1. Introduction
Flavonoids, which occur in plants, vegetables and fruits, are generally considered as efficient antioxidants and chemoprotectants. They are a plentiful but a structurally homogeneous group of plant natural products, whose structure is derived from phenylchromene hydroxylated or methoxylated at various positions. A number of flavonoids are present in natural sources in the form of glycosides. The flavonol quercetin is one of the most common and abundant flavonoids in the plant kingdom. It is a prominent bioactive flavonol in the human diet and its daily intake has increased considerably due to its use as a food supplement [
1] and mainly due to the general medical recommendation to “eat five servings of fruits and vegetables a day” [
2].
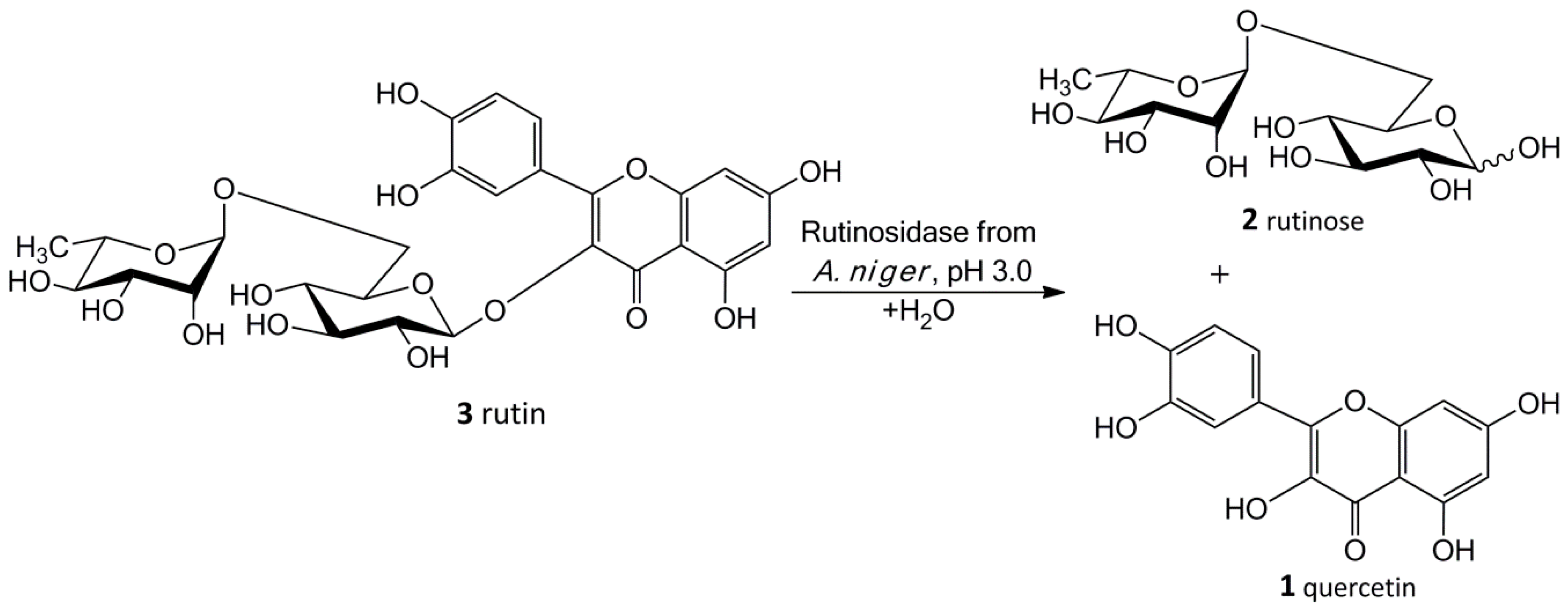
Flavonol quercetin (3,2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4
H-1-benzopyran-4-one;
1) frequently occurs in the human diet (typically in apples, onions, buckwheat etc.) in the form of glycosides—the most prominent being quercetin-3-
O-β-
d-glucopyranoside (isoquercitrin) [
3] and quercetin-3-
O-rutinoside (rutin;
3) (
Figure 1) [
4]. They have a broad range of positive effects in humans [
5] and they are used in a number of medicines and over-the-counter preparations and often in food supplements. Quercetin is largely utilized as a phytochemical remedy for a variety of diseases [
5], such as diabetes/obesity and circulatory dysfunctions, inflammation and mood disorders or as an adaptogen. Furthermore, it may be used as an efficient and affordable radioprotectant (e.g., for a large population in case of attack with a “dirty bomb”—radiological dispersal device that combines radioactive material with conventional explosives) [
6]. Quercetin used to be considered potentially harmful, since it gave a positive reaction in the Ames’ test, which implied its mutagenicity and potential cancerogenicity [
7]. Nevertheless, these doubts have recently been dispelled and the Federal Drug Agency in the US issued GRAS notice for pure quercetin [
8]. This certification led to the recent commercial boom in quercetin marketing.
The structure of quercetin is quite unique in terms of antioxidant and antiradical activity: the catechol moiety together with the free OH group at C-3 in conjugation with the C-4 oxo group ensures electron delocalization on ring B. Furthermore, the configuration of OH groups at C-3, -5 and -7 together with the C-4 oxo group make quercetin one of the most potent antioxidants ever discovered [
5]. The free C-3 OH group, often glycosylated in natural quercetin glycosides (e.g., rutin, quercitrin, isoquercitrin), is crucial for the high antioxidant activity of quercetin [
9,
10]. This is the reason why free quercetin has a considerably higher antioxidant activity than its glycosides.
The direct extraction of quercetin from natural sources is impractical, mostly due to the complexity of the matrix and its occurrence in various (glycosylated) forms. An alternative method described in the literature is the oxidation of taxifolin (dihydroquercetin), obtained from Siberian woods, with NaNO
2 [
11]. This multistep method does not yield high-purity quercetin and uses relatively toxic chemicals.
A common starting material for quercetin production is rutin (
3)—a commodity chemical obtained from various plant materials by extraction (typically from the Brazilian tree Fava d’anta,
Dimorphandra mollis). Quercetin is currently prepared by acid hydrolysis of rutin, for example, by boiling with diluted HCl [
12,
13]. This process yields quercetin and the monosaccharides
d-glucose and
l-rhamnose because rutinose bound to quercetin via the β-glycosidic linkage is completely hydrolyzed. Boiling in a strong acid may lead to a partial decomposition of all reactants, which deteriorates the purity and quality of the final product. Such a product can also hardly be declared to be of “bio” quality.
There are also enzymatic methods for producing quercetin from rutin [
13]. These methods typically employ mixtures of enzymes (e.g., β-
d-glucosidases and α-
l-rhamnosidases) that yield mixtures of the respective monosaccharides and quercetin. Since rutin has a relatively low water solubility (5 g/L at pH 5; 15 g/L at pH 8) and all enzymatic methods described so far require a completely dissolved rutin, they employ cosolvents such as MeOH, EtOH or DMSO [
14,
15]. The use of cosolvents complicates the procedure as they are flammable, toxic and their residues may contaminate the final product (especially DMSO, which is not easy to remove by evaporation). Moreover, co-solvents often inactivate the enzymes employed and thus decrease the final yields and extend reaction times. Therefore, their use must always be a compromise between the increase in rutin solubility and enzyme denaturation. The low rutin solubility also limits the possibilities of enzyme recycling or the use of immobilized enzymes-cf. [
16].
