Bacillus anthracis Edema Toxin Inhibits Efferocytosis in Human Macrophages and Alters Efferocytic Receptor Signaling

 , and
, and
Abstract
:1. Introduction
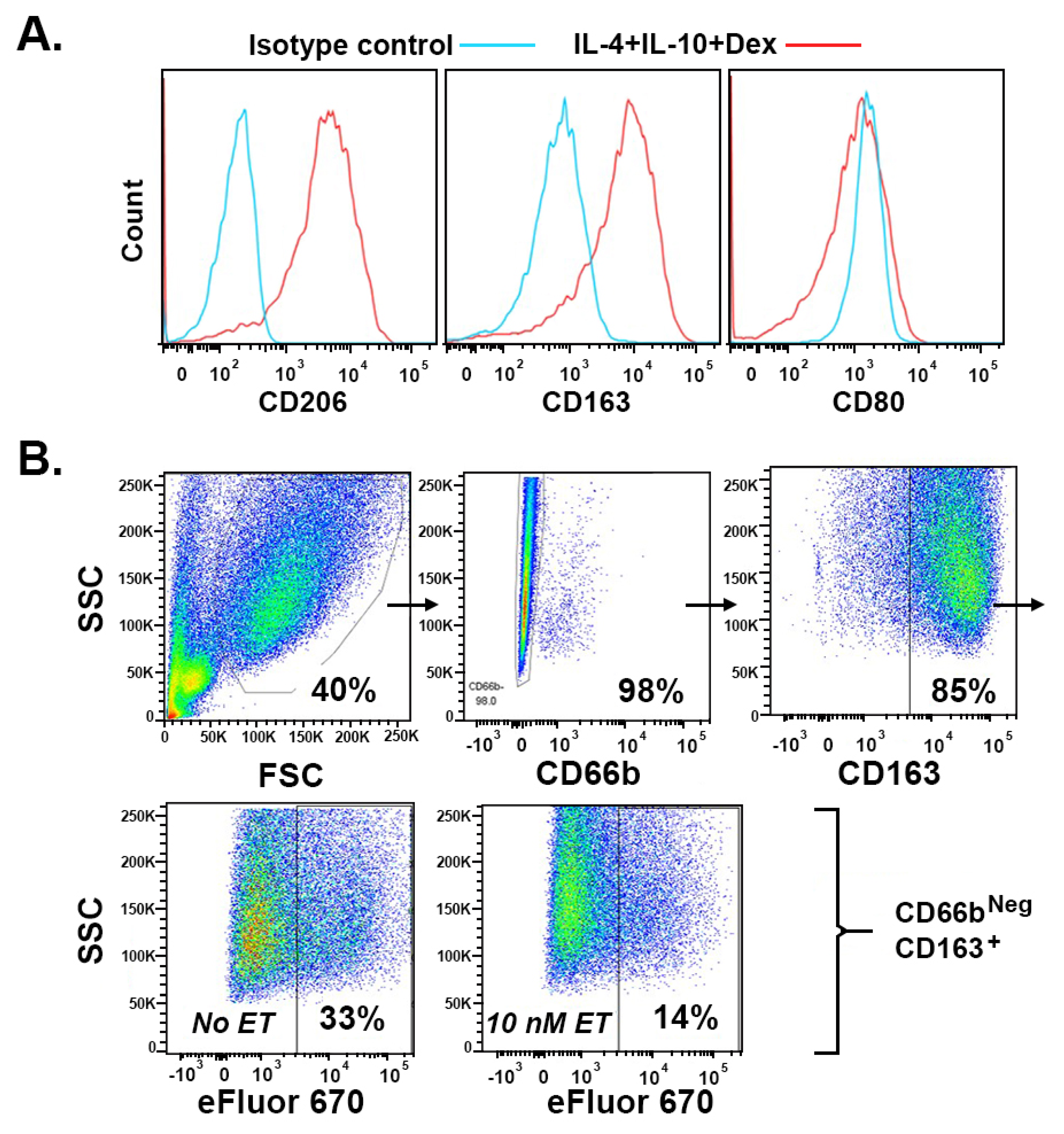
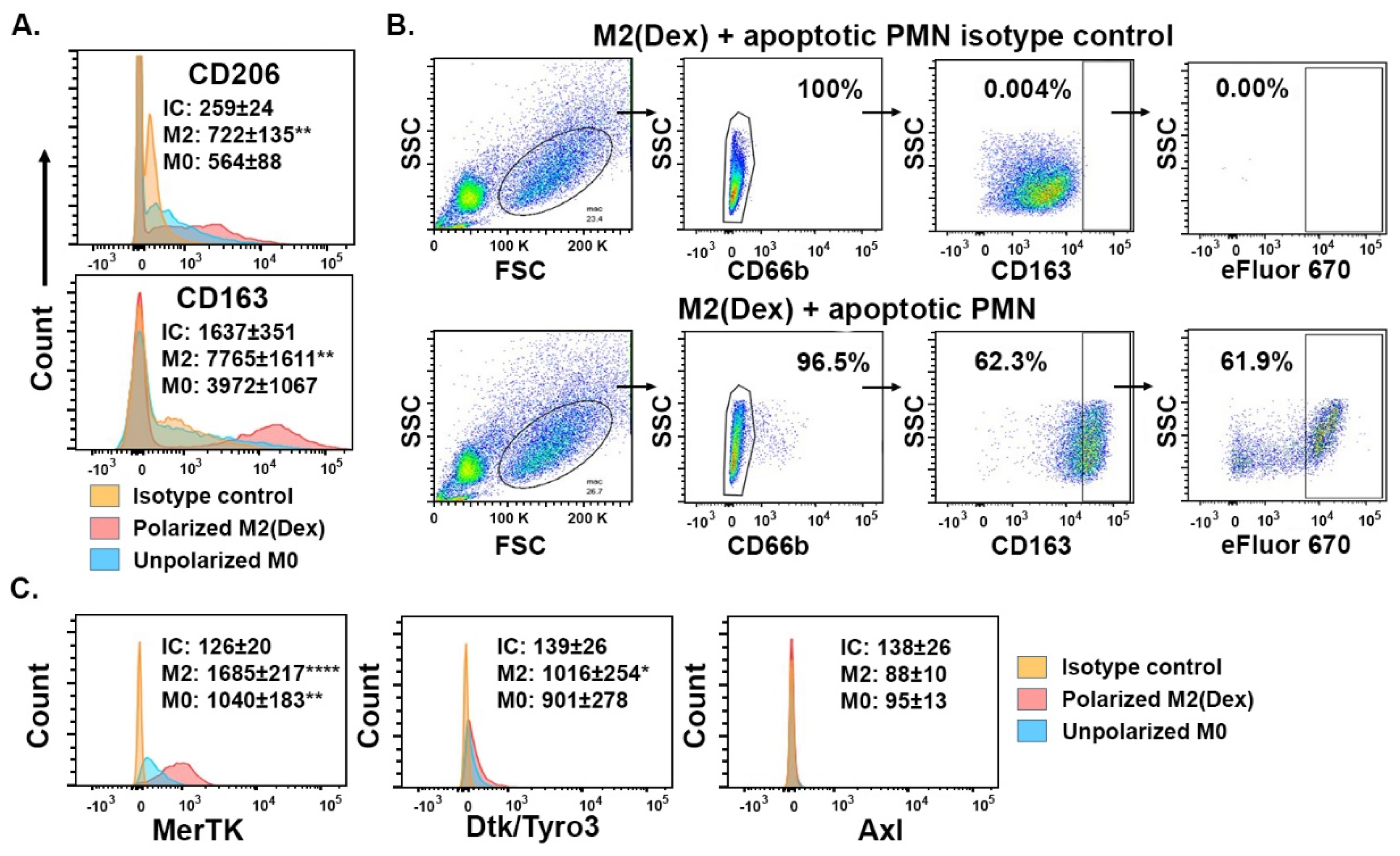
2. Results
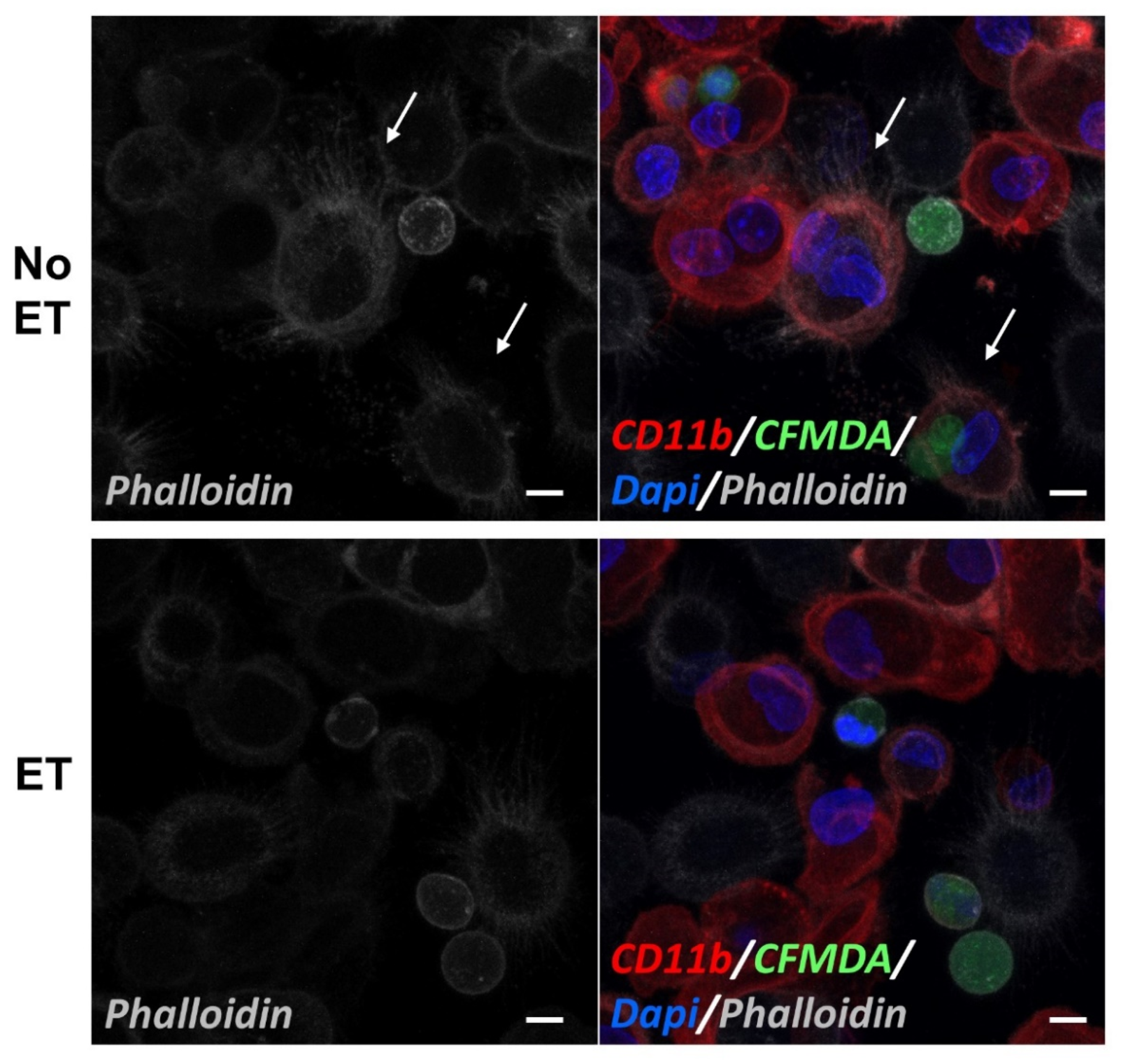
2.1. Bacillus anthracis Edema Toxin (ET) Inhibits Efferocytosis in a Dose-Dependent Manner
2.2. Efferocytosis by Monocyte-Derived Macrophages Polarized by Dexamethasone (Dex) Depends on Protein S in the Absence of Serum
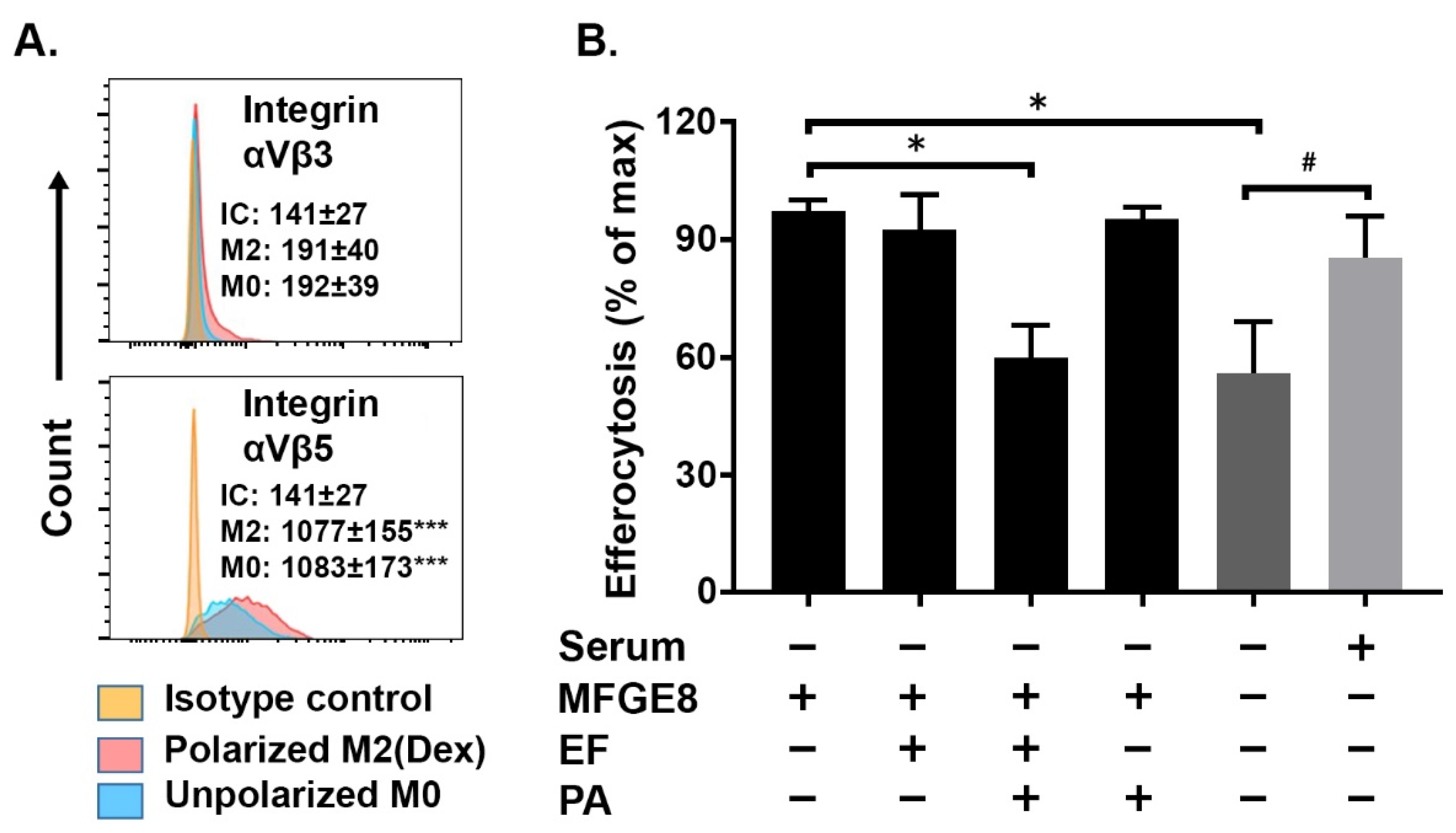
2.3. MFGE8-Enhanced Efferocytosis by Human M2(Dex) Macrophages Is Associated with Integrin αVβ5 Expression and Inhibited by Edema Toxin (ET)
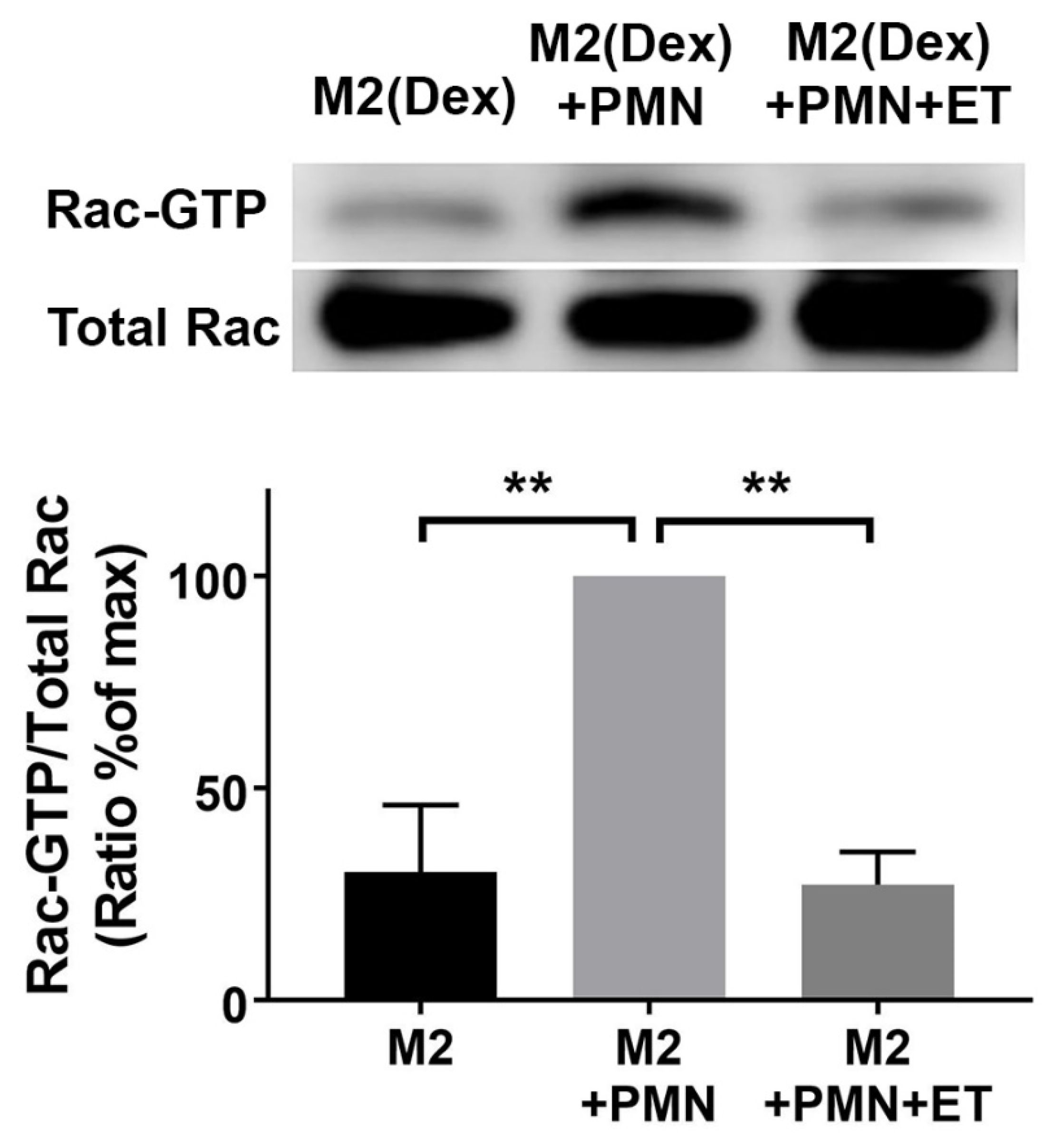
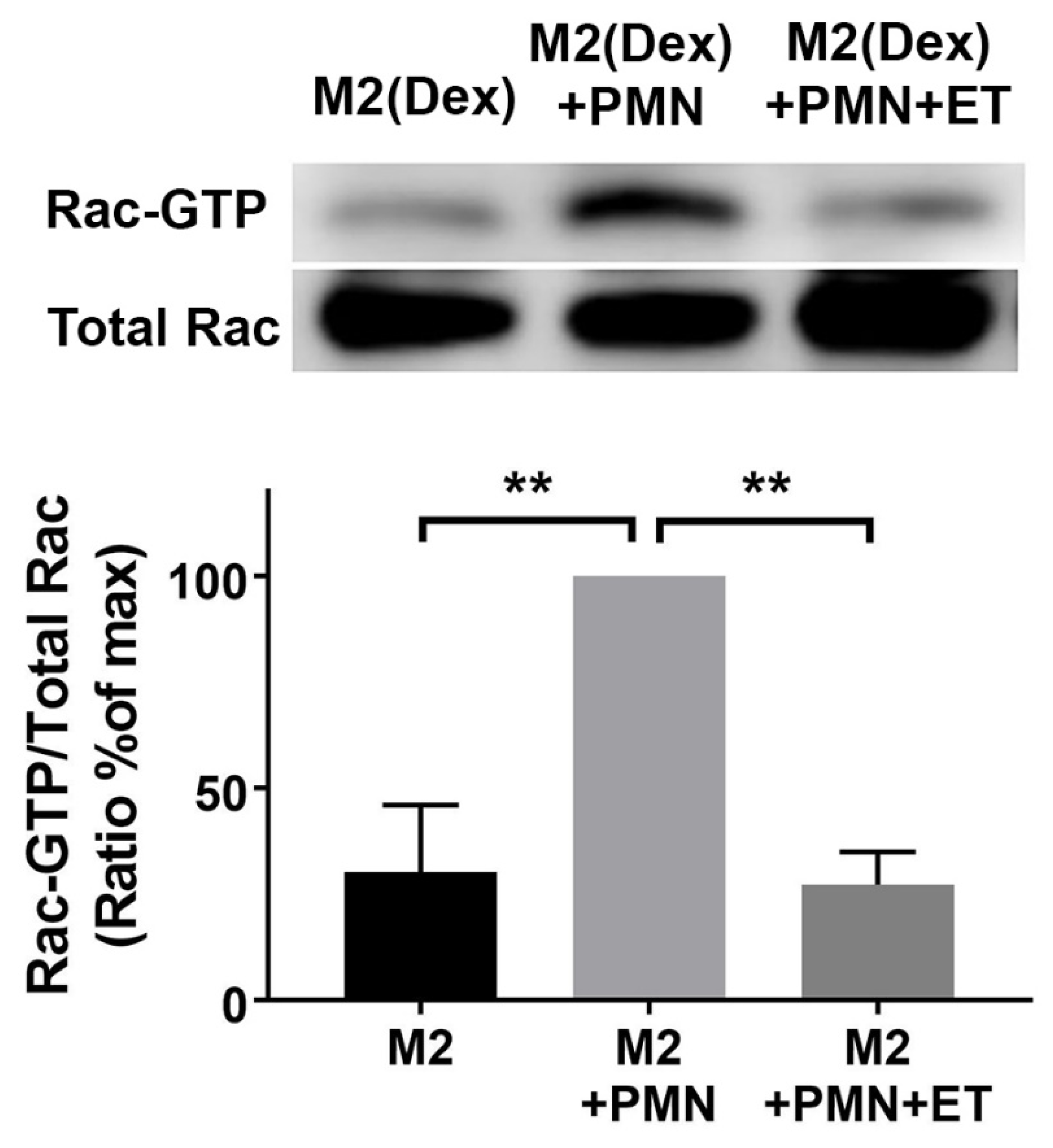
2.4. Edema Toxin (ET) Inhibits Rac1 Activation and Alters Cytoskeletal Phosphoprotein Signaling in M2(Dex) Macrophages Induced by Protein S-Opsonized Apoptotic PMN
3. Discussion
4. Materials and Methods
4.1. Isolation, Differentiation, and Polarization of Human Monocytes
4.2. Generation of Early Apoptotic Human Polymorphonuclear Neutrophils (PMN)
4.3. Flow Cytometry
4.4. Efferocytosis Assay
4.5. MerTK Inhibition Efferocytosis Assay
4.6. Rac Pull-Down Assay
4.7. Phosphoprotein Profiling Assay
4.8. Confocal Microscopy
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| cAMP | cyclic adenosine monophosphate |
| CamK | Ca2+/calmodulin-dependent protein kinase |
| EF | edema factor |
| ET | edema toxin |
| FAK | focal adhesion kinase |
| FBS | fetal bovine serum |
| PA | protective antigen |
| M2(Dex) | dexamethasone-polarized M2 macrophages |
| M2(IL-4+IL-10+Dex) | M2 macrophages polarized with a mixture of IL-4, IL-10, and dexamethasone |
| M-CSF | macrophage colony stimulating factor |
| MerTK | Mer proto-oncogene tyrosine kinase |
| MFGE8 | milk fat globulin protein E8 |
| MKK3 | mitogen-activated protein kinase 3 |
| PIP5K | phosphoinositide kinase, FYVE-type zinc finger containing |
| PMN | polymorphonuclear neutrophils |
| VASP | vasodilator-stimulated phosphoprotein |
References
