Histone Deacetylase Inhibitor, Trichostatin A, Synergistically Enhances Paclitaxel-Induced Cytotoxicity in Urothelial Carcinoma Cells by Suppressing the ERK Pathway

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. TSA Enhances the Cytotoxicity of Paclitaxel and Reduces Viability in Human UC Cells
2.2. TSA Potentiates the Apoptotic Effect of Paclitaxel on UC Cells
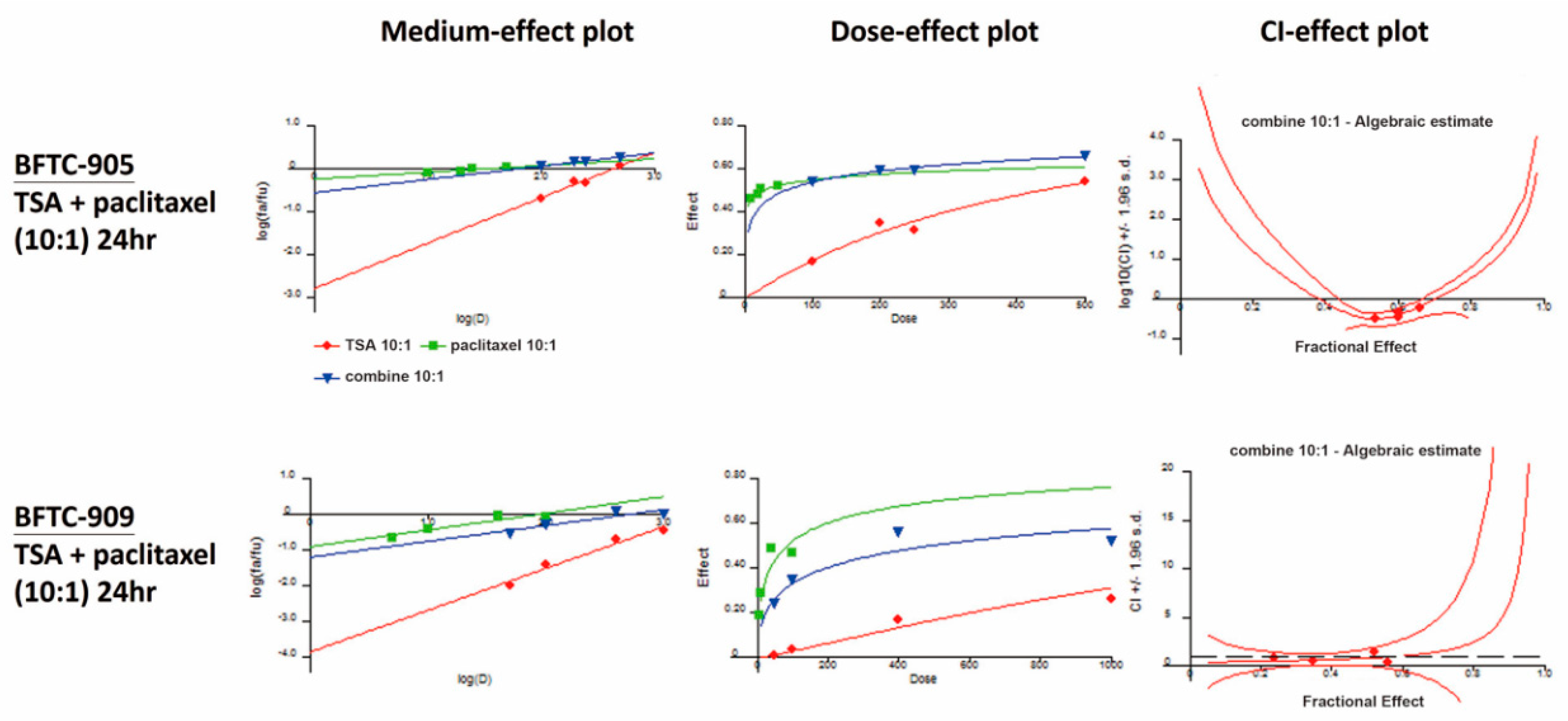
2.3. Paclitaxel in Combination with TSA Synergistically Inhibits Viability in Human UC Cells
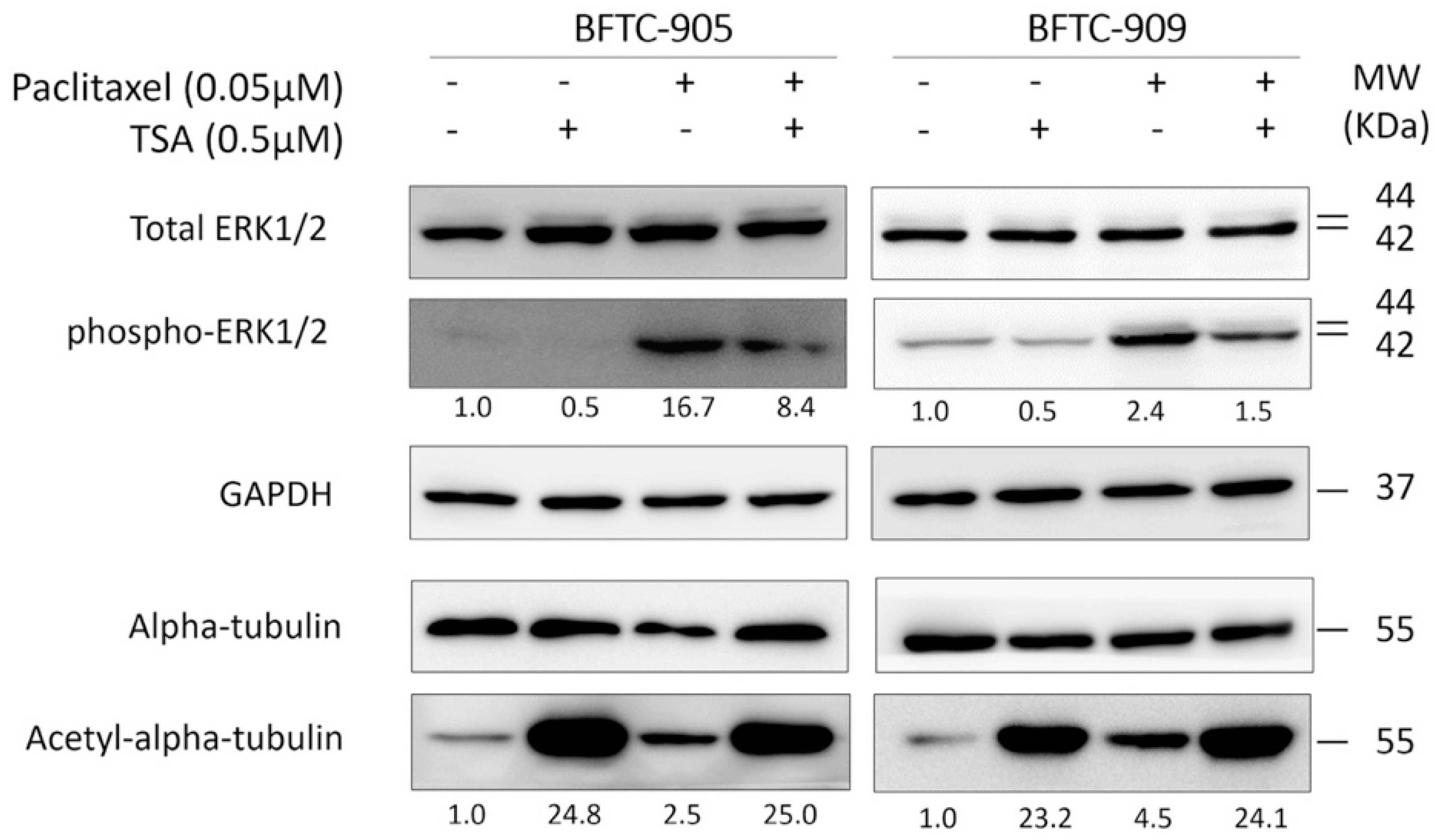
2.4. TSA Suppression of ERK Pathway Activation Following Paclitaxel Treatment in Human UC Cells
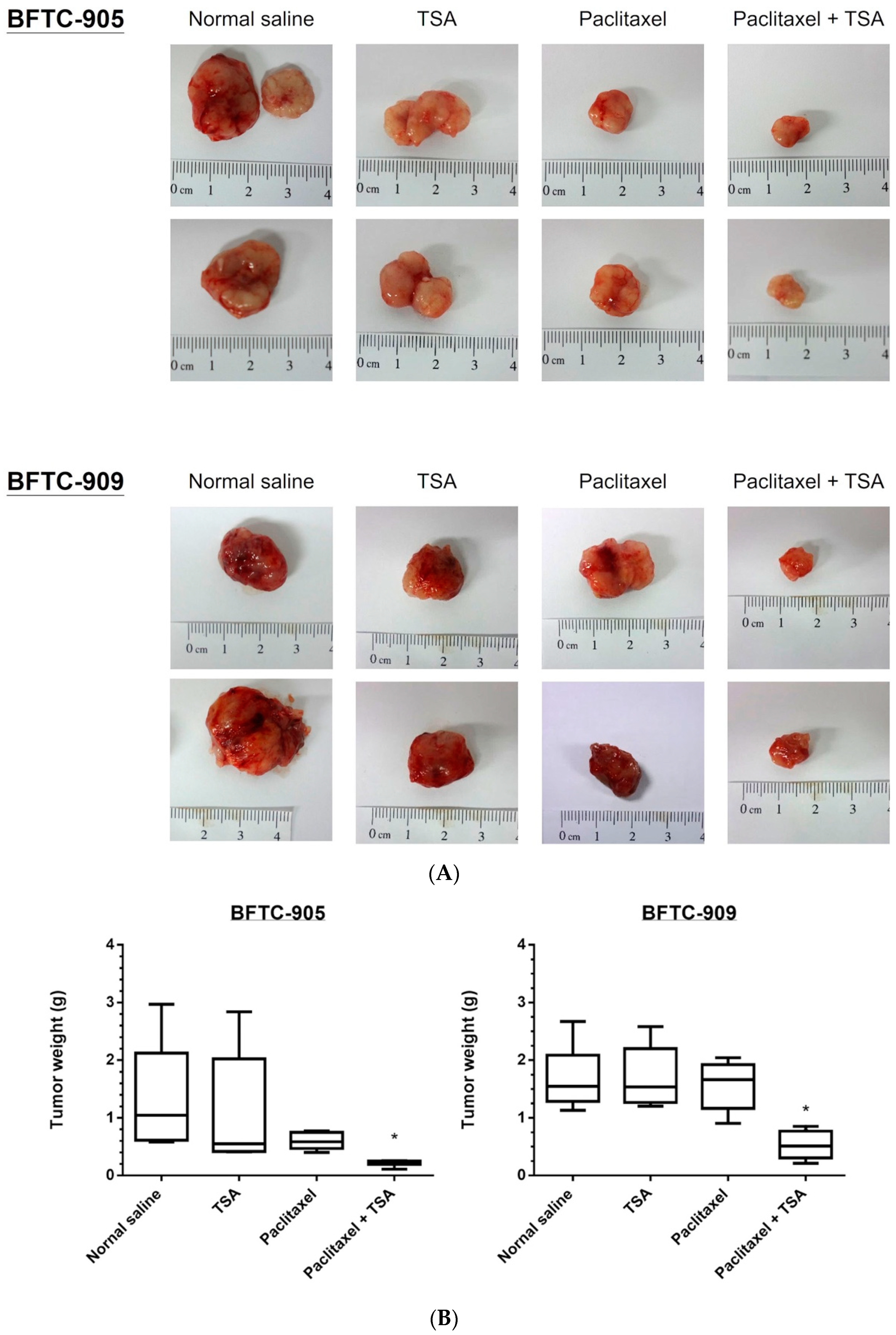
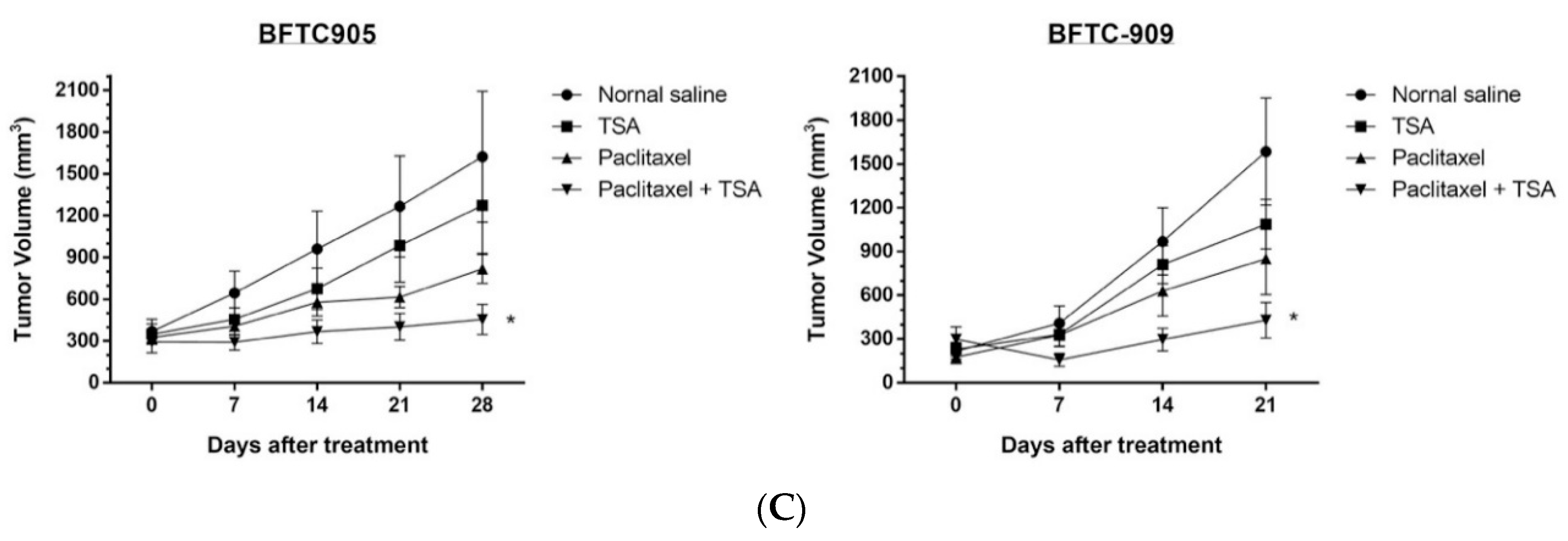
2.5. Paclitaxel-Induced Antitumor Effects Were Enhanced by TSA in a Xenograft Mouse Model
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. Reagents and Chemicals
4.3. Antibodies
4.4. Cell Viability Assay
4.5. Combination Index
4.6. Western Blot Analysis
4.7. Apoptosis Assay
4.8. In Vivo Xenograft
4.9. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gueritte-Voegelein, F.; Guenard, D.; Dubois, J.; Wahl, A.; Potier, P. Chemical and biological studies on Taxol (Paclitaxel) and Taxotere (Docetaxel), new antineoplastic agents. J. Pharm. Belg. 1994, 49, 193–205. [Google Scholar] [PubMed]
- Hu, C.Y.; Tsai, Y.C.; Wang, S.M.; Huang, C.Y.; Tai, H.C.; Chen, C.H.; Pu, Y.S.; Lin, W.C.; Huang, K.H. Ureteral involvement and diabetes increase the risk of subsequent bladder recurrence after nephroureterectomy for upper urinary tract urothelial carcinoma. BioMed Res. Int. 2015, 2015, 527976. