Regulation of Chemokines and Cytokines by Histone Deacetylases and an Update on Histone Decetylase Inhibitors in Human Diseases


 ,
,
Abstract
1. Introduction
1.1. Histone Deacetylases
1.2. Cytokines and Chemokines
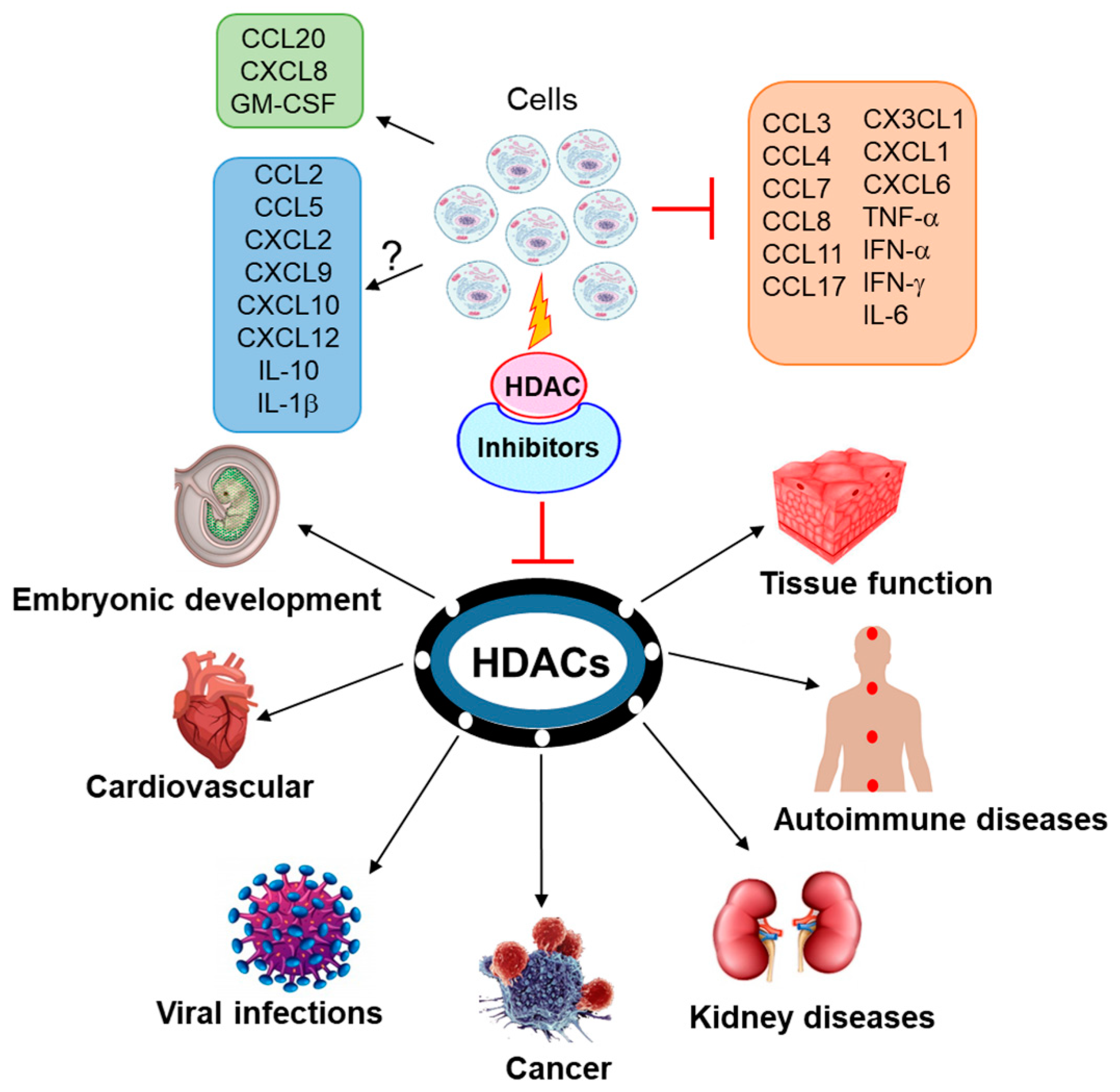
2. Cytokines, HDACs and HDAC Inhibitors
2.1. IFN-α
2.2. IFN-γ
2.3. IL-1β
2.4. IL-6
2.5. IL-10
2.6. TNF-α
2.7. GM-CSF
3. Chemokines, HDACs and HDAC Inhibitors
3.1. CCL2
3.2. CCL3
3.3. CCL4
3.4. CCL5
3.5. CCL7
3.6. CCL8
3.7. CCL11
3.8. CCL17
3.9. CCL20
3.10. CX3CL1
3.11. CXCL1
3.12. CXCL2
3.13. CXCL6
3.14. CXCL8
3.15. CXCL9
3.16. CXCL10
3.17. CXCL12
4. HDAC Inhibitors in Cancer
5. HDAC Inhibitors in Viral Infections
5.1. HIV
5.2. DNA Virus
5.3. RNA Virus
6. HDAC Inhibitors in Cardiovascular Diseases
6.1. Cardiac Hypertrophy
6.2. Cardiac Fibrosis
6.3. Myocardial Infarction
6.4. Atherosclerosis
6.5. Cardiac Arrhythmia
7. HDAC inhibitors in Autoimmune Diseases
7.1. Rheumatoid Arthritis
7.2. Multiple Sclerosis
7.3. Ulcerative Colitis
7.4. Systemic Lupus Erythematous (SLE)
8. HDAC Inhibitors in Kidney Diseases
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HAT | Histone acetyltransferases |
| HDAC | Histone deacetylases |
| IL | Interleukin |
| TNF | Tumor necrosis family |
| LPS | Lipopolysaccharide |
| IFN | Interferon |
| MMP | Matrix metalloproteases |
| PTCL | Peripheral T-cell lymphoma |
| CTCL | Cutaneous T-cell lymphoma |
| ART | Antiretroviral therapy |
| HBV | Hepatitis B virus |
| HCMV | Human cytomegalovirus |
| IAV | Influenza A virus |
| HFpEF | Heart failure with preserved ejection fractions |
| RA FLSs | Rheumatoid arthritis fibroblast-like synoviocytes |
| DSS | Dextran sulfate sodium |
| SLE | Systemic lupus erythematous |
| RCC | Renal cell carcinoma |
References
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Galdieri, L.; Gatla, H.; Vancurova, I.; Vancura, A. Activation of AMP-activated protein kinase by metformin induces protein acetylation in prostate and ovarian cancer cells. J. Biol. Chem. 2016, 291, 25154–25166. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Koneru, B.; Nguyen, T.H.; Kang, M.H.; Reynolds, C.P. Reactive oxygen species-mediated synergism of fenretinide and romidepsin in preclinical models of T-cell lymphoid malignancies. Mol. Cancer Ther. 2017, 16, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Chun, P. Therapeutic effects of histone deacetylase inhibitors on kidney disease. Arch. Pharm. Res. 2018, 41, 162–183. [Google Scholar] [CrossRef] [PubMed]
- Arcidiacono, O.; Krejčí, J.; Suchánková, J.; Bártová, E. Deacetylation of Histone H4 Accompanying Cardiomyogenesis is Weakened in HDAC1-Depleted ES Cells. Int. J. Mol. Sci. 2018, 19, 2425. [Google Scholar] [CrossRef] [PubMed]
- Večeřa, J.; Bártová, E.; Krejčí, J.; Legartová, S.; Komůrková, D.; Rudá-Kučerová, J.; Štark, T.; Dražanová, E.; Kašpárek, T.; Šulcová, A.; et al. HDAC1 and HDAC3 underlie dynamic H3K9 acetylation during embryonic neurogenesis and in schizophrenia-like animals. J. Cell. Physiol. 2018, 233, 530–548. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.L.; Roskams, A.J. Histone deacetylases 1 and 2 are expressed at distinct stages of neuro-glial development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2008, 237, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Hsieh, J. HDAC3 controls gap 2/mitosis progression in adult neural stem/progenitor cells by regulating CDK1 levels. Proc. Natl. Acad. Sci. USA 2014, 111, 13541–13546. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.W.; Carpio, L.R.; Van Wijnen, A.J.; McGee-Lawrence, M.E.; Westendorf, J.J. Histone deacetylases in bone development and skeletal disorders. Physiol. Rev. 2015, 95, 1359–1381. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Vancurova, I.; Uddin, M.M.; Zou, Y.; Vancura, A. Combination therapies targeting HDAC and IKK in solid tumors. Trends Pharmacol. Sci. 2018, 39, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Nguyen, T.H.; Koneru, B.; Hindle, A.; Chen, W.-H.; Verlekar, D.U.; Kang, M.H.; Reynolds, C.P. Vorinostat and Fenretinide Synergize in Preclinical Models of T-Cell Lymphoid Malignancies via Reactive Oxygen Species; AACR: Chicago, IL, USA, 2018. [Google Scholar]
- Bixler, S.; Goff, A. The role of cytokines and chemokines in filovirus infection. Viruses 2015, 7, 5489–5507. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Historical insights into cytokines. Eur. J. Immunol. 2007, 37, S34–S45. [Google Scholar] [CrossRef] [PubMed]
- Gulati, K.; Guhathakurta, S.; Joshi, J.; Rai, N.; Ray, A. Cytokines and their Role in Health and Disease: A Brief Overview. MOJ Immunol. 2016, 4, 00121. [Google Scholar]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Singha, B.; Gatla, H.R.; Vancurova, I. Transcriptional regulation of chemokine expression in ovarian cancer. Biomolecules 2015, 5, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Horuk, R. Chemokine receptor antagonists: Overcoming developmental hurdles. Nat. Rev. Drug Discov. 2009, 8, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Baba, T. Chemokines in tumor development and progression. Exp. Cell Res. 2012, 318, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Singha, B.; Gatla, H.R.; Manna, S.; Chang, T.