- Dixon, T.C.; Meselson, M.; Guillemin, J.; Hanna, P.C. Anthrax. N. Engl. J. Med. 1999, 341, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Coggeshall, K.M.; Lupu, F.; Ballard, J.; Metcalf, J.P.; James, J.A.; Farris, A.D.; Kurosawa, S. The sepsis model: An emerging hypothesis for the lethality of inhalation anthrax. J. Cell. Mol. Med. 2013, 17, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Lupu, F.; Taylor, F.B., Jr.; Kinasewitz, G.; Kurosawa, S. Sepsis and pathophysiology of anthrax in a nonhuman primate model. Am. J. Pathol. 2006, 169, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.L. Sepsis and septic shock. Nat. Rev. Dis. Prim. 2016, 2, 16045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotchkiss, R.S.; Nicholson, D.W. Apoptosis and caspases regulate death and inflammation in sepsis. Nat. Rev. 2006, 6, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Zeerleder, S.; Stephan, F.; Emonts, M.; de Kleijn, E.D.; Esmon, C.T.; Varadi, K.; Hack, C.E.; Hazelzet, J.A. Circulating nucleosomes and severity of illness in children suffering from meningococcal sepsis treated with protein c. Crit. Care Med. 2012, 40, 3224–3229. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ye, L.; Jin, Y.; Zhang, N.; Lou, T.; Qiu, Z.; Cheng, B.; Fang, X. Circulating nucleosomes as a predictor of sepsis and organ dysfunction in critically ill patients. Int. J. Infect. Dis. 2012, 16, e558–e564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blander, J.M. The many ways tissue phagocytes respond to dying cells. Immunol. Rev. 2017, 277, 158–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baratin, M.; Simon, L.; Jorquera, A.; Ghigo, C.; Dembele, D.; Nowak, J.; Gentek, R.; Wienert, S.; Klauschen, F.; Malissen, B.; et al. T cell zone resident macrophages silently dispose of apoptotic cells in the lymph node. Immunity 2017, 47, 349–362. [Google Scholar] [CrossRef] [PubMed]
- McGaha, T.L.; Chen, Y.; Ravishankar, B.; van Rooijen, N.; Karlsson, M.C. Marginal zone macrophages suppress innate and adaptive immunity to apoptotic cells in the spleen. Blood 2011, 117, 5403–5412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.G.; McCutcheon, J.C.; Roy, N.; Chilvers, E.R.; Haslett, C.; Dransfield, I. Regulation of macrophage phagocytosis of apoptotic cells by camp. J. Immunol. 1998, 160, 3562–3568. [Google Scholar] [PubMed]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis signaling in the regulation of macrophage inflammatory responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Zent, C.S.; Elliott, M.R. Maxed out macs: Physiologic cell clearance as a function of macrophage phagocytic capacity. FEBS J. 2017, 284, 1021–1039. [Google