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.S.; Su, C.H.; Huang, K.H. A Comprehensive Review of US FDA-Approved Immune Checkpoint Inhibitors in Urothelial Carcinoma. J. Immunol. Res. 2017, 2017, 6940546. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.M.; Suzman, D.; Maher, V.E.; Zhang, L.; Tang, S.; Ricks, T.; Palmby, T.; Fu, W.; Liu, Q.; Goldberg, K.B.; et al. FDA Approval Summary: Atezolizumab for the Treatment of Patients with Progressive Advanced Urothelial Carcinoma after Platinum-Containing Chemotherapy. Oncologist 2017, 22, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Brower, V. Anti-PD-L1 inhibitor durvalumab in bladder cancer. Lancet Oncol. 2016, 17, e275. [Google Scholar] [CrossRef]
- Hunt, C.R.; Ramnarain, D.; Horikoshi, N.; Iyengar, P.; Pandita, R.K.; Shay, J.W.; Pandita, T.K. Histone modifications and DNA double-strand break repair after exposure to ionizing radiations. Radiat. Res. 2013, 179, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Abmayr, S.M.; Workman, J.L. Holding on through DNA replication: Histone modification or modifier? Cell 2012, 150, 875–877. [Google Scholar] [CrossRef] [PubMed]
- Kamieniarz, K.; Izzo, A.; Dundr, M.; Tropberger, P.; Ozretic, L.; Kirfel, J.; Scheer, E.; Tropel, P.; Wisniewski, J.R.; Tora, L.; et al. A dual role of linker histone H1.4 Lys 34 acetylation in transcriptional activation. Genes Dev. 2012, 26, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, D.; Murugan, A.K.; Liu, Z.; Xing, M. Histone deacetylation of NIS promoter underlies BRAF V600E-promoted NIS silencing in thyroid cancer. Endocr. Relat. Cancer 2014, 21, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, N.; Kobayashi, M.; Nagashima, K.; Wakisaka, Y.; Koizumi, K. A new antifungal antibiotic, trichostatin. J. Antibiot. 1976, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Dickinson, M. Romidepsin for cutaneous T-cell lymphoma. Clin. Cancer Res. 2012, 18, 3509–3515. [Google Scholar] [CrossRef] [PubMed]