-P.; Sanacora, S.; Poltoratsky, V.; Vancura, A.; Vancurova, I. Proteasome inhibition increases recruitment of IκB kinase β (IKKβ), S536P-p65, and transcription factor EGR1 to interleukin-8 (IL-8) promoter, resulting in increased IL-8 production in ovarian cancer cells. J. Biol. Chem. 2014, 289, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Singha, B.; Gatla, H.R.; Phyo, S.; Patel, A.; Chen, Z.-S.; Vancurova, I. IKK inhibition increases bortezomib effectiveness in ovarian cancer. Oncotarget 2015, 6, 26347–26358. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.J.; Lolis, E. Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 469–499. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.; Shi, W.; Jankowski, K.; Kan, C.; Cluse, L.; Martin, B.; MacKenzie, K.; Smyth, G.; Johnstone, R. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013, 4, e519. [Google Scholar] [CrossRef] [PubMed]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC inhibitors as epigenetic regulators of the immune system: Impacts on cancer therapy and inflammatory diseases. BioMed Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef] [PubMed]
- Marié, I.J.; Chang, H.-M.; Levy, D.E. HDAC stimulates gene expression through BRD4 availability in response to IFN and in interferonopathies. J. Exp. Med. 2018, 215, 3194–3212. [Google Scholar] [CrossRef] [PubMed]
- Génin, P.; Lin, R.; Hiscott, J.; Civas, A. Recruitment of histone deacetylase 3 to the interferon-A gene promoters attenuates interferon expression. PLoS ONE 2012, 7, e38336. [Google Scholar] [CrossRef] [PubMed]
- Nusinzon, I.; Horvath, C.M. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc. Natl. Acad. Sci. USA 2003, 100, 14742–14747. [Google Scholar] [CrossRef] [PubMed]
- Vlasáková, J.; Nováková, Z.; Rossmeislová, L.; Kahle, M.; Hozák, P.; Hodný, Z. Histone deacetylase inhibitors suppress IFNα-induced up-regulation of promyelocytic leukemia protein. Blood 2007, 109, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Dahllöf, M.S.; Christensen, D.P.; Harving, M.; Wagner, B.K.; Mandrup-Poulsen, T.; Lundh, M. HDAC inhibitor-mediated beta-cell protection against cytokine-induced toxicity is STAT1 Tyr701 phosphorylation independent. J. Interferon Cytokine Res. 2015, 35, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Dahllöf, M.S.; Christensen, D.P.; Lundh, M.; Dinarello, C.A.; Mascagni, P.; Grunnet, L.G.; Mandrup-Poulsen, T. The lysine deacetylase inhibitor givinostat inhibits β-cell IL-1β induced IL-1β transcription and processing. Islets 2012, 4, 417–422. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Mattarollo, S.R.; Shortt, J.; Cluse, L.A.; Christiansen, A.J.; Smyth, M.J.; Johnstone, R.W. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res. 2013, 73, 7265–7276. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Zhang, P.; Liu, W.; Zeng, X.; Ma, X.; Shi, L.; Wang, T.; Yin, Y.; Chang, W.; Zhang, P.; et al. HDAC is indispensable for IFN-γ-induced B7-H1 expression in gastric cancer. Clin. Epigenet. 2018, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Reilly, C.M.; Brown, D.R.; Ruiz, P.; Gilkeson, G.S. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Investig. 2003, 111, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Carta, S.; Tassi, S.; Semino, C.; Fossati, G.; Mascagni, P.; Dinarello, C.A.; Rubartelli, A. Histone deacetylase inhibitors prevent exocytosis of interleukin-1β-containing secretory lysosomes: Role of microtubules. Blood 2006, 108, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Stammler, D.; Eigenbrod, T.; Menz, S.; Frick, J.S.; Sweet, M.J.; Shakespear, M.R.; Jantsch, J.; Siegert, I.; Wölfle, S.; Langer, J.D.; et al. Inhibition of Histone Deacetylases Permits Lipopolysaccharide-Mediated Secretion of Bioactive IL-1β via a Caspase-1–Independent Mechanism. J. Immunol. 2015, 195, 5421–5431. [Google Scholar] [CrossRef] [PubMed]
- Di Liddo, R.; Valente, S.; Taurone, S.; Zwergel, C.; Marrocco, B.; Turchetta, R.; Conconi, M.T.; Scarpa, C.; Bertalot, T.; Schrenk, S.; et al. Histone deacetylase inhibitors restore IL-10 expression in lipopolysaccharide-induced cell inflammation and reduce IL-1β and IL-6 production in breast silicone implant in C57BL/6J wild-type murine model. Autoimmunity 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Komiyama, T.; Miyazawa, S.I.; Shen, Z.N.; Furumatsu, T.; Doi, H.; Yoshida, A.; Yamana, J.; Yamamura, M.; Ninomiya, Y.; et al. Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21WAF1/Cip1 expression. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2004, 50, 3365–3376. [Google Scholar] [CrossRef] [PubMed]
- Makki, M.S.; Haqqi, T.M. Histone Deacetylase Inhibitor Vorinostat (SAHA) Suppresses IL-1β–Induced Matrix Metallopeptidase-13 Expression by Inhibiting IL-6 in Osteoarthritis Chondrocyte. Am. J. Pathol. 2016, 186, 2701–2708. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, Y.; Jacobi, J.L.; Tuan, R.S. Inhibition of histone deacetylases antagonized FGF2 and IL-1β effects on MMP expression in human articular chondrocytes. Growth Factors 2009, 27, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Susick, L.; Senanayake, T.; Veluthakal, R.; Woster, P.M.; Kowluru, A. A novel histone deacetylase inhibitor prevents IL-1β induced metabolic dysfunction in pancreatic β-cells. J. Cell. Mol. Med. 2009, 13, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Glauben, R.; Sonnenberg, E.; Wetzel, M.; Mascagni, P.; Siegmund, B. Histone deacetylase (HDAC) inhibitors modulate IL-6-dependent CD4+ T cell polarization in vitro and in vivo. J. Biol. Chem. 2014, 289, 6142–6151. [Google Scholar] [CrossRef] [PubMed]