Scholar] [CrossRef] [PubMed]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Meer, J.H.; van der Poll, T.; van’t Veer, C. Tam receptors, gas6, and protein s: Roles in inflammation and hemostasis. Blood 2014, 123, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Hanayama, R.; Tanaka, M.; Miwa, K.; Shinohara, A.; Iwamatsu, A.; Nagata, S. Identification of a factor that links apoptotic cells to phagocytes. Nature 2002, 417, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Akakura, S.; Singh, S.; Spataro, M.; Akakura, R.; Kim, J.I.; Albert, M.L.; Birge, R.B. The opsonin mfg-e8 is a ligand for the alphavbeta5 integrin and triggers dock180-dependent rac1 activation for the phagocytosis of apoptotic cells. Exp. Cell Res. 2004, 292, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Kim, K.H.; Lau, L.F. The matricellular protein ccn1 mediates neutrophil efferocytosis in cutaneous wound healing. Nat. Commun. 2015, 6, 7386. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Valladeau-Guilemond, J.; Donnadieu, M.H.; Sastre-Garau, X.; Soumelis, V.; Amigorena, S. Characterization of resident and migratory dendritic cells in human lymph nodes. J. Exp. Med. 2012, 209, 653–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annane, D.; Bellissant, E.; Bollaert, P.E.; Briegel, J.; Keh, D.; Kupfer, Y. Corticosteroids for treating sepsis. Cochrane Database Syst. Rev. 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Gibbison, B.; Lopez-Lopez, J.A.; Higgins, J.P.; Miller, T.; Angelini, G.D.; Lightman, S.L.; Annane, D. Corticosteroids in septic shock: A systematic review and network meta-analysis. Crit. Care 2017, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- McColl, A.; Bournazos, S.; Franz, S.; Perretti, M.; Morgan, B.P.; Haslett, C.; Dransfield, I. Glucocorticoids induce protein s-dependent phagocytosis of apoptotic neutrophils by human macrophages. J. Immunol. 2009, 183, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires m2c polarization and mertk induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef] [PubMed]
- Mock, M.; Fouet, A. Anthrax. Annu. Rev. Microbiol. 2001, 55, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Moayeri, M.; Leppla, S.H. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 2014, 22, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Rainey, G.J.; Bradley, K.A.; Young, J.A. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic amp concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [PubMed]