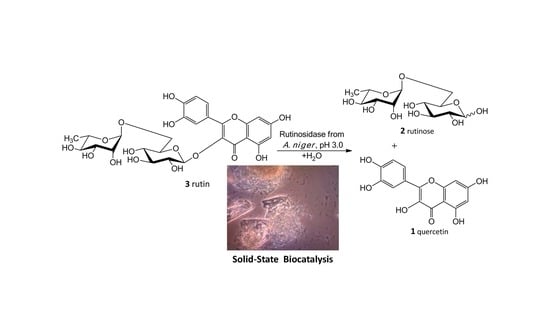
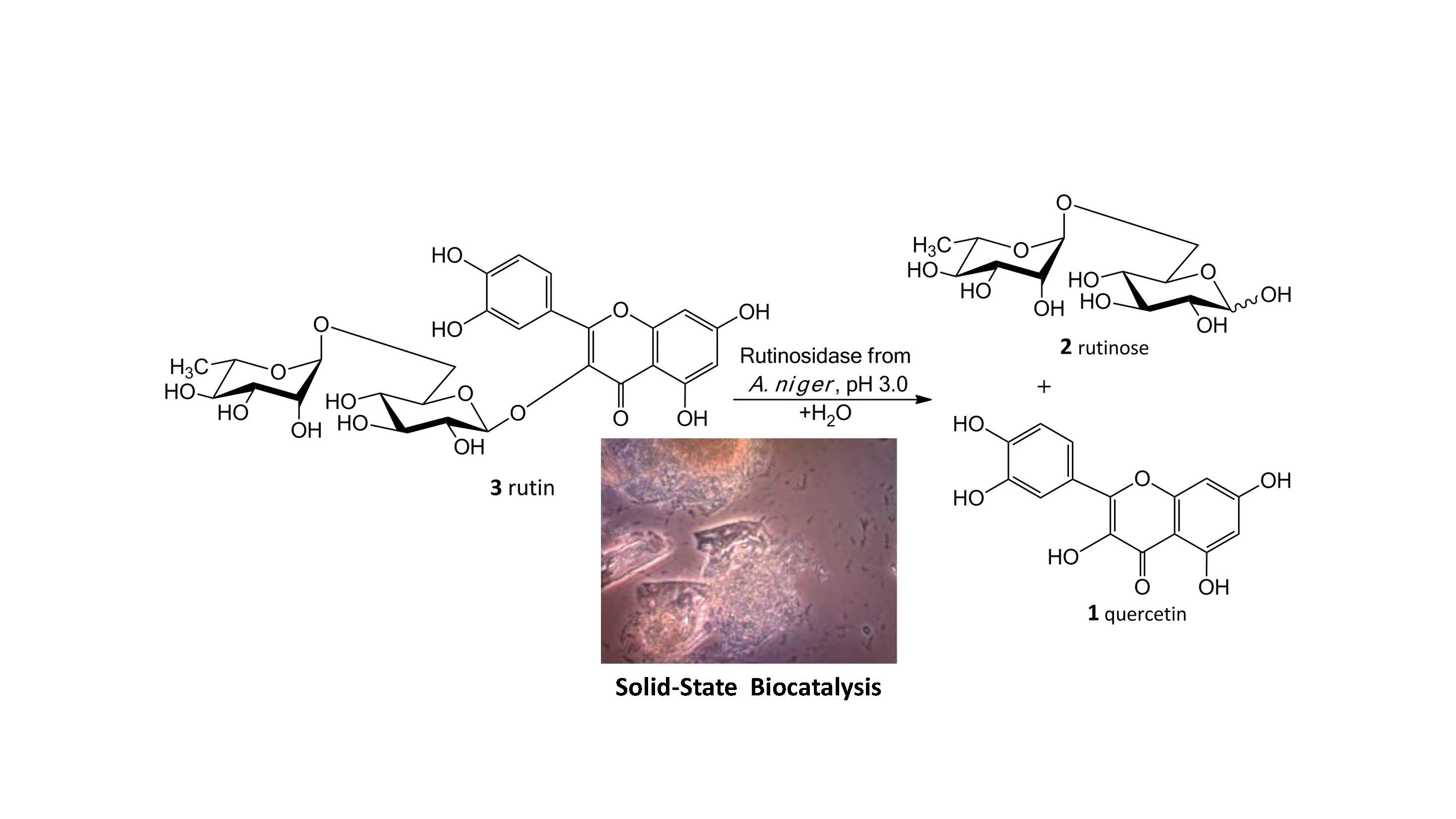
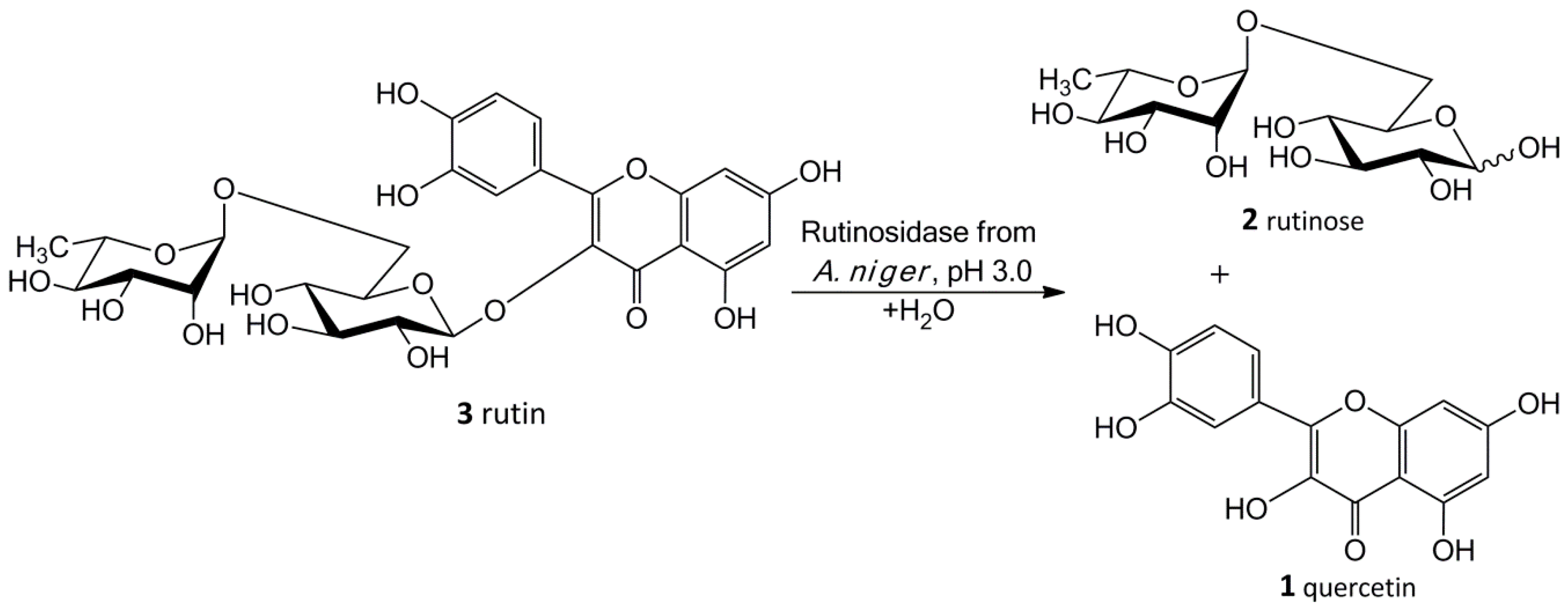
Our new biosynthetic method allows working with rutin in highly concentrated aqueous suspensions (up to ca 300 g/L, ca 0.5 M) without any co-solvents. This method directly yields a precipitated product (
1) in a virtually quantitative yield that can easily be filtrated and purified just by washing at the filter. The filtrate contains a still active enzyme, which can be reused despite the presence of rutinose from the first reaction—rutinose has no inhibitory activity on the enzyme. The rare and so far unexplored sugar rutinose (
2) is produced as a side product and can be crystallized from the concentrated filtrate in a high purity (
Figure 1). This disaccharide has a potential application in cosmetics. We present here a novel concept of “Solid State Biocatalysis” that enables an exceptionally high space-time-yield.