- Nural-Guvener, H.; Zakharova, L.; Feehery, L.; Sljukic, S.; Gaballa, M. Anti-fibrotic effects of class I HDAC inhibitor, mocetinostat is associated with IL-6/Stat3 signaling in ischemic heart failure. Int. J. Mol. Sci. 2015, 16, 11482–11499. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, S.; Iyer, L.R.; Muley, M.; Singh, P.K.; Shastry, A.; Saxena, A.; Kulathingal, J.; Vijaykanth, G.; Raghul, J.; Rajesh, N.; et al. Tubastatin, a selective histone deacetylase 6 inhibitor shows anti-inflammatory and anti-rheumatic effects. Int. Immunopharmacol. 2013, 16, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Makki, M.S.; Haqqi, T.M. Histone deacetylase inhibitor vorinostat (SAHA, MK0683) perturb miR-9-MCPIP1 axis to block IL-1β-induced IL-6 expression in human OA chondrocytes. Connect. Tissue Res. 2017, 58, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Durham, B.S.; Grigg, R.; Wood, I.C. Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J. Neurochem. 2017, 143, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Villagra, A.; Cheng, F.; Wang, H.-W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, M.; Rossato, M.; Calzetti, F.; Tamassia, N.; Zeminian, S.; Cassatella, M.A.; Bazzoni, F. IL-10 disrupts the Brd4-docking sites to inhibit LPS-induced CXCL8 and TNF-α expression in monocytes: Implications for chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cheng, F.; Woan, K.; Sahakian, E.; Merino, O.; Rock-Klotz, J.; Vicente-Suarez, I.; Pinilla-Ibarz, J.; Wright, K.L.; Seto, E.; et al. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J. Immunol. 2011, 186, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Lienlaf, M.; Wang, H.-W.; Perez-Villarroel, P.; Lee, C.; Woan, K.; Rock-Klotz, J.; Sahakian, E.; Woods, D.; Pinilla-Ibarz, J.; et al. A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J. Immunol. 2014, 193, 2850–2862. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, Y.; Jiao, J.; Huang, Q. Mycobacterium tuberculosis infection induces IL-10 gene expression by disturbing histone deacetylase 6 and histonedeacetylase 11 equilibrium in macrophages. Tuberculosis 2018, 108, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Licciardi, P.V.; Karagiannis, T.C. Regulation of immune responses by histone deacetylase inhibitors. ISRN Hematol. 2012, 2012, 690901. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, K.; Makena, M.; Bhowmick, K.; Pandey, M. Advancement of NF-κB Signaling Pathway: A Novel Target in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3890. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shan, L.; Schiller, P.W.; Mai, A.; Peng, T. Histone deacetylase-3 activation promotes tumor necrosis factor-α (TNF-α) expression in cardiomyocytes during lipopolysaccharide stimulation. J. Biol. Chem. 2010, 285, 9429–9436. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Lin, Z.; SenBanerjee, S.; Jain, M.K. Tumor necrosis factor alpha-mediated reduction of KLF2 is due to inhibition of MEF2 by NF-κB and histone deacetylases. Mol. Cell. Biol. 2005, 25, 5893–5903. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Nishida, K.; Takasugi, K.; Ogawa, H.; Sada, K.; Kadota, Y.; Inagaki, J.; Hirohata, S.; Ninomiya, Y.; Makino, H. Increased activity and expression of histone deacetylase 1 in relation to tumor necrosis factor-alpha in synovial tissue of rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, R133. [Google Scholar] [CrossRef] [PubMed]
- Symanowski, J.; Vogelzang, N.; Zawel, L.; Atadja, P.; Pass, H.; Sharma, S. A histone deacetylase inhibitor LBH589 downregulates XIAP in mesothelioma cell lines which is likely responsible for increased apoptosis with TRAIL. J. Thorac. Oncol. 2009, 4, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Han, B.R.; Park, W.H. Suberoylanilide hydroxamic acid increases anti-cancer effect of tumor necrosis factor-α through up-regulation of TNF receptor 1 in lung cancer cells. Oncotarget 2017, 8, 17726–17737. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tang, X.; Zhou, Z.; Huang, Q. Histone deacetylase 6 inhibitor enhances resistance to Mycobacterium tuberculosis infection through innate and adaptive immunity in mice. Pathog. Dis. 2018, 76, fty064. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Giembycz, M.; Ito, K.; Lim, S.; Jazrawi, E.; Barnes, P.J.; Adcock, I.; Erdmann, E.; Chung, K.F. Mitogen-activated protein kinase modulation of nuclear factor-κB–induced granulocyte macrophage–colony-stimulating factor release from human alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2004, 30, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Stapnes, C.; Ryningen, A.; Hatfield, K.; Øyan, A.M.; Eide, G.E.; Corbascio, M.; Kalland, K.-H.; Gjertsen, B.T.; Bruserud, Ø. Functional characteristics and gene expression profiles of primary acute myeloid leukaemia cells identify patient subgroups that differ in susceptibility to histone deacetylase inhibitors. Int. J. Oncol. 2007, 31, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Riddle, S.R.; Frid, M.G.; El Kasmi, K.C.; McKinsey, T.A.; Sokol, R.J.; Strassheim, D.; Meyrick, B.; Yeager, M.E.; Flockton, A.R.; et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J. Immunol. 2011, 187, 2711–2722. [Google Scholar] [CrossRef] [PubMed]
