 OE: ↓ pJNK, ↓ neuronal death [68]. |
| DUSP4/MKP2 | ERK, JNK > p38 Nuclear | -SH-SY5Y cells: ↓ DUSP4 mRNA upon AgNP treatment [61]. -Mouse ESCs: DUSP4 KD: ↓ neuronal differentiation, ↑ pERK1/2; ↑ DUSP4 in RA-induced differentiation [69]. -SH-SY5Y, KCNR cells: ↓ DUSP4 upon RA-induced differentiation [67]. -SK-N-AS cells: ↓ DUSP4 mRNA upon ALK inhibition, ↑ DUSP4 mRNA upon mutant ALK OE [70]. |
| DUSP5 | ERK Nuclear | -SiMa, Kelly, SH-SY5Y, CHP-134 cells: ↑ DUSP5P1/DUSP5 ratio compared to normal cells [71]. -SH-SY5Y, SMS-KCNR, IMR-32 cells: ↑ DUSP5 mRNA upon RA-induced neuronal differentiation; SH-SY5Y cells: ↑ DUSP5 mRNA upon EGF and PMA stimulation; DUSP5 KD:↑ proliferation [67]. -SK-N-AS cells: ↓ DUSP5 mRNA upon ALK inhibition, ↑ DUSP5 mRNA upon mutant ALK OE [70]. -NB tumors: DUSP5 protein expression associated with poor prognosis and pERK1/2 expression [67]. |
| DUSP6/MKP3 | ERK Cytoplasmic | -PC12 cells: ↑ DUSP6 mRNA in FGF and NGF-mediated neuronal differentiation [72,73,74]. -P19 cells: ↑ DUSP6 protein upon RA differentiation, dependent on ERK1/2 activation [64]. -SH-SY5Y, BE(2)-C cells: ↑ DUSP6 mRNA upon RA-induced differentiation; DUSP6 KD + RGS16 KD in SH-SY5Y cells: ↓ RA-mediated proliferation arrest [75]. -IMR-32 cells: ↑DUSP6 mRNA upon RA-induced differentiation [67]. -SH-SY5Y cells: ↓ DUSP6 protein upon H2O2 stimulation [76]. -SK-N-AS cells: ↓ DUSP6 mRNA upon ALK inhibition, ↑ DUSP6 mRNA upon mutant ALK OE [70]. -BE(2)-C, LAN-1 cells: ↓ DUSP6 by a MYCN-SIRT1 complex [77]. |
| DUSP7/MKPX | ERK Cytoplasmic | -NB tumors: Positive immunostaining, no clinical associations [67]. |
| DUSP8 | JNK, p38 Cytoplasmic/nuclear | -Mouse J1 ESCs: ↑ DUSP8 in RA-induced neuronal differentiation [78]. |
| DUSP9/MKP4 | ERK > p38 Cytoplasmic | -Mouse J1 ESCs:↓ DUSP9 in RA-induced neuronal differentiation [78]. -SH-SY5Y, SMS-KCNR cells: ↓ DUSP9 mRNA upon RA-induced differentiation [67]. -NB tumors: Mostly negative immunostaining, no clinical associations [67]. |
| DUSP10/MKP5 | JNK, p38 Cytoplasmic/nuclear | -Mouse J1 ESCs: ↑ DUSP10 in RA-induced neuronal differentiation [78]. |
| DUSP16/MKP7 | JNK, p38 Cytoplasmic/nuclear | -DUSP16 -/- mice: hydrocephalus and brain overgrowth [79]. -SMS-KCNR, IMR-32 cells: ↑ DUSP16 mRNA upon RA-induced differentiation [67]. |
| DUSP3/VHR | (ERK, JNK) Cytoplasmic/nuclear | -SH-SY5Y cells: ↓ DUSP3 mRNA upon AgNP treatment [61]. |
| DUSP13A/MDSP | Cytoplasmic | -SK-N-SH cells: physical association with pro-apoptotic ASK1, independent of phosphatase activity; DUSP13A KD: ↓ ASK1 kinase activity [80]. |
| DUSP23/VHZ | (ERK, JNK) Cytoplasmic/nuclear | -Mouse J1 ESCs: ↑ DUSP23 in RA-induced neuronal differentiation; DUSP23 KD:↓ neuronal differentiation, ↑ pp38, ↓ pERK1/2, ↓ pJNK [78]. -NB tumors: ↑ Methylation in MYCN-amplified tumors, ↓ DUSP23 mRNA in patients with poor outcome [81]. |
| DUSP26/MKP8 | (p38) Nuclear | -Mouse J1 ESCs: ↑ DUSP26 in RA-induced neuronal differentiation [78]. -PC12 cells: ↑ DUSP26 mRNA in NGF-induced differentiation; DUSP26 KD: ↑ EGFR, ↑ NGF-induced differentiation; DUSP26 OE: ↓ EGF-induced cell growth, ↓ NGF-induced differentiation, ↑ cisplatin-induced apoptosis, ↓ pAKT, ↓ EGFR [82,83]. -Human NB cell lines: ↓ DUSP26 mRNA compared to normal adrenal gland [84]. -IMR-32 cells: DUSP26 KD: ↑ doxorubicin-induced apoptosis, ↓ proliferation, ↑ pp53, ↑ pp38; SH-SY5Y cells: DUSP26 KD: ↓proliferation; SK-N-SH cells: DUSP26 OE: ↓ doxorubicin-induced apoptosis, ↓ pp53 [85,86]. -NB tumors: ↑ DUSP26 protein in high-risk NB tumors [85]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunes-Xavier, C.E.; Zaldumbide, L.; Aurtenetxe, O.; López-Almaraz, R.; López, J.I.; Pulido, R. Dual-Specificity Phosphatases in Neuroblastoma Cell Growth and Differentiation. Int. J. Mol. Sci. 2019, 20, 1170. https://doi.org/10.3390/ijms20051170
Nunes-Xavier CE, Zaldumbide L, Aurtenetxe O, López-Almaraz R, López JI, Pulido R. Dual-Specificity Phosphatases in Neuroblastoma Cell Growth and Differentiation. International Journal of Molecular Sciences. 2019; 20(5):1170. https://doi.org/10.3390/ijms20051170
Chicago/Turabian StyleNunes-Xavier, Caroline E., Laura Zaldumbide, Olaia Aurtenetxe, Ricardo López-Almaraz, José I. López, and Rafael Pulido. 2019. "Dual-Specificity Phosphatases in Neuroblastoma Cell Growth and Differentiation" International Journal of Molecular Sciences 20, no. 5: 1170. https://doi.org/10.3390/ijms20051170
APA StyleNunes-Xavier, C. E., Zaldumbide, L., Aurtenetxe, O., López-Almaraz, R., López, J. I., & Pulido, R. (2019). Dual-Specificity Phosphatases in Neuroblastoma Cell Growth and Differentiation. International Journal of Molecular Sciences, 20(5), 1170. https://doi.org/10.3390/ijms20051170