3. Discussion
The above results clearly demonstrate that the rutinosidase from
Aspergillus niger is a highly attractive enzyme for biotechnological application. Its heterologous expression elegantly solved the problem of contaminating α-
l-rhamnosidase and β-
d-glucosidase activities co-produced with the wild-type enzyme, and, at the same time, allowed a multiple-litre production scale-up, which can be further upscaled if convenient. Moreover, due to the production selectivity, the robust crude medium can directly be applied for a one-step bioproduction of a GRAS-classified phytochemical quercetin with a high space-time yield. Here, we must highlight the necessity of a thorough search for the optimum reaction conditions. As previously confirmed with other glycosidases, the temperature optimum may not copy the temperature stability [
20]. Admittedly, the mere activity measurement profiles under given conditions do not always reflect the real situation during the biocatalytical process. There, the enzyme active site is protected against the detrimental effect of extreme conditions such as high temperature by the proceeding biocatalytical reaction. This was clearly visible comparing the results of stability measurements and the real reaction outcome at 50 °C (
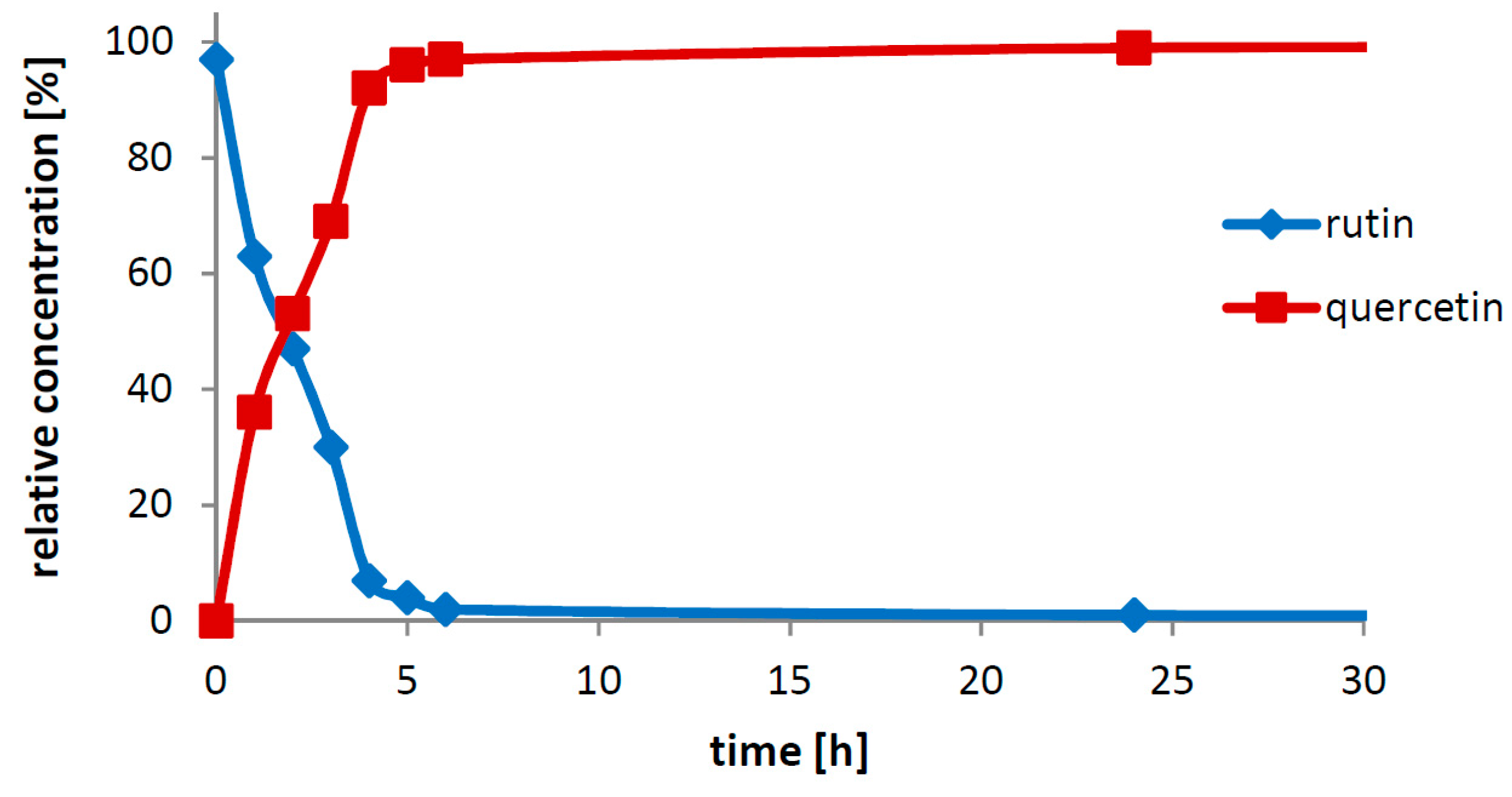
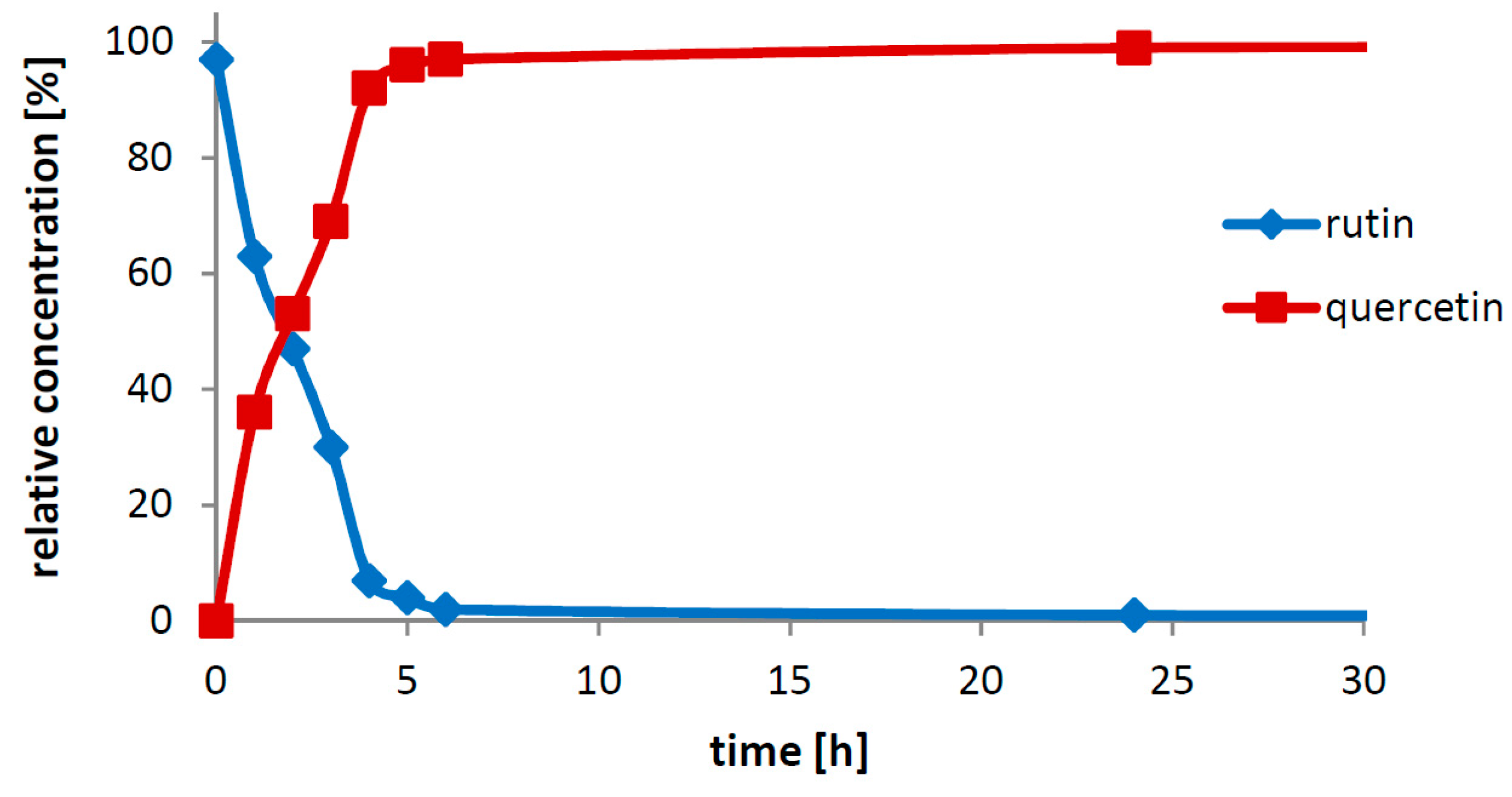
Section 2.2.2). Interestingly, this effect is observed even though no inhibition by the released hydrolytic product (rutinose) occurred. Rutinosidase inhibition with rutinose has been tested and up to 0.5 M rutinose (corresponding to ca 300 g/L starting concentration of rutin) the rutinosidase activity remains unchanged (data not shown). Apparently, since the enzyme active site is constantly occupied by the incoming reaction substrate in excess, its architecture is well protected against the impact of detrimental conditions. Thus, the enzyme is obviously more stable in a bioconversion reaction with a high rutin concentration than the enzyme incubated under the same conditions in a mere buffer.