- Elsharkawy, A.M.; Oakley, F.; Lin, F.; Packham, G.; Mann, D.A.; Mann, J. The NF-κB p50: p50: HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J. Hepatol. 2010, 53, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Barnes, P.J.; Adcock, I.M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1β-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 2000, 20, 6891–6903. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2–mediated deacetylation of the glucocorticoid receptor enables NF-κB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Gonneaud, A.; Gagné, J.M.; Turgeon, N.; Asselin, C. The histone deacetylase Hdac1 regulates inflammatory signalling in intestinal epithelial cells. J. Inflamm. 2014, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, K.; Park, D.; Lee, E.; Lee, H.; Lee, Y.-S.; Choe, J.; Jeoung, D. Histone deacetylase 3 mediates allergic skin inflammation by regulating expression of MCP1 protein. J. Biol. Chem. 2012, 287, 25844–25859. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Telles, E.; Karl, M.; Cheng, F.; Luetteke, N.; Sotomayor, E.M.; Miller, R.H.; Seto, E. Loss of HDAC11 ameliorates clinical symptoms in a multiple sclerosis mouse model. Life Sci. Alliance 2018, 1, e201800039. [Google Scholar] [CrossRef] [PubMed]
- Safronova, O.S.; Nakahama, K.-I.; Morita, I. Acute hypoxia affects P-TEFb through HDAC3 and HEXIM1-dependent mechanism to promote gene-specific transcriptional repression. Nucleic Acids Res. 2014, 42, 8954–8969. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Kirkwood, J.; Dent, P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget 2017, 8, 83155–83170. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peubez, C.; Smith, V.; Xiong, S.; Kocsis-Fodor, G.; Kennedy, B.; Wagner, S.; Balotis, C.; Jayne, S.; Dyer, M.J.; et al. CUDC-907 blocks multiple pro-survival signals and abrogates microenvironment protection in CLL. J. Cell. Mol. Med. 2019, 23, 340–380. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.W.; Song, J.W.; Ha, N.; Choi, Y.I.; Kim, S. CKD-506, a novel HDAC6-selective inhibitor, improves renal outcomes and survival in a mouse model of systemic lupus erythematosus. Sci. Rep. 2018, 8, 17297. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Zhou, X.; Zheng, X.; Cui, Y.; Tsien, J.Z.; Li, C.; Wang, H. Histone deacetylase inhibitor alleviates the neurodegenerative phenotypes and histone dysregulation in presenilins-deficient mice. Front. Aging Neurosci. 2018, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; Shields, C.E.D.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell 2017, 171, 1284–1300. [Google Scholar] [CrossRef] [PubMed]
- Aung, H.T.; Schroder, K.; Himes, S.R.; Brion, K.; van Zuylen, W.; Trieu, A.; Suzuki, H.; Hayashizaki, Y.; Hume, D.A.; Sweet, M.J.; et al. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J. 2006, 20, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.-Y.; Ho, P.C.; Tanaka, Y.; Lin, H.-S. The histone deacetylase inhibitors MS-275 and SAHA suppress the p38 mitogen-activated protein kinase signaling pathway and chemotaxis in rheumatoid arthritic synovial fibroblastic E11 cells. Molecules 2013, 18, 14085–14095. [Google Scholar] [CrossRef] [PubMed]
- Toki, S.; Goleniewska, K.; Reiss, S.; Zhou, W.; Newcomb, D.C.; Bloodworth, M.H.; Stier, M.T.; Boyd, K.L.; Polosukhin, V.V.; Subramaniam, S.; et al. The histone deacetylase inhibitor trichostatin A suppresses murine innate allergic inflammation by blocking group 2 innate lymphoid cell (ILC2) activation. Thorax 2016, 71, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Buglio, D.; Georgakis, G.V.; Hanabuchi, S.; Arima, K.; Khaskhely, N.M.; Liu, Y.-J.; Younes, A. Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood 2008, 112, 1424–1433. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.-R.; Kang, S.-S.; Lee, D.; Yun, C.-H.; Han, S.H. Killed Whole-cell Oral cholera Vaccine induces ccl20 secretion by human intestinal epithelial cells in the Presence of the short-chain Fatty acid, Butyrate. Front. Immunol. 2018, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Novitskaya, T.; McDermott, L.; Zhang, K.X.; Chiba, T.; Paueksakon, P.; Hukriede, N.A.; de Caestecker, M.P. A PTBA small molecule enhances recovery and reduces postinjury fibrosis after aristolochic acid-induced kidney injury. Am. J. Physiol. Ren. Physiol. 2013, 306, F496–F504. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Gong, A.-Y.; Chen, D.; Miller, R.E.; Eischeid, A.N.; Chen, X.-M. Histone deacetylases and NF-κB signaling coordinate expression of CX3CL1 in epithelial cells in response to microbial challenge by suppressing miR-424 and miR-503. PLoS ONE 2013, 8, e65153. [Google Scholar]