- Rossi Paccani, S.; Tonello, F.; Patrussi, L.; Capitani, N.; Simonato, M.; Montecucco, C.; Baldari, C.T. Anthrax toxins inhibit immune cell chemotaxis by perturbing chemokine receptor signalling. Cell. Microbiol. 2007, 9, 924–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeager, L.A.; Chopra, A.K.; Peterson, J.W. Bacillus anthracis edema toxin suppresses human macrophage phagocytosis and cytoskeletal remodeling via the protein kinase a and exchange protein activated by cyclic amp pathways. Infect. Immun. 2009, 77, 2530–2543. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Greten, F.R.; Wong, A.; Westrick, R.J.; Arthur, J.S.; Otsu, K.; Hoffmann, A.; Montminy, M.; Karin, M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis—Creb and nf-kappab as key regulators. Immunity 2005, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Friedlander, A.; Dreier, T.; Ezzell, J.; Leppla, S. Effects of anthrax toxin components on human neutrophils. Infect. Immun. 1985, 47, 306–310. [Google Scholar] [PubMed]
- Wright, G.G.; Mandell, G.L. Anthrax toxin blocks priming of neutrophils by lipopolysaccharide and by muramyl dipeptide. J. Exp. Med. 1986, 164, 1700–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szarowicz, S.E.; During, R.L.; Li, W.; Quinn, C.P.; Tang, W.J.; Southwick, F.S. Bacillus anthracis edema toxin impairs neutrophil actin-based motility. Infect. Immun. 2009, 77, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Tournier, J.N.; Quesnel-Hellmann, A.; Mathieu, J.; Montecucco, C.; Tang, W.J.; Mock, M.; Vidal, D.R.; Goossens, P.L. Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J. Immunol. 2005, 174, 4934–4941. [Google Scholar] [CrossRef] [PubMed]
- Cleret-Buhot, A.; Mathieu, J.; Tournier, J.N.; Quesnel-Hellmann, A. Both lethal and edema toxins of bacillus anthracis disrupt the human dendritic cell chemokine network. PLoS ONE 2012, 7, e43266. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Arocho, F.J.; Bradley, K.A. Anthrax edema toxin induces maturation of dendritic cells and enhances chemotaxis towards macrophage inflammatory protein 3beta. Infect. Immun. 2009, 77, 2036–2042. [Google Scholar] [CrossRef] [PubMed]