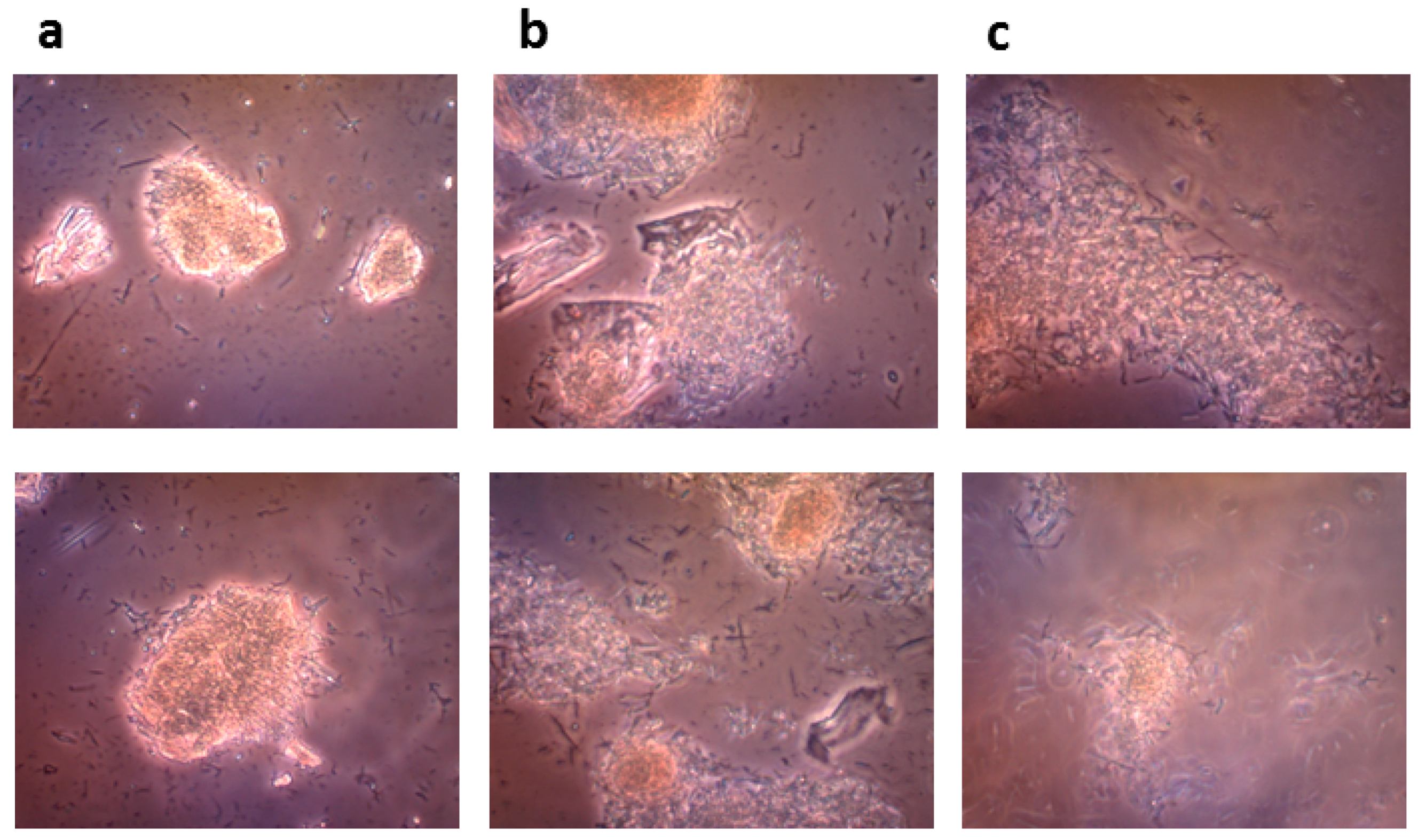
The crucial finding presented in this work is the particular biocatalytic set-up that we denoted as “Solid-State-Biocatalysis.” From the above data, it is obvious that the bioconversion of rutin into quercetin and rutinose catalyzed by rutinosidase proceeds in a very thick suspension (tested up to 450 g/L ≈ 0.7 M), where both the starting material and product remain primarily in the solid phase (the typical solubility of both reactants is ca 1–3 g/L, with quercetin being less soluble than rutin). Numerous authors tested this and other similar reactions, typically the partial hydrolysis of rutin into isoquercitrin with α-
l-rhamnosidase, with the addition of various cosolvents [
21]. In these reaction set-ups, authors barely achieved concentrations of dissolved rutin higher than approx. 10 g/L, following the commonly accepted scheme that enzymatic reactions proceed solely or at least in majority in the liquid phase. In the present case, in contrast, the majority of substrate is present in the solid phase. We suppose that the reaction proceeds in the (over)saturated microenvironment solution of the substrate crystals. The formed product, also badly water soluble, precipitates quickly out of this microenvironment, thereby constantly shifting the reaction equilibrium to the full conversion.
There are obviously two major driving forces of the reaction: (i) the high affinity of the biocatalyst for the substrate, enabling its conversion at a low concentration and (ii) the thermodynamic shift of equilibrium towards product formation caused by continuous product “removal” due to its precipitation. This type of reaction apparently runs well when both reactants are poorly soluble and the catalyst is efficient. Interestingly, when a cosolvent (e.g., 5% DMSO) is added into the reaction mixture, the reaction is slower and the conversion is not complete. This corroborates the above hypothesis because with the addition of the cosolvent, the product occurs in higher concentrations (inhibition with a higher concentration of quercetin) in the reaction mixture due to its increased solubility. A partial inactivation of the enzyme by the cosolvent cannot be excluded either.
Another advantage of the “Solid-State Biocatalysis” setup is the resistance of rutinosidase towards product inhibition by the disaccharide rutinose. On the contrary, the accumulation of rutinose in the reaction mixture and the running catalytic reaction stabilize the enzyme, as observed in our stability studies (see
Section 2.2.2 and
Figure S3, Supplementary Material). Rutinose in a higher concentration may also diminish the solubility of quercetin, lowering thus its concentration.
The main product of this enzymatic process is pure quercetin that does not involve any harmful and irritant chemicals, which makes it applicable in the cosmetics and food industries as a “Bio-quality” product. This procedure also yields the valuable side product rutinose, which has not been available in multigram/kilogram scale for a reasonable price so far. Rutinose can be obtained from the filtrate of the reaction mixture after short boiling with charcoal, calcium hydroxide and Celite (each 0.5 g/L), which removes protein and phosphate impurities and decolorizes the solution. After filtration and evaporation, pure rutinose (>97%) is obtained by crystallization or lyophilization. This opens possibilities for research and potential application of this disaccharide for example, in cosmetic applications because α-
l-rhamnose and/or α-
l-rhamnosides were found to interact with specific receptors on keratinocytes, which play an important role in cell and (skin) tissue aging [
22].
4. Materials and Methods
4.1. Materials
The enzymes and buffers for DNA manipulations were obtained from New England Biolabs (Ipswich, MA, USA). Media components were from Oxoid (Basingstoke, UK) or Carl-ROTH (Karlsruhe, Germany). Rutin was from Alchimica (Prague, Czech Republic) and other chemicals were purchased from Sigma-Aldrich.
4.2. Biological Material
An EasySelect Pichia pastoris KM71H Expression Kit was purchased from Invitrogen (Waltham, MA, USA). This expression system employs the AOX1 (alcohol oxidase 1) promoter, inducible by methanol.
The culture of Aspergillus niger K2 CCIM is deposited in the Collection of Microorganisms of the Institute of Microbiology of the Czech Academy of Sciences, Prague. The culture is maintained on slants [g/L]: agar-agar, 20; bacto-peptone, 5; and malt extract, 35.
4.3. Media
The inoculum for P. pastoris cultivation was prepared in BMGY medium (Buffered Glycerol-Complex Medium) [g/L]: yeast extract, 10; peptone, 20; 100 mM potassium phosphate, pH 6.0; Yeast Nitrogen Base (YNB, Oxoid cz., Brno, Czech Republic), 13.4; biotin 0.0004; glycerol, 10.
Buffered Methanol-Complex Medium (BMMY) has the same composition as BMGY but instead of 1% (w/v) glycerol methanol (0.5%, v/v) is added.
For large-scale productions, minimal media were used. BMGH medium (Buffered Minimal Glycerol Medium) [g/L]: 100 mM potassium phosphate pH 6.0; YNB, 13.4; biotin 0.0004; glycerol, 10 for overnight preculture and BMMH medium (Buffered Minimal Methanol Medium) [g/L]: 100 mM potassium phosphate pH 6.0; YNB, 13.4; biotin 0.0004; methanol, 5).
Fed-batch fermentations were carried out in BSM medium (Basal Salt Medium) [g/L]: 85% H3PO4, 26.7 mL; CaSO4·2H2O, 1.17; K2SO4, 18.2; MgSO4·7H2O, 14.9; KOH, 4.13; and glycerol, 40; and supplemented with 4.35 mL/L of PTM1 (trace salts solution) [g/L]: CuSO4·5H2O, 6; NaI, 0.08; MnSO4·H2O, 3; Na2MoO4·2H2O, 0.2; H3BO3, 0.02; CoCl2, 0.5; ZnCl2, 20; FeSO4·7H2O, 65; biotin, 0.2; H2SO4 conc. 9.2 g). Methanol added in fed-batch experiments was also supplemented with PTM1 (1.2 mL/L pure methanol).
The production medium for
A. niger cultivation of consisted of [g/L]: rutin, 5.0; KH
2PO
4, 15.0; NH
4Cl, 4.0; KCl, 0.5; yeast extract, 5.0; casein hydrolysate, 1.0; and 1.0 mL of trace element Vishniac solution [
23] at pH 5.0. The pH of the medium was adjusted to 5.0. After sterilization, each flask was supplemented with 1.0 mL of sterile 10% MgSO
4·7H
2O (
w/
v).
4.4. Preparation of Enzymes
4.4.1. Cultivation of Aspergillus niger
The production medium (200 mL) was inoculated with a suspension of spores in a 0.1% (v/v) Tween 80 solution and cultivated in 500-mL Erlenmeyer flasks on a rotary shaker at 28 °C and 250 rpm. The mycelium of A. niger K2 CCIM was cultivated for 12 days at 28 °C and 250 rpm in Erlenmeyer flasks in the presence of 0.5% w/v rutin as the rutinosidase inducer. The mycelium was subsequently removed by filtration through an asbestos-cellulose filter (C10, Vertex CZ, Prague, Czech Republic). The filtrate was directly used (as a crude wild rutinosidase) for rutin conversion.
4.4.2. Heterologous Expression of Rutinosidase in Pichia pastoris
The expression vector pPICZαA-RUT, obtained as described previously [
17] was linearized with restriction endonuclease
SacI and electroporated to competent
P. pastoris cells according to the manufacturer’s instructions (EasySelect
Pichia Expression Kit, Invitrogen; Waltham, MA, USA). The electroporated cells were grown at various concentrations of zeocin (100 µg/mL) on YPD (Yeast Extract Peptone Dextrose Medium; yeast extract OXOID 1%, bacteriological peptone OXOID 2%, glucose 2% (LACHNER, Neratovice, Czech Republic)) agar plates for 2 days at 28 °C.