- Giordano, T.J. The cancer genome atlas research network: A sight to behold. Endocr. Pathol. 2014, 25, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Poyet, C.; Jentsch, B.; Hermanns, T.; Schweckendiek, D.; Seifert, H.H.; Schmidtpeter, M.; Sulser, T.; Moch, H.; Wild, P.J.; Kristiansen, G. Expression of histone deacetylases 1, 2 and 3 in urothelial bladder cancer. BMC Clin. Pathol. 2014, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.T.; Yoon, J.; Yee, H.; Chiriboga, L.; Liebes, L.; Ara, G.; Qian, X.; Bajorin, D.F.; Sun, T.T.; Wu, X.R.; et al. The histone deacetylase inhibitor belinostat (PXD101) suppresses bladder cancer cell growth in vitro and in vivo. J. Transl. Med. 2007, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Vallo, S.; Xi, W.; Hudak, L.; Juengel, E.; Tsaur, I.; Wiesner, C.; Haferkamp, A.; Blaheta, R.A. HDAC inhibition delays cell cycle progression of human bladder cancer cells in vitro. Anticancer Drugs 2011, 22, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Kang, Y.D.; Zhou, M.S.; Fu, L.L.; Hua, Z.H.; Wang, L.M. Experimental study on inhibitory effects of histone deacetylase inhibitor MS-275 and TSA on bladder cancer cells. Urol. Oncol. 2010, 28, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, A.; Tanji, N.; Kikugawa, T.; Sasaki, T.; Yanagihara, Y.; Miura, N.; Yokoyama, M. Inhibition of bladder tumour growth by histone deacetylase inhibitor. BJU Int. 2010, 105, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Li, D.R.; Zhang, H.; Peek, E.; Wang, S.; Du, L.; Li, G.; Chin, A.I. Synergy of