- Comer, J.E.; Chopra, A.K.; Peterson, J.W.; Konig, R. Direct inhibition of t-lymphocyte activation by anthrax toxins in vivo. Infect. Immun. 2005, 73, 8275–8281. [Google Scholar] [CrossRef] [PubMed]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D’Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress t lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Rossi Paccani, S.; Benagiano, M.; Capitani, N.; Zornetta, I.; Ladant, D.; Montecucco, C.; D’Elios, M.M.; Baldari, C.T. The adenylate cyclase toxins of bacillus anthracis and bordetella pertussis promote th2 cell development by shaping t cell antigen receptor signaling. PLoS Pathog. 2009, 5, e1000325. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Jung, M.Y.; Lee, S.J.; Kang, K.B.; Gratchev, A.; Riabov, V.; Kzhyshkowska, J.; Kim, I.S. Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J. Cell Sci. 2009, 122, 3365–3373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambarus, C.A.; Krausz, S.; van Eijk, M.; Hamann, J.; Radstake, T.R.; Reedquist, K.A.; Tak, P.P.; Baeten, D.L. Systematic validation of specific phenotypic markers for in vitro polarized human macrophages. J. Immunol. Methods 2012, 375, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Ritter, M.; Orso, E.; Langmann, T.; Klucken, J.; Schmitz, G. Regulation of scavenger receptor cd163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J. Leukoc. Biol. 2000, 67, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Jaguin, M.; Houlbert, N.; Fardel, O.; Lecureur, V. Polarization profiles of human m-csf-generated macrophages and comparison of m1-markers in classically activated macrophages from gm-csf and m-csf origin. Cell. Immunol. 2013, 281, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, W.; Stashko, M.A.; Deryckere, D.; Cummings, C.T.; Hunter, D.; Yang, C.; Jayakody, C.N.; Cheng, N.; Simpson, C.; et al. Unc1062, a new and potent mer inhibitor. Eur. J. Med. Chem. 2013, 65, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Robinson, D.R.; Kung, H.J. Signal pathways in up-regulation of chemokines by tyrosine kinase mer/nyk in prostate cancer cells. Cancer Res. 2004, 64, 7311–7320. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.R.; deCathelineau, A.M.; Ogden, C.A.; Leverrier, Y.; Bratton, D.L.; Daleke, D.L.; Ridley, A.J.; Fadok, V.A.; Henson, P.M. Phosphatidylserine (ps) induces ps receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J. Cell Biol. 2001, 155, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Haribabu, B.; Hook, S.S.; Selbert, M.A.; Goldstein, E.G.; Tomhave, E.D.; Edelman, A.M.; Snyderman, R.; Means, A.R. Human