- Ferrari, R.; Gou, D.; Jawdekar, G.; Johnson, S.A.; Nava, M.; Su, T.; Yousef, A.F.; Zemke, N.R.; Pellegrini, M.; Kurdistani, S.K.; et al. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 2014, 16, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Ziesche, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Müller, H.; Newel, D.; Kronich, P.; Schneider, H.; et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-κB. Nucleic Acids Res. 2012, 41, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Lundh, M.; Christensen, D.; Nielsen, M.D.; Richardson, S.; Dahllöf, M.; Skovgaard, T.; Berthelsen, J.; Dinarello, C.; Stevenazzi, A.; Mascagni, P.; et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia 2012, 55, 2421–2431. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.; Singha, B.; Phyo, S.A.; Gatla, H.R.; Chang, T.-P.; Sanacora, S.; Ramaswami, S.; Vancurova, I. Proteasome inhibition by bortezomib increases IL-8 expression in androgen-independent prostate cancer cells: The role of IKKα. J. Immunol. 2013, 191, 2837–2846. [Google Scholar] [CrossRef] [PubMed]
- Singha, B.; Phyo, S.A.; Gatla, H.R.; Vancurova, I. Quantitative analysis of bortezomib-induced IL-8 gene expression in ovarian cancer cells. In Cytokine Bioassays; Springer: New York, NY, USA, 2014; pp. 295–304. [Google Scholar]
- Gatla, H.R.; Singha, B.; Persaud, V.; Vancurova, I. Evaluating cytoplasmic and nuclear levels of inflammatory cytokines in cancer cells by western blotting. In Cytokine Bioassays; Springer: New York, NY, USA, 2014; pp. 271–283. [Google Scholar]
- Uddin, M.M.; Zou, Y.; Sharma, T.; Gatla, H.R.; Vancurova, I. Proteasome inhibition induces IKK-dependent interleukin-8 expression in triple negative breast cancer cells: Opportunity for combination therapy. PLoS ONE 2018, 13, e0201858. [Google Scholar] [CrossRef] [PubMed]
- Gatla, H.R.; Zou, Y.; Uddin, M.M.; Singha, B.; Bu, P.; Vancura, A.; Vancurova, I. Histone deacetylase (HDAC) inhibition induces IκB kinase (IKK)-dependent interleukin-8/CXCL8 expression in ovarian cancer cells. J. Biol. Chem. 2017, 292, 5043–5054. [Google Scholar] [CrossRef] [PubMed]
- Vancurova, I.; Gatla, H.R.; Vancura, A. HDAC/IKK inhibition therapies in solid tumors. Oncotarget 2017, 8, 34030–34031. [Google Scholar] [CrossRef] [PubMed]
- Chavey, C.; Muhlbauer, M.; Bossard, C.; Freund, A.; Durand, S.; Jorgensen, C.; Jobin, C.; Lazennec, G. Interleukin-8 expression is regulated by histone deacetylases through the NF-κB pathway in breast cancer. Mol. Pharmacol. 2008, 74, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Gatla, H.R.; Zou, Y.; Uddin, M.M.; Vancurova, I. Epigenetic regulation of interleukin-8 expression by class I HDAC and CBP in ovarian cancer cells. Oncotarget 2017, 8, 70798–70810. [Google Scholar] [CrossRef] [PubMed]
- Bartling, T.R.; Drumm, M.L. Loss of CFTR results in reduction of histone deacetylase 2 in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L35–L43. [Google Scholar] [CrossRef] [PubMed]
- Angiolilli, C.; Kabala, P.A.; Grabiec, A.M.; Van Baarsen, I.M.; Ferguson, B.S.; García, S.; Fernandez, B.M.; McKinsey, T.A.; Tak, P.P.; Fossati, G.; et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann. Rheum. Dis. 2017, 76, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Angiolilli, C.; Grabiec, A.M.; Ferguson, B.S.; Ospelt, C.; Fernandez, B.M.; van Es, I.E.; van Baarsen, L.G.; Gay, S.; McKinsey, T.A.; Tak, P.P.; et al. Inflammatory cytokines epigenetically regulate rheumatoid arthritis fibroblast-like synoviocyte activation by suppressing HDAC5 expression. Ann. Rheum. Dis. 2016, 75, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Bosisio, D.; Vulcano, M.; Prete, A.; Sironi, M.; Salvi, V.; Salogni, L.; Riboldi, E.; Leoni, F.; Dinarello, C.A.; Girolomoni, G.; et al. Blocking TH17-polarizing cytokines by histone deacetylase inhibitors in vitro and in vivo. J. Leukoc. Biol. 2008, 84, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Y.; Wu, J.; Roizman, B.; Zhou, G.G. Innate responses to gene knockouts impact overlapping gene networks and vary with respect to resistance to viral infection. Proc. Natl. Acad. Sci. USA 2018, 115, E3230–E3237. [Google Scholar] [CrossRef] [PubMed]
- Coward, W.R.; Watts, K.; Feghali-Bostwick, C.A.; Jenkins, G.; Pang, L. Repression of IP-10 by interactions between histone deacetylation and hypermethylation in idiopathic pulmonary fibrosis. Mol. Cell. Biol. 2010, 30, 2874–2886. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Bhasin, M.; Libermann, T.; Barth, S.; Michaelis, M.; Cinatl, J.; Jones, J.; Hudak, L.; Jonas, D.; Blaheta, R.A. Alterations of the gene expression profile in renal cell carcinoma after treatment with the histone deacetylase-inhibitor valproic acid and interferon-alpha. World J. Urol. 2011, 29, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-J.; Li, Q.-L.; Zhang, J.; Huang, A.-L. Histone deacetylation is involved in activation of CXCL10 upon IFNgamma stimulation. Mol. Cells 2006, 22, 163–167. [Google Scholar] [PubMed]
- Orecchia, A.; Scarponi, C.; Di Felice, F.; Cesarini, E.; Avitabile, S.; Mai, A.; Mauro, M.L.; Sirri, V.; Zambruno, G.; Albanesi, C.; et al. Sirtinol treatment reduces inflammation in human dermal microvascular endothelial cells. PLoS ONE 2011, 6, e24307. [Google Scholar] [CrossRef]