Histone-Deacetylase Inhibitor AR-42 with Cisplatin in Bladder Cancer. J. Urol. 2015, 194, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.Y.; Park, M.J.; Lee, J.S.; Lee, S.C.; Oh, J.J.; Park, H.; Chung, C.W.; Abdullajanov, M.M.; Jeong, S.J.; Hong, S.K.; et al. The histone deacetylase inhibitor trichostatin A synergistically resensitizes a cisplatin resistant human bladder cancer cell line. J. Urol. 2011, 185, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Yeh, B.W.; Li, W.M.; Li, C.C.; Kang, W.Y.; Huang, C.N.; Hour, T.C.; Liu, Z.M.; Wu, W.J.; Huang, H.S. Histone deacetylase inhibitor trichostatin A resensitizes gemcitabine resistant urothelial carcinoma cells via suppression of TG-interacting factor. Toxicol. Appl. Pharmacol. 2016, 290, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.L.; Steelman, L.S.; Shelton, J.G.; Wong, E.W.; Chappell, W.H.; Basecke, J.; Stivala, F.; Donia, M.; Nicoletti, F.; Libra, M.; et al. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle 2010, 9, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Chappell, W.H.; Russo, S.; Ove, R.; Milella, M.; Tafuri, A.; Lunghi, P.; Bonati, A.; et al. Emerging MEK inhibitors. Expert Opin. Emerg. Drugs 2010, 15, 203–223. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Franklin, R.A.; Montalto, G.; Cervello, M.; Libra, M.; Candido, S.; Malaponte, G.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: How mutations can result in therapy resistance and how to overcome resistance. Oncotarget 2012, 3, 1068–1111. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Hsu, F.S.; Kuo, K.L.; Liu, S.H.; Shun, C.T.; Shi, C.S.; Chang, H.C.; Tsai, Y.C.; Lin, M.C.; Wu, J.T.; et al. Trichostatin A, a histone deacetylase inhibitor, induces synergistic cytotoxicity with chemotherapy via suppression of Raf/MEK/ERK pathway in urothelial carcinoma. J. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, C.C.; Liu, H.S.; Li, C.; Jin, Y.T.; Chen, R.M.; Yang, W.H.; Lin, J.S. Characterization of two urothelium cancer cell lines derived from a blackfoot disease endemic area in Taiwan. Anticancer Res. 1996, 16, 1797–1804. [Google Scholar] [PubMed]
- Ho, I.L.; Kuo, K.L.; Liu, S.H.; Chang, H.C.; Hsieh, J.T.; Wu, J.T.; Chiang, C.K.; Lin, W.C.; Tsai, Y.C.; Chou, C.T.; et al. MLN4924 Synergistically Enhances Cisplatin-induced Cytotoxicity via JNK and Bcl-xL Pathways in Human Urothelial Carcinoma. Sci. Rep. 2015, 5, 16948. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Huang, H.S.; Su, H.Y.; Li, P.H.; Chiang, P.H.; Huang, C.H.; Chen, C.H.; Hsieh, M.C. Prognostic impact of tumor infiltrating lymphocytes on patients with metastatic urothelial carcinoma receiving platinum based chemotherapy. Sci. Rep. 2018, 8, 7485. [Google Scholar] [CrossRef] [PubMed]
- Sonpavde, G.; Pond, G.R.; Choueiri, T.K.; Mullane, S.; Niegisch, G.; Albers, P.; Necchi, A.; Di Lorenzo, G.; Buonerba, C.; Rozzi, A.; et al. Single-agent Taxane Versus Taxane-containing Combination Chemotherapy as Salvage Therapy for Advanced Urothelial Carcinoma. Eur. Urol. 2016, 69, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Terakawa, T.; Miyake, H.; Yokoyama, N.; Miyazaki, A.; Tanaka, H.; Inoue, T.; Fujisawa, M. Clinical outcome of paclitaxel and carboplatin as second-line chemotherapy for advanced urothelial carcinoma resistant to first-line therapy with gemcitabine and cisplatin. Urol. Int. 2014, 92, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Brousell, S.C.; Fantony, J.J.; Van