calcium-calmodulin dependent protein kinase i: Cdna cloning, domain structure and activation by phosphorylation at threonine-177 by calcium-calmodulin dependent protein kinase i kinase. EMBO J. 1995, 14, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.A.; Anderson, K.A.; Noeldner, P.K.; Means, A.R. The autonomous activity of calcium/calmodulin-dependent protein kinase iv is required for its role in transcription. J. Biol. Chem. 2005, 280, 20530–20538. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Noeldner, P.K.; Reece, K.; Wadzinski, B.E.; Means, A.R. Regulation and function of the calcium/calmodulin-dependent protein kinase iv/protein serine/threonine phosphatase 2a signaling complex. J. Biol. Chem. 2004, 279, 31708–31716. [Google Scholar] [CrossRef] [PubMed]
- Berwick, D.C.; Dell, G.C.; Welsh, G.I.; Heesom, K.J.; Hers, I.; Fletcher, L.M.; Cooke, F.T.; Tavare, J.M. Protein kinase b phosphorylation of pikfyve regulates the trafficking of glut4 vesicles. J. Cell Sci. 2004, 117, 5985–5993. [Google Scholar] [CrossRef] [PubMed]
- Comerford, K.M.; Lawrence, D.W.; Synnestvedt, K.; Levi, B.P.; Colgan, S.P. Role of vasodilator-stimulated phosphoprotein in pka-induced changes in endothelial junctional permeability. FASEB J. 2002, 16, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Brautigan, D.L. Cytokine activation of p38 mitogen-activated protein kinase and apoptosis is opposed by alpha-4 targeting of protein phosphatase 2a for site-specific dephosphorylation of mek3. Mol. Cell. Biol. 2007, 27, 4217–4227. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.Z.; Romer, L.; Miyamoto, S.; Volberg, T.; Matsumoto, K.; Cukierman, E.; Geiger, B.; Yamada, K.M. Targeting membrane-localized focal adhesion kinase to focal adhesions: Roles of tyrosine phosphorylation and src family kinases. J. Biol. Chem. 2003, 278, 29115–29120. [Google Scholar] [CrossRef] [PubMed]
- Deramaudt, T.B.; Dujardin, D.; Hamadi, A.; Noulet, F.; Kolli, K.; De Mey, J.; Takeda, K.; Ronde, P. Fak phosphorylation at tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol. Biol. Cell 2011, 22, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, S.L.; Ramsey, S.; Aitchison, M.; Renne, T.; Evans, T.J. Modulation of lamellipodial structure and dynamics by no-dependent phosphorylation of vasp ser239. J. Cell Sci. 2007, 120, 3011–3021. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.; Proff, J.; Havemeier, A.; Ladwein, M.; Rottner, K.; Barlag, B.; Pich, A.; Tatge, H.; Just, I.; Gerhard, R. Serine-71 phosphorylation of rac1 modulates downstream signaling. PLoS ONE 2012, 7, e44358. [Google Scholar] [CrossRef] [PubMed]