- Romain, B.; Benbrika-Nehmar, R.; Marisa, L.; Legrain, M.; Lobstein, V.; Oravecz, A.; Poidevin, L.; Bour, C.; Freund, J.-N.; Duluc, I.; et al. Histone hypoacetylation contributes to CXCL12 downregulation in colon cancer: Impact on tumor growth and cell migration. Oncotarget 2017, 8, 38351–38366. [Google Scholar] [CrossRef] [PubMed]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Gloghini, A.; Buglio, D.; Khaskhely, N.M.; Georgakis, G.; Orlowski, R.Z.; Neelapu, S.S.; Carbone, A.; Younes, A. Expression of histone deacetylases in lymphoma: Implication for the development of selective inhibitors. Br. J. Haematol. 2009, 147, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC inhibitor: From cancer to cardiovascular diseases. Chonnam Med J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.J.; Demierre, M.-F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Foss, F.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Belt, R.J.; Jacobsen, E.D.; et al. A Phase II trial of Belinostat (PXD 101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 2015, 168, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the treatment of multiple myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Biophys. Acta 2018. [Google Scholar] [CrossRef] [PubMed]
- Wightman, F.; Lu, H.K.; Solomon, A.E.; Saleh, S.; Harman, A.N.; Cunningham, A.L.; Gray, L.; Churchill, M.; Cameron, P.U.; Dear, A.E.; et al. Entinostat is a histone deacetylase inhibitor selective for class 1 histone deacetylases and activates HIV production from latently infected primary T cells. AIDS 2013, 27, 2853–2863. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.K.; Gray, L.R.; Wightman, F.; Ellenberg, P.; Khoury, G.; Cheng, W.-J.; Mota, T.M.; Wesselingh, S.; Gorry, P.R.; Cameron, P.U.; et al. Ex vivo response to histone deacetylase (HDAC) inhibitors of the HIV long terminal repeat (LTR) derived from HIV-infected patients on antiretroviral therapy. PLoS ONE 2014, 9, e113341. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Procopio, F.A.; Ruggiero, A.; Noto, A.; Ohmiti, K.; Cavassini, M.; Corpataux, J.-M.; Paxton, W.A.; Pollakis, G.; Perreau, M. Blood CXCR3+ CD4 T Cells Are Enriched in Inducible Replication Competent HIV in Aviremic Antiretroviral Therapy-Treated Individuals. Front. Immunol. 2018, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Marsden, M.D.; Zack, J.A. Experimental approaches for eliminating latent HIV. In Forum on Immunopathological Diseases and Therapeutics; Begel House Inc.: Danbury, CT, USA, 2015. [Google Scholar]
- Margolis, D.M. Histone deacetylase inhibitors and HIV latency. Curr. Opin. Hiv Aids 2011, 6, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Lucera, M.; Tilton, C.A.; Mao, H.; Dobrowolski, C.; Tabler, C.; Haqqani, A.A.; Karn, J.; Tilton, J.C. The histone deacetylase inhibitor vorinostat (SAHA) increases the susceptibility of uninfected CD4+ T cells to HIV by increasing the kinetics and efficiency of post-entry viral events. J. Virol. 2014, 88, 10803–10812. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Kirchherr, J.L.; Sung, J.A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba, A.D.; Kuruc, J.D.; et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Investig. 2017, 127, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Tolstrup, M.; Møller, H.J.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Laursen, A.L.; Østergaard, L.; Søgaard, O.S. Activation of latent human immunodeficiency virus by the histone deacetylase inhibitor panobinostat: A pilot study to assess effects on the central nervous system. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Wollebo, H.S.; Woldemichaele, B.; Khalili, K.; Safak, M.; White, M.K. Epigenetic regulation of polyomavirus JC. Virol. J. 2013, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.-S.; Brent, M.M.; Getachew, R.; Jayakumar, P.; Chen, L.-F.; Schnolzer, M.; McBurney, M.W.; Marmorstein, R.; Greene, W.C.; Ott, M. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe 2008, 3, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.J.; Jiang, M.; Milner, J. Oncogenic viral protein HPV E7 up-regulates the SIRT1 longevity protein in human cervical cancer cells. Aging 2009, 1, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Chandra, A.; Dittrich, T.; Welte, K.; Chandra, P. Inhibition of SIRT1 by HIV-1 viral protein Tat results in activation of p53 pathway. Biochem. Biophys. Res. Commun. 2012, 424, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-B.; Jiang, H.; Cheng, S.-T.; Hu, Z.-W.; Ren, J.-H.; Chen, J. AGK2, A SIRT2 Inhibitor, Inhibits Hepatitis B Virus Replication In Vitro And In Vivo. Int. J. Med. Sci. 2018, 15, 1356. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Zhou, J.; Wiedmer, A.; Madden, K.; Yuan, Y.; Lieberman, P.M. Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 2003, 77, 11425–11435. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.S.; Avdiushko, S.A.; Kryscio, R.J.; Danaher, R.J.; Jacob, R.J. Effect of prophylactic valacyclovir on the presence of human herpesvirus DNA in saliva of healthy individuals after dental treatment. J. Clin. Microbiol. 2005, 43, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, J.; Sissons, P. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Kaufmann, J.K.; Wang, P.-Y.; Nguyen, T.; Speranza, M.-C.; Kasai, K.; Okemoto, K.; Otsuki, A.; Nakano, I.; Fernandez, S.; et al. Histone deacetylase 6 inhibition enhances oncolytic viral replication in glioma. J. Clin. Investig. 2015, 125, 4269–4280. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, E.; Budayeva, H.G.; Miteva, Y.V.; Ricci, D.P.; Silhavy, T.J.; Shenk, T.; Cristea, I.M. Sirtuins are evolutionarily conserved viral restriction factors. mBio 2014, 5, e02249-14. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Cheung, C.-Y. Histone deacetylase 6 inhibits influenza A virus release by downregulating the trafficking of viral components to the plasma membrane via its substrate acetylated microtubules. J. Virol. 2014, 88, 11229–11239. [Google Scholar] [CrossRef] [PubMed]