The production of recombinant rutinosidase was performed according to the manufacturer’s instructions as follows: the colonies were inoculated into 100 mL of BMGY medium pH 6.0 and incubated overnight with shaking at 28 °C. The cells were collected by centrifugation (5000×
g, 10 min, 20 °C) and the pellet was resuspended in 30 mL of BMMY medium (see
Section 4.3 Media) in a 300 mL baffled flask. The production of rutinosidase was induced by the addition of methanol (0.5%
v/
v) every 24 h for 4 days. The flasks were incubated at 28 °C and 220 rpm.
For the large-scale production of rutinosidase, we used 1 L of BMGH medium for overnight preculture. The cells were then collected and resuspended in 200 mL of BMMH medium and incubated at 28 °C on a rotary shaker with methanol induction (0.5% v/v) every 24 h for 4 days.
4.4.3. Fermentation Scale-Up
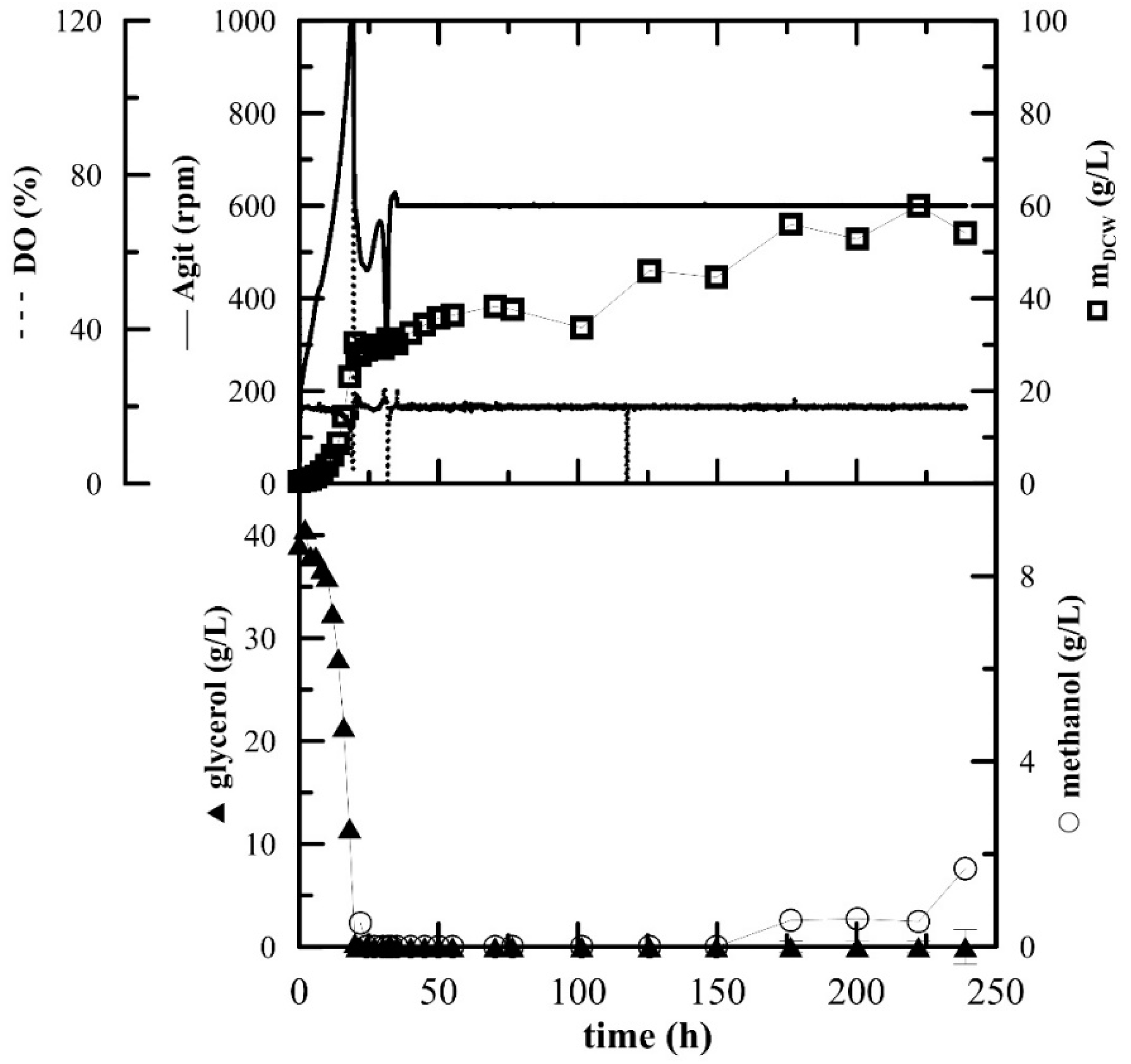
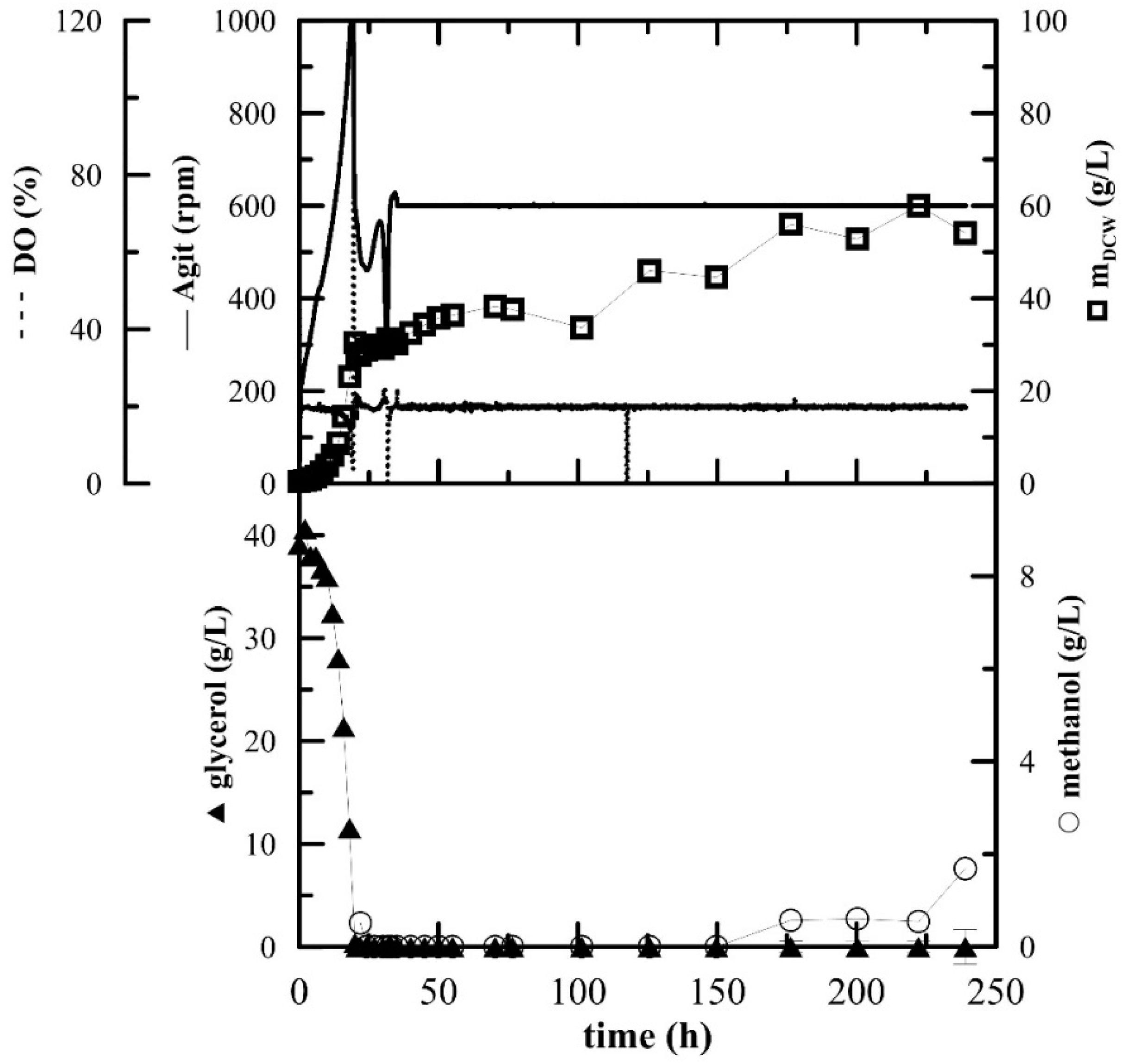
Bioreactor Cultivation
The inoculum for the fermentation cultivation was prepared in 100 mL of BMGY medium. The fermentation was essentially performed as described previously [
20] in 3-L laboratory fermenters (Brunswick BioFlo
® 115, Eppendorf, Hamburg, Germany). Then, 1.5 L of BSM media supplemented with 6.53 mL of PTM
1 was inoculated with inoculum (OD
600 approx. 10–12) to a concentration of 5%
v/
v. The fermentation conditions were as follows: 30 °C, pH 5 maintained with ammonium solution (28–30%), DO (dissolved oxygen) 20% maintained by agitation cascade from 50 to 1000 rpm, aeration 0.66 vvm with the addition of 200 µL of Struktol J650 (Schill + Seilacher “Struktol” GmbH, Hamburg, Germany) as an antifoaming agent. After the complete depletion of glycerol (approx. 20 h), two methanol pulses of 3 g/L were added after 20 and 31 h. After the depletion of the second pulse, the agitation was fixed at 600 rpm, the agitation cascade was stopped and additional methanol (3 g/L) was added. The methanol feed was connected to the actual level of dissolved oxygen as described in Reference [
20]. Whenever the level of DO rose above 20%, the methanol feed was turned on by an automated program and when the DO level rose above 30%, signalling an excess of methanol and the inability of the culture to utilize it, the pump was turned off again.