- Hutt, J.A.; Lovchik, J.A.; Drysdale, M.; Sherwood, R.L.; Brasel, T.; Lipscomb, M.F.; Lyons, C.R. Lethal factor, but not edema factor, is required to cause fatal anthrax in cynomolgus macaques after pulmonary spore challenge. Am. J. Pathol. 2014, 184, 3205–3216. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.E.; Gallegos-Candela, M.; Quinn, C.P.; Woolfitt, A.R.; Brumlow, J.O.; Isbell, K.; Hoffmaster, A.R.; Lins, R.C.; Barr, J.R. High-sensitivity maldi-tof ms quantification of anthrax lethal toxin for diagnostics and evaluation of medical countermeasures. Anal. Bioanal. Chem. 2015, 407, 2847–2858. [Google Scholar] [CrossRef] [PubMed]
- Molin, F.D.; Fasanella, A.; Simonato, M.; Garofolo, G.; Montecucco, C.; Tonello, F. Ratio of lethal and edema factors in rabbit systemic anthrax. Toxicon 2008, 52, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Singh, S.; Georgescu, M.M.; Birge, R.B. A role for mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J. Cell Sci. 2005, 118, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L.; Kim, J.I.; Birge, R.B. Alphavbeta5 integrin recruits the crkii-dock180-rac1 complex for phagocytosis of apoptotic cells. Nat. Cell Biol. 2000, 2, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Thapa, B.; Koo, B.H.; Kim, Y.H.; Kwon, H.J.; Kim, D.S. Plasminogen activator inhibitor-1 regulates infiltration of macrophages into melanoma via phosphorylation of fak-tyr(9)(2)(5). Biochem. Biophys. Res. Commun. 2014, 450, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Coppolino, M.G.; Krause, M.; Hagendorff, P.; Monner, D.A.; Trimble, W.; Grinstein, S.; Wehland, J.; Sechi, A.S. Evidence for a molecular complex consisting of fyb/slap, slp-76, nck, vasp and wasp that links the actin cytoskeleton to fcgamma receptor signalling during phagocytosis. J. Cell Sci. 2001, 114, 4307–4318. [Google Scholar] [PubMed]