- Nagesh, P.T.; Husain, M. Influenza A virus dysregulates host histone deacetylase 1 that inhibits viral infection in lung epithelial cells. J. Virol. 2016, 90, 4614–4625. [Google Scholar] [CrossRef] [PubMed]
- Nagesh, P.T.; Hussain, M.; Galvin, H.D.; Husain, M. Histone Deacetylase 2 Is a Component of Influenza A Virus-Induced Host Antiviral Response. Front. Microbiol. 2017, 8, 1315. [Google Scholar] [CrossRef] [PubMed]
- Delgado, F.; Cárdenas, P.; Castellanos, J. Valproic Acid Downregulates Cytokine Expression in Human Macrophages Infected with Dengue Virus. Diseases 2018, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.; Roe, K.; Orillo, B.; Shi, P.-Y.; Verma, S. Combined treatment of adenosine nucleoside inhibitor NITD008 and histone deacetylase inhibitor vorinostat represents an immunotherapy strategy to ameliorate West Nile virus infection. Antivir. Res. 2015, 122, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Pagidipati, N.J.; Gaziano, T.A. Estimating deaths from cardiovascular disease: A review of global methodologies of mortality measurement. Circulation 2013, 127, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Sannino, A.; Toscano, E.; Cattaneo, F.; Trimarco, B.; Esposito, G.; Perrino, C. Cardiovascular effects of histone deacetylase inhibitors epigenetic therapies: Systematic review of 62 studies and new hypotheses for future research. Int. J. Cardiol. 2016, 219, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Olson, E. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [PubMed]
- Morales, C.R.; Li, D.L.; Pedrozo, Z.; May, H.I.; Jiang, N.; Kyrychenko, V.; Cho, G.W.; Kim, S.Y.; Wang, Z.V.; Rotter, D.; et al. Inhibition of class I histone deacetylases blunts cardiac hypertrophy through TSC2-dependent mTOR repression. Sci. Signal. 2016, 9, ra34. [Google Scholar] [CrossRef] [PubMed]
- Ooi, J.Y.; Tuano, N.K.; Rafehi, H.; Gao, X.-M.; Ziemann, M.; Du, X.-J.; El-Osta, A. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics 2015, 10, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Bae, E.H.; Park, S.; Lee, K.E.; Suh, S.H.; Kim, S.W.; Jeong, M.H. HDAC inhibition suppresses cardiac hypertrophy and fibrosis in DOCA-salt hypertensive rats via regulation of HDAC6/HDAC8 enzyme activity. Kidney Blood Press. Res. 2013, 37, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Rawal, S.; Manning, P.; Katare, R. Cardiovascular microRNAs: As modulators and diagnostic biomarkers of diabetic heart disease. Cardiovasc. Diabetol. 2014, 13, 44. [Google Scholar] [CrossRef] [PubMed]
- Rawal, S.; Munasinghe, P.E.; Thevakar, P.; Lew, J.K.S.; Jones, G.T.; Willams, M.J.; Davis, P.; Bunton, R.W.; Galvin, I.F.; Manning, P.; et al. Downregulation of miR-15a/b accelerates fibrotic remodelling in the type-2 diabetic human and mouse heart. Clin. Sci. 2017, 131, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Milan, M.; Pace, V.; Maiullari, F.; Chirivì, M.; Baci, D.; Maiullari, S.; Madaro, L.; Maccari, S.; Stati, T.; Marano, G.; et al. Givinostat reduces adverse cardiac remodeling through regulating fibroblasts activation. Cell Death Dis. 2018, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Du, J.; Zhao, Y.T.; Zhang, L.; Lv, G.; Zhuang, S.; Qin, G.; Zhao, T.C. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc. Diabetol. 2015, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Hill, J.A. Inhibition of Hypertrophy Is a Good Therapeutic Strategy in Ventricular Pressure OverloadResponse to Schiattarella and Hill. Circulation 2015, 131, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Nural-Guvener, H.F.; Zakharova, L.; Nimlos, J.; Popovic, S.; Mastroeni, D.; Gaballa, M.A. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair 2014, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-M.; Lin, M.-S.; Chang, N.-C. Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H968–H977. [Google Scholar] [CrossRef] [PubMed]
- Granger, A.; Abdullah, I.; Huebner, F.; Stout, A.; Wang, T.; Huebner, T.; Epstein, J.A.; Gruber, P.J. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008, 22, 3549–3560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, B.; Zhao, Y.; Dubielecka-Szczerba, P.; Wei, L.; Qin, G.J.; Chin, E.Y.; Wang, Y.; Zhao, T.C. Inhibition of histone deacetylases-induced myocardial repair is mediated by c-kit in infarcted hearts. J. Biol. Chem. 2012, 287, 39338–39348. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-X.; Zhou, T.; Wang, X.-A.; Tong, X.-H.; Ding, J.-W. Histone deacetylases and atherosclerosis. Atherosclerosis 2015, 240, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.-G. Cardiac arrhythmias: Diagnosis, symptoms, and treatments. Cell Biochem. Biophys. 2015, 73, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Kook, H.; Lepore, J.J.; Gitler, A.D.; Lu, M.M.; Yung, W.W.-M.; Mackay, J.; Zhou, R.; Ferrari, V.; Gruber, P.; Epstein, J.A. Cardiac hypertrophy and histone deacetylase–dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J. Clin. Investig. 2003, 112, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Regna, N.; Reillya, C. Isoform-selective HDAC inhibition in autoimmune disease. J. Clin. Cell Immunol. 2014, 5, 206–213. [Google Scholar]