4.5. Purification of Recombinant Rutinosidase
Recombinant rutinosidase was purified from the culture medium of P. pastoris after 6 days of cultivation with methanol induction. The cells were harvested by centrifugation (5000× g, 10 min at 4 °C). The supernatant was dialyzed against 6 L of 10 mM sodium acetate buffer, pH 3.6, for 2 h (dialysis tubing cellulose membrane, Sigma-Aldrich, cut-off 10 kDa). The pH of the solution was then adjusted to 3.6 with 10% acetic acid and filtered. This solution was loaded into a Fractogel EMD SO3− column (15 × 100 mm) in 10 mM sodium acetate buffer, pH 3.6. The protein was eluted using a linear gradient of 0–1 M NaCl (5 mL/min). Fractions were collected and then analyzed for rutinosidase activity using p-nitrophenol rutinoside as a substrate. The fractions containing rutinosidase activity were concentrated by ultrafiltration using cellulose membranes with a 10 kDa cut-off (Millipore, Merck, Darmstadt, Germany). The concentrated protein was then purified to homogeneity by gel filtration in a Superdex 200 10/300 GL column (10 × 300 mm, 10 mM citrate-phosphate buffer, pH 5.0, 150 mM NaCl).
Protein concentrations were determined by Bradford assay calibrated for bovine serum albumin. The purity of recombinant rutinosidase was checked by 12% SDS-PAGE.
4.6. Storage of Enzymes
The purified or crude rutinosidase (as a cultivation medium from transformed P. pastoris) could be stored with no notable loss of activity in a fridge (ca 6 °C) for a minimum of 6 months. For lyophilization, the crude medium was rapidly frozen in a liquid nitrogen bath (shock freezing) or frozen in a deep freezer (−80 °C, slow freezing). The material was then lyophilized to dryness. The loss of specific activity of rutinosidase was in both cases under 10%. The lyophilized enzyme can be stored in a fridge in a tightly closed vessel for a minimum of 6 months with no loss of activity.
4.7. Enzyme Activity Assay
The rutinosidase activity was measured spectrophotometrically using p-nitrophenyl rutinoside as a substrate at a starting concentration of 2 mM. The reaction mixture contained substrate (10 mM solution, 10 µL), 50 mM citrate-phosphate buffer pH 5.0 (10 µL) and the enzyme solution (30 µL). After incubation of the reaction mixture at 35 °C for 10 min with shaking at 850 rpm, 0.1 M Na2CO3 (1 mL) was added to the reaction mixture. The released p-nitrophenol was determined spectrophotometrically at 420 nm. One unit of enzymatic activity was defined as the amount of enzyme releasing 1 µmol of p-nitrophenol per minute in 50 mM citrate-phosphate buffer at pH 5.0.