Noord, M.G.; Harrison, M.R.; Inman, B.A. Vinflunine for the treatment of advanced or metastatic transitional cell carcinoma of the urothelial tract: An evidence-based review of safety, efficacy, and place in therapy. Core Evid. 2018, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Rink, M.; Oechsle, K.; Seidel, C.; von Amsberg, G.; Bokemeyer, C. Second Line Chemotherapy for Advanced and Metastatic Urothelial Carcinoma: Vinflunine and Beyond-A Comprehensive Review of the Current Literature. J. Urol. 2016, 195, 254–263. [Google Scholar] [CrossRef] [PubMed]
- De Santis, M.; Bellmunt, J.; Mead, G.; Kerst, J.M.; Leahy, M.; Maroto, P.; Gil, T.; Marreaud, S.; Daugaard, G.; Skoneczna, I.; et al. Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin-based chemotherapy: EORTC study 30986. J. Clin. Oncol. 2012, 30, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.C.; Jiang, S.; Zhou, X.C.; Hou, X.; Jin, F.; Podratz, K.C.; Jiang, S.W. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol. Cancer Ther. 2006, 5, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Jebar, A.H.; Hurst, C.D.; Tomlinson, D.C.; Johnston, C.; Taylor, C.F.; Knowles, M.A. FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene 2005, 24, 5218–5225. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BFTC-905 (24 h) | Paclitaxel (nM) | Fraction Affected (Fa) | TSA:Paclitaxel = 10:1 |
| TSA (nM) | Combination Index (CI) | ||
| 100 | 10 | 0.54 | 0.330 |
| 200 | 20 | 0.60 | 0.369 |
| 250 | 25 | 0.60 | 0.461 |
| 500 | 50 | 0.66 | 0.640 |
| BFTC-909 (24 h) | Paclitaxel (nM) | Fraction Affected (Fa) | TSA:Paclitaxel = 10:1 |
| TSA (nM) | Combination Index (CI) | ||
| 50 | 5 | 0.24 | 0.798 |
| 100 | 10 | 0.35 | 0.552 |