- Chung, Y.-L.; Lee, M.-Y.; Wang, A.-J.; Yao, L.-F. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol. Ther. 2003, 8, 707–717. [Google Scholar] [CrossRef]
- Chen, H.; Pan, J.; Wang, J.-D.; Liao, Q.-M.; Xia, X.-R. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, induces apoptosis in rheumatoid arthritis fibroblast-like synoviocytes. Inflammation 2016, 39, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, B. Trichostatin A, an Inhibitor of Histone Deacetylase, Inhibits the Viability and Invasiveness of Hypoxic Rheumatoid Arthritis Fibroblast-Like Synoviocytes via PI3K/Akt Signaling. J. Biochem. Mol. Toxicol. 2016, 30, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.R.; Suh, D.-H.; Bae, D.; Ha, N.; Choi, Y.I.; Yoo, H.J.; Park, J.K.; Lee, E.Y.; Lee, E.B.; Song, Y.W. Therapeutic effect of a novel histone deacetylase 6 inhibitor, CKD-L, on collagen-induced arthritis in vivo and regulatory T cells in rheumatoid arthritis in vitro. Arthritis Res. Ther. 2017, 19, 154. [Google Scholar] [CrossRef] [PubMed]
- Angiolilli, C.; Kabala, P.A.; Grabiec, A.M.; Rossato, M.; Lai, W.S.; Fossati, G.; Mascagni, P.; Steinkühler, C.; Blackshear, P.J.; Reedquist, K.A.; et al. Control of cytokine mRNA degradation by the histone deacetylase inhibitor ITF2357 in rheumatoid arthritis fibroblast-like synoviocytes: Beyond transcriptional regulation. Arthritis Res. Ther. 2018, 20, 148. [Google Scholar] [CrossRef] [PubMed]
- Grabiec, A.M.; Korchynskyi, O.; Tak, P.P.; Reedquist, K.A. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann. Rheum. Dis. 2012, 71, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Peedicayil, J. Epigenetic drugs for multiple sclerosis. Curr. Neuropharmacol. 2016, 14, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Camelo, S.; Iglesias, A.H.; Hwang, D.; Due, B.; Ryu, H.; Smith, K.; Gray, S.G.; Imitola, J.; Duran, G.; Assaf, B.; et al. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 164, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, S.; Polak, P.E.; Lin, S.X.; Braun, D.; Guizzetti, M.; Zhang, X.; Rubinstein, I.; Feinstein, D.L. Dimethyl fumarate regulates histone deacetylase expression in astrocytes. J. Neuroimmunol. 2013, 263, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Da, Y.; Xue, Z.; Zhang, K.; Zhuang, H.; Peng, M.; Li, Y.; Li, W.; Simard, A.; Hao, J.; et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp. Neurol. 2013, 241, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Felice, C.; Lewis, A.; Armuzzi, A.; Lindsay, J.; Silver, A. selective histone deacetylase isoforms as potential therapeutic targets in inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2015, 41, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.N.; Choijookhuu, N.; Takagi, H.; Srisowanna, N.; Huynh, M.N.N.; Yamaguchi, Y.; Oo, P.S.; Kyaw, M.T.H.; Sato, K.; Yamaguchi, R.; et al. The HDAC Inhibitor, SAHA, Prevents Colonic Inflammation by Suppressing Pro-inflammatory Cytokines and Chemokines in DSS-induced Colitis. Acta Histochem. Cytochem. 2018, 51, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Glauben, R.; Batra, A.; Stroh, T.; Erben, U.; Fedke, I.; Lehr, H.A.; Leoni, F.; Mascagni, P.; Dinarello, C.A.; Zeitz, M.; et al. Histone deacetylases: Novel targets for prevention of colitis-associated cancer in mice. Gut 2008, 57, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, R.; Xu, H.; Song, Y.; Qi, Y. A Highly Potent and Selective Histone Deacetylase 6 Inhibitor Prevents DSS-Induced Colitis in Mice. Biol. Pharm. Bull. 2017, 40, 936–940. [Google Scholar] [CrossRef] [PubMed]
- De Zoeten, E.F.; Wang, L.; Sai, H.; Dillmann, W.H.; Hancock, W.W. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 2010, 138, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Reilly, C.M.; Regna, N.; Mishra, N. HDAC inhibition in lupus models. Mol. Med. 2011, 17, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Regna, N.L.; Vieson, M.D.; Gojmerac, A.M.; Luo, X.M.; Caudell, D.L.; Reilly, C.M. HDAC expression and activity is upregulated in diseased lupus-prone mice. Int. Immunopharmacol. 2015, 29, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Panther, E.; Liao, X.; Grammer, A.; Lipsky, P.; Reilly, C. The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2018, 19, 4007. [Google Scholar] [CrossRef] [PubMed]
- Regna, N.L.; Vieson, M.D.; Luo, X.M.; Chafin, C.B.; Puthiyaveetil, A.G.; Hammond, S.E.; Caudell, D.L.; Jarpe, M.B.; Reilly, C.M. Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in NZB/W mice. Clin. Immunol. 2016, 162, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Hadden, M.; Advani, A. Histone deacetylase inhibitors and diabetic kidney disease. Int. J. Mol. Sci. 2018, 19, 2630. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Yoshimura, K.; Shin, T.; Verheul, H.; Hammers, H.; Sanni, T.B.; Salumbides, B.C.; Van Erp, K.; Schulick, R.; Pili, R. Synergistic