Enzymatic Preparation of Colorimetric Substrate p-Nitrophenyl Rutinoside
The colorimetric substrate
p-nitrophenyl rutinoside (not commercially available) was prepared by the glycosylation of
p-nitrophenyl glucopyranoside using α-
l-rhamnosidase (
A. terreus) and a free rhamnose yielding the title substrate, however with very low yields (ca 3%) [
17]. Therefore, we developed another method for the preparation of this compound, taking advantage of the high transglycosylation activity of rutinosidase from
A. niger towards phenolic substances [
17,
24]. The following protocol was used:
p-nitrophenol (100 mg) and rutin (200 mg) were dissolved in DMSO (1.1 mL) and crude rutinosidase (cultivation medium of transformed
P. pastoris; 6.4 mL, 0.4 U/mL) was added. The pH was adjusted to 3.0 and the mixture was incubated at 40 °C for 24 h. The reaction was monitored by TLC and HPLC. The reaction was stopped by boiling for 10 min, filtered, the pH was adjusted to 7.5–7.7 and unreacted
p-nitrophenol was removed by extraction (3 × 30 mL EtOAc). The aqueous phase was evaporated, the residue was dissolved in 20%
v/
v methanol in water (2 mL) and loaded into a Sephadex LH-20 column (eluted with 20%
v/
v methanol). Fractions containing the product (TLC, Silicagel 60 F
254, Merck;
n-propanol/H
2O/NH
4OH = 7:2:1,
v/
v/
v) were pooled and evaporated to yield 47 mg (32% related to rutin) of the title compound.
4.8. Bioconversion of Rutin to Quercetin
4.8.1. Analytical Scale
Purified recombinant rutinosidase from P. pastoris was used for the optimization of biotransformation conditions.
pH Optimum
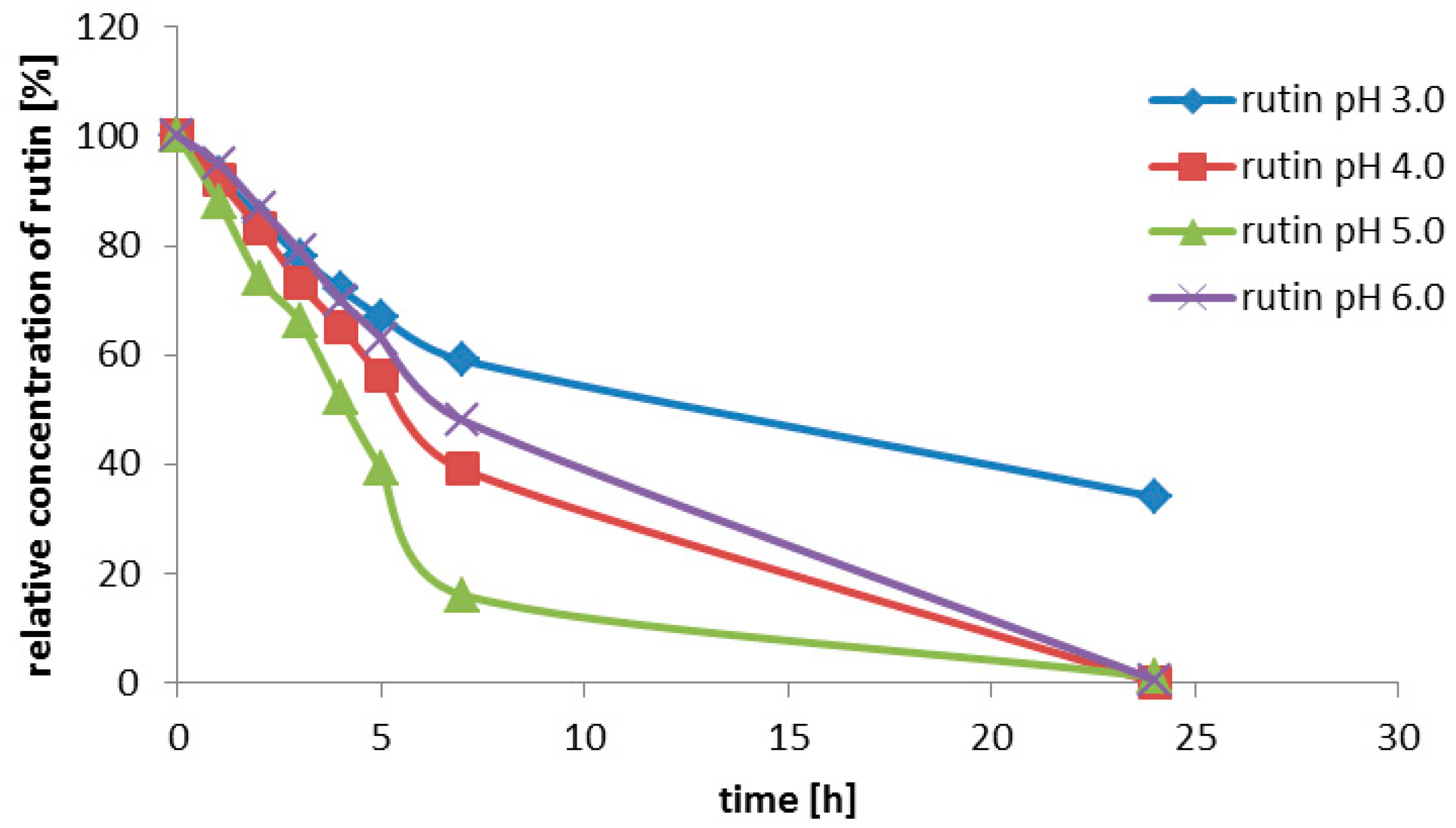
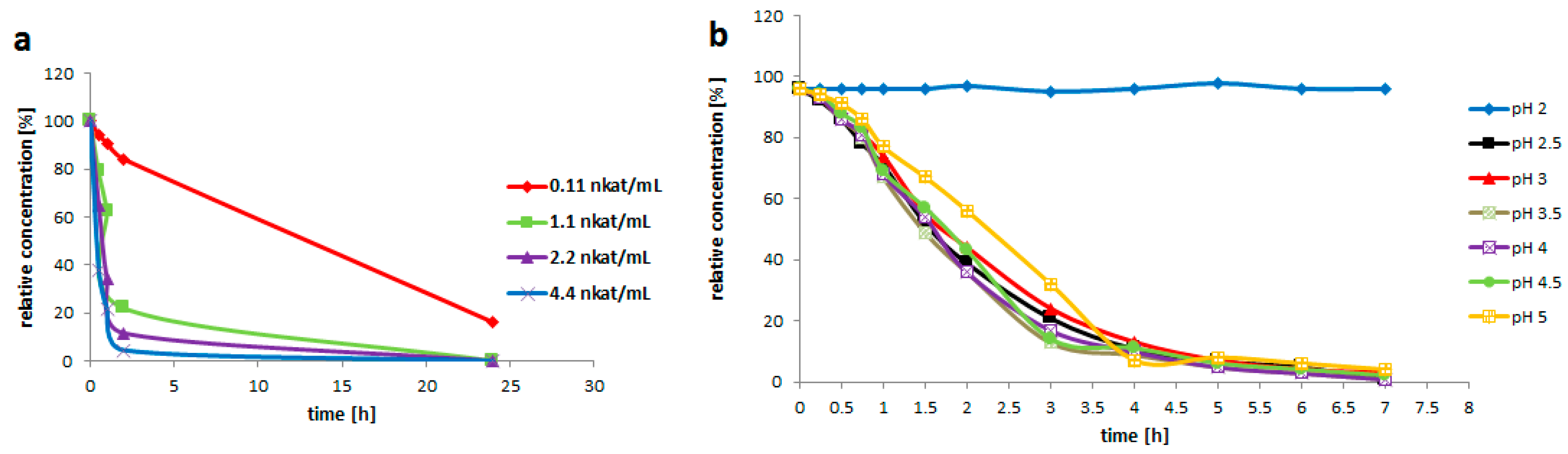
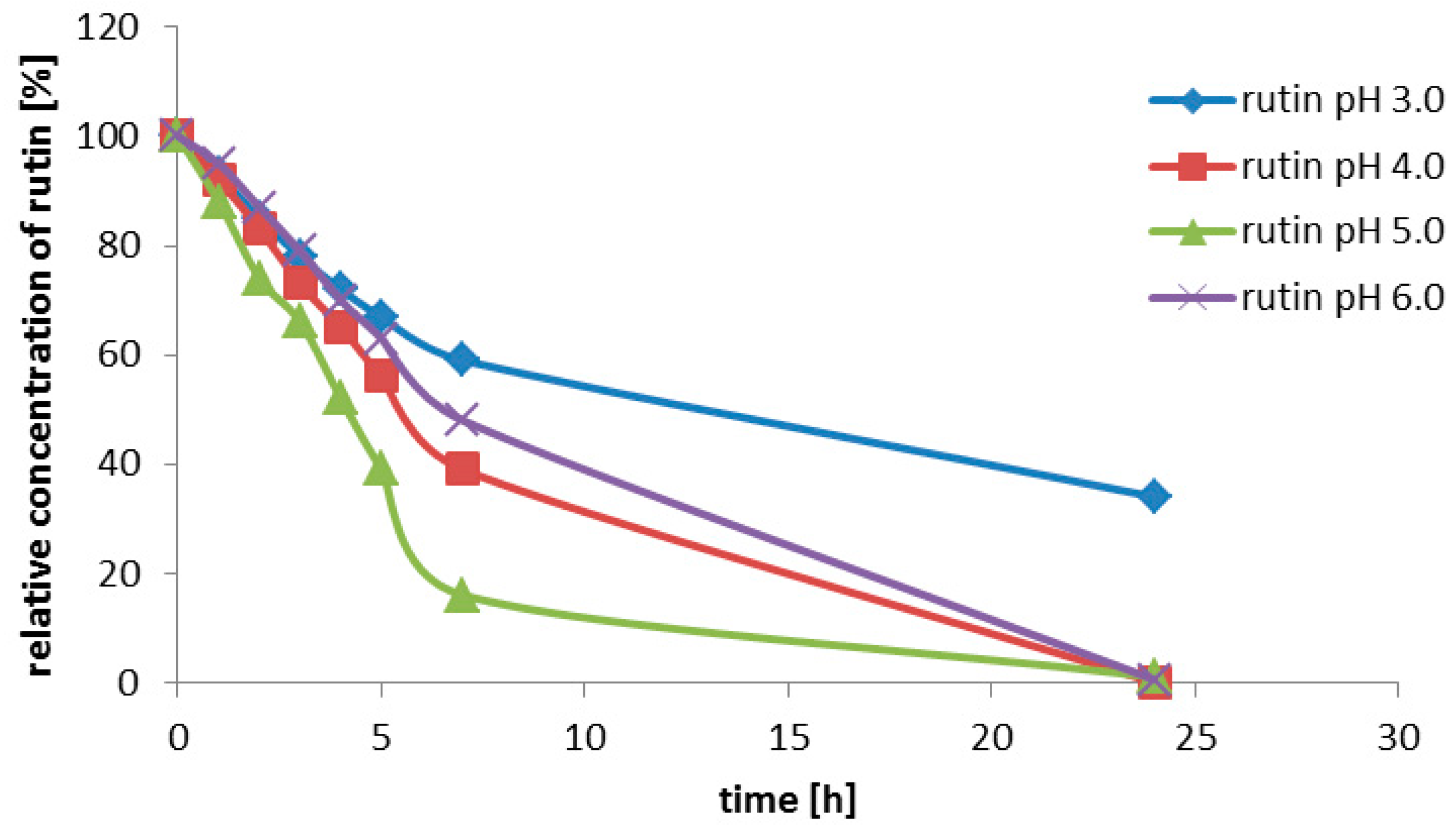
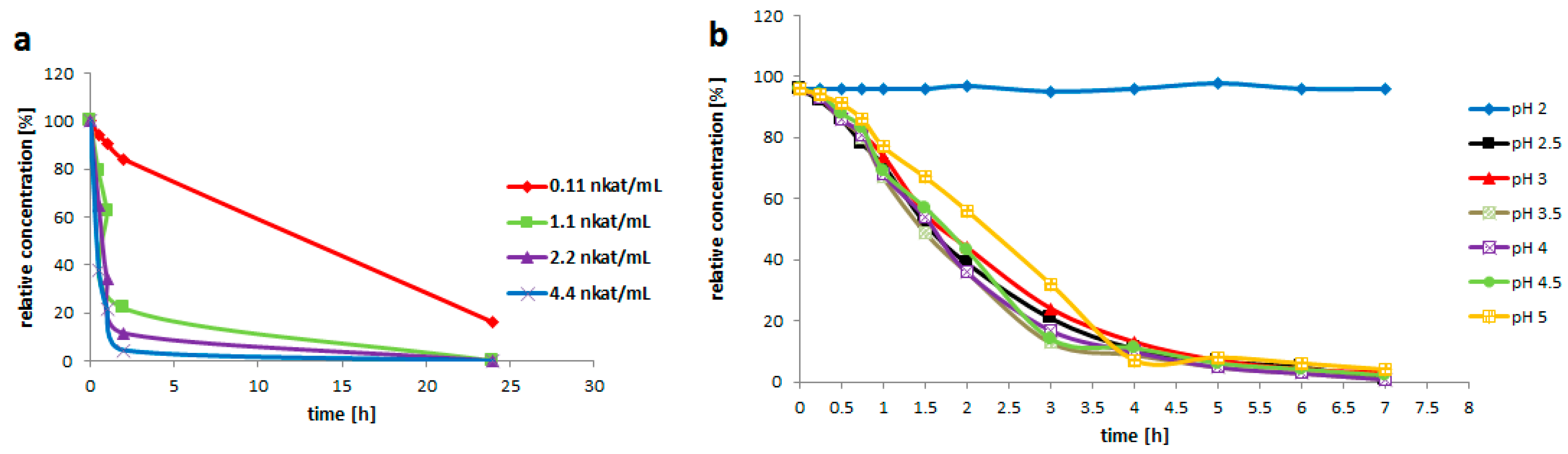
Rutin (3, 200 g/L) was suspended in the enzyme solution, which contained 0.2 M glycine-buffer and the pure enzyme (0.2 U/mL). The buffer pH was adjusted to 2.0; 2.5; 3.0; 3.5; 4.0; 4.5 or 5.0. The solution (3 mL) was incubated at 35 °C with shaking (750 rpm) for 7 h. Samples were taken every 30 min for the HPLC analysis.