| 400 | 40 | 0.56 | 0.460 |
| 1000 | 100 | 0.52 | 1.516 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, F.-S.; Wu, J.-T.; Lin, J.-Y.; Yang, S.-P.; Kuo, K.-L.; Lin, W.-C.; Shi, C.-S.; Chow, P.-M.; Liao, S.-M.; Pan, C.-I.; et al. Histone Deacetylase Inhibitor, Trichostatin A, Synergistically Enhances Paclitaxel-Induced Cytotoxicity in Urothelial Carcinoma Cells by Suppressing the ERK Pathway. Int. J. Mol. Sci. 2019, 20, 1162. https://doi.org/10.3390/ijms20051162
Hsu F-S, Wu J-T, Lin J-Y, Yang S-P, Kuo K-L, Lin W-C, Shi C-S, Chow P-M, Liao S-M, Pan C-I, et al. Histone Deacetylase Inhibitor, Trichostatin A, Synergistically Enhances Paclitaxel-Induced Cytotoxicity in Urothelial Carcinoma Cells by Suppressing the ERK Pathway. International Journal of Molecular Sciences. 2019; 20(5):1162. https://doi.org/10.3390/ijms20051162
Chicago/Turabian StyleHsu, Fu-Shun, June-Tai Wu, Jing-Yi Lin, Shao-Ping Yang, Kuan-Lin Kuo, Wei-Chou Lin, Chung-Sheng Shi, Po-Ming Chow, Shih-Ming Liao, Chun-I Pan, and et al. 2019. "Histone Deacetylase Inhibitor, Trichostatin A, Synergistically Enhances Paclitaxel-Induced Cytotoxicity in Urothelial Carcinoma Cells by Suppressing the ERK Pathway" International Journal of Molecular Sciences 20, no. 5: 1162. https://doi.org/10.3390/ijms20051162
APA StyleHsu, F.-S., Wu, J.-T., Lin, J.-Y., Yang, S.-P., Kuo, K.-L., Lin, W.-C., Shi, C.-S., Chow, P.-M., Liao, S.-M., Pan, C.-I., Hong, J.-Y., Chang, H.-C., & Huang, K.-H. (2019). Histone Deacetylase Inhibitor, Trichostatin A, Synergistically Enhances Paclitaxel-Induced Cytotoxicity in Urothelial Carcinoma Cells by Suppressing the ERK Pathway. International Journal of Molecular Sciences, 20(5), 1162. https://doi.org/10.3390/ijms20051162