- Chen, X.J.; Squarr, A.J.; Stephan, R.; Chen, B.; Higgins, T.E.; Barry, D.J.; Martin, M.C.; Rosen, M.K.; Bogdan, S.; Way, M. Ena/vasp proteins cooperate with the wave complex to regulate the actin cytoskeleton. Dev. Cell. 2014, 30, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Jersmann, H.P.; Ross, K.A.; Vivers, S.; Brown, S.B.; Haslett, C.; Dransfield, I. Phagocytosis of apoptotic cells by human macrophages: Analysis by multiparameter flow cytometry. Cytom. Part A 2003, 51, 7–15. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein b | Phospho Site | Mean ± SEM No ET | Mean ± SEM ET | p-Value c | % Change d |
|---|---|---|---|---|---|
| CamK1α | Thr 177 | 0.897 ± 0.043 | 0.533 ± 0.054 | 0.006 | 40.6 ↓ |
| Rac1 | Ser 71 | 2.990 ± 0.251 | 1.750 ± 0.056 | 0.009 | 41.5 ↓ |
| CamK4 | Thr 196/200 | 1.657 ± 0.102 | 1.160 ± 0.017 | 0.009 | 30.0 ↓ |
| PIP5K | Ser 307 | 0.563 ± 0.023 | 0.657 ± 0.003 | 0.017 | 16.7 ↑ |
| VASP | Ser 157 | 1.283 ± 0.166 | 0.630 ± 0.135 | 0.038 | 50.9 ↓ |
| MKK3 | Thr 222 | 0.960 ± 0.058 | 0.787 ± 0.019 | 0.046 | 18.0 ↓ |
| FAK | Tyr 925 | 1.230 ± 0.086 | 0.987 ± 0.018 | 0.051 | 19.8 ↓ |
| VASP | Ser 238 | 1.903 ± 0.133 | 1.267 ± 0.200 | 0.057 | 33.0 ↓ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Z.; Dumas, E.K.; Lawrence, C.; Pate, L.; Longobardi, S.; Wang, X.; James, J.A.; Kovats, S.; Farris, A.D. Bacillus anthracis Edema Toxin Inhibits Efferocytosis in Human Macrophages and Alters Efferocytic Receptor Signaling. Int. J. Mol. Sci. 2019, 20, 1167. https://doi.org/10.3390/ijms20051167
Pan Z, Dumas EK, Lawrence C, Pate L, Longobardi S, Wang X, James JA, Kovats S, Farris AD. Bacillus anthracis Edema Toxin Inhibits Efferocytosis in Human Macrophages and Alters Efferocytic Receptor Signaling. International Journal of Molecular Sciences. 2019; 20(5):1167. https://doi.org/10.3390/ijms20051167
Chicago/Turabian StylePan, Zijian, Eric K. Dumas, Christina Lawrence, Lance Pate, Sherri Longobardi, Xiaodong Wang, Judith A. James, Susan Kovats, and A. Darise Farris. 2019. "Bacillus anthracis Edema Toxin Inhibits Efferocytosis in Human Macrophages and Alters Efferocytic Receptor Signaling" International Journal of Molecular Sciences 20, no. 5: 1167. https://doi.org/10.3390/ijms20051167
APA StylePan, Z., Dumas, E. K., Lawrence, C., Pate, L., Longobardi, S., Wang, X., James, J. A., Kovats, S., & Farris, A. D. (2019). Bacillus anthracis Edema Toxin Inhibits Efferocytosis in Human Macrophages and Alters Efferocytic Receptor Signaling. International Journal of Molecular Sciences, 20(5), 1167. https://doi.org/10.3390/ijms20051167