Enzyme Activity
Rutin (3, 200 mg/mL) was suspended in the solution (3 mL) containing 0.2 M glycine-buffer pH 3.5 and the pure enzyme of various activities (0.021; 0.2; 0.4 or 0.8 U/mL). The reaction mixture was incubated at 35 °C with shaking (750 rpm) for 24 h. The reaction was monitored by HPLC. 1 U is defined as the amount of enzyme converting 1 µM of substrate within 1 min under the given conditions.
Temperature Optimum
Rutin (3, 200 g/L) was suspended in a solution (10 mL) containing 0.2 M glycine-buffer pH 3.5 and pure enzyme (0.2 U/mL). The reaction mixture was incubated at 10 °C; 20 °C; 35 °C; 40 °C or 50 °C with shaking (750 rpm) for 24 h. Samples were taken every 30 min for the HPLC analysis.
Thermostability
The crude medium (P. pastoris) containing rutinosidase was adjusted to pH 3.0 and was incubated at 50 °C for 150 min. Every 10 min, samples (30 µL) were taken and the enzyme activity was measured using the above procedure.
4.8.2. Scale-Up of Bioconversion of Rutin to Quercetin
Bioconversion Scale-Up Using Crude Wild-Type Rutinosidase (A. niger)
For the scale-up process, the centrifuged crude medium from the cultivation of the production strain A. niger containing rutinosidase (0.3 U/mL) was used. The crude medium was diluted twice with water and the pH was adjusted to 3.5. The medium itself had a good buffering capacity. Rutin (3; 50 g/L; 100 g/L; 150 g/L or 200 g/L) was suspended in the buffered enzyme solution, incubated at 40 °C with shaking (750 rpm) and the reaction conversion was monitored by HPLC.
Bioconversion Scale-Up Using Crude Recombinant Rutinosidase (P. pastoris)
For the scale-up process, the crude medium (prepared in a fermenter—see scale up) from transformed P. pastoris (containing typically 0.4 U/mL) was adjusted to 0.2 U/mL with water and the pH was adjusted to 3.5 with H3PO4. The medium itself had a good buffering capacity. Rutin (3; 200 g/L; 300 g/L or 450 g/L) was suspended in the buffered enzyme solution, incubated at 40 °C with shaking (750 rpm) and the reaction conversion was monitored by HPLC. Rutin was completely consumed after 24 h; then the reaction was stopped by heating to 99 °C for 5 min. The reaction mixture was then centrifuged (5000× g, 10 min at 20 °C) or filtered.
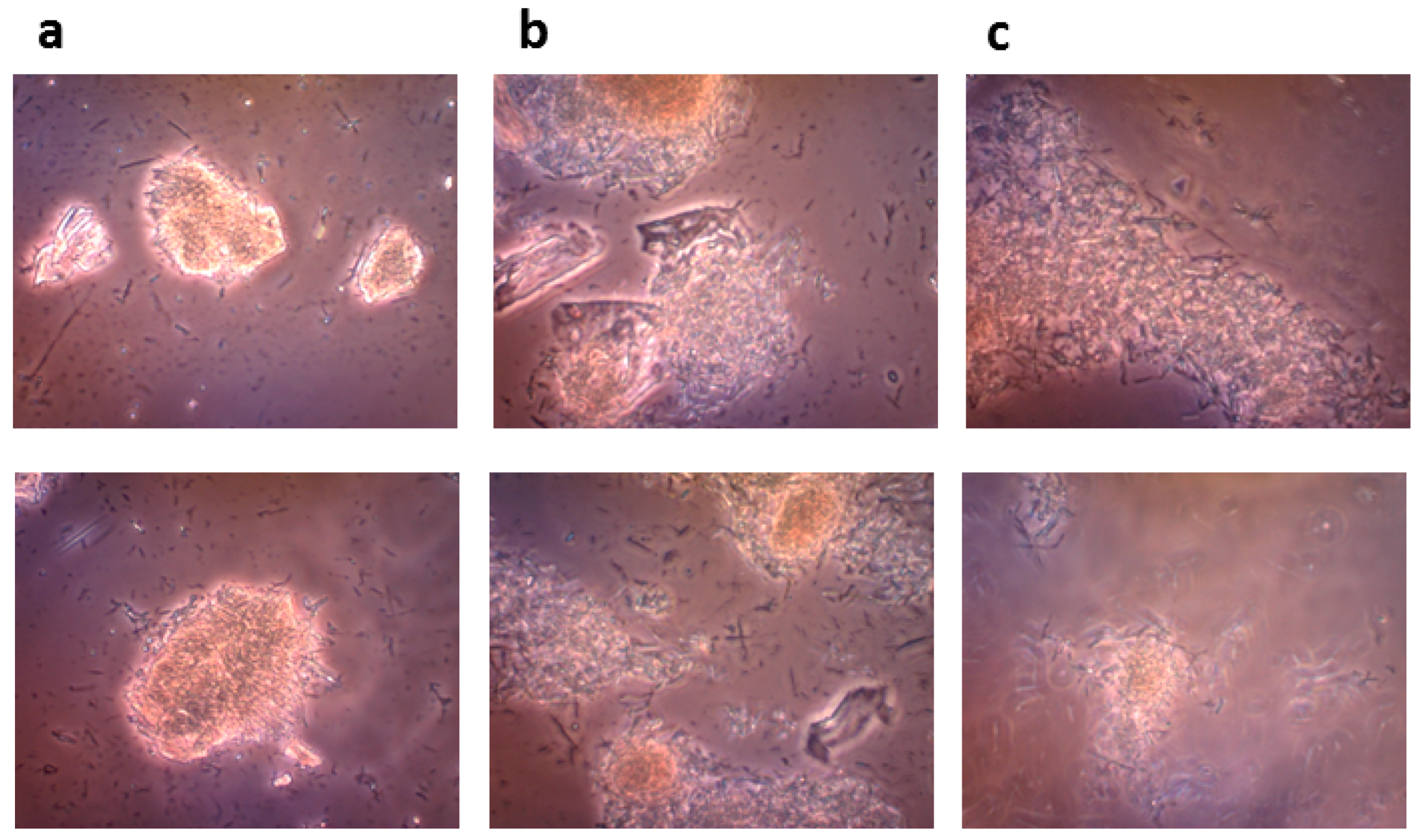
4.8.3. Microscopic Monitoring of the Solid State Biocatalysis
The progress of bioconversion of rutin to quercetin catalyzed by the crude recombinant rutinosidase (rutin 100 g/L) was monitored with an OLYMPUS CX41 optical microscope equipped with an OLYMPUS U-CMAD3 digital camera (Olympus Europa SE & Co., KG, Hamburg, Germany).
4.9. Analytical methods
Analytical HPLC
HPLC analyses were performed in a Shimadzu Prominence LC analytical system (Shimadzu, Kyoto, Japan) consisting of the following Shimadzu components: LC-20AD binary HPLC pump, CTO-10AS column oven, SIL-20ACHT cooling autosampler, CBM-20A system controller and SPD-20MA diode array detector. The sample (20 μL) was dissolved in DMSO (150 μL) and analyzed by a Chromolith Performance RP-18e column (100 × 3 mm, Merck, Germany) coupled with an RP-18e guard cartridge kit (5 × 4.6 mm, Merck, Darmstadt, Germany). Binary gradient elution was used: mobile phase A (
v/
v): 5% acetonitrile, 0.1% formic acid; mobile phase B (
v/
v): 80% acetonitrile, 0.1% formic acid; gradient: 7–25% B for 0–3 min, 30% B for 3–5 min; 7% B for 5–7.5 min. The flow rate was 1.5 mL/min at 25 °C and the injection volume was 0.1 μL; peaks were detected at 360 nm. Retention times [min]: rutin, 2.380; quercetin, 3.964. The authenticity of the compounds, for example, rutin and quercetin, was re-confirmed by MS spectra using LC-MS with commercial samples. Structure of rutinose was confirmed by
1H and
13C NMR [
17].
Glycerol and methanol concentrations were measured by HPLC with an Agilent Technologies 1220 Infinity LC apparatus combined with an Agilent Technologies 1260 Infinity RI detector (Agilent Technologies, Waldbronn, Germany), a WATREX Polymer IEX H form 8 μm, 250 × 8 mm as the main column and a WATREX Polymer IEX H form 8 μm, 40 × 8 mm (WATREX, Prague, Czech Republic) as a guard column, at a flow rate of 0.8 mL/min of 9 mM sulfuric acid at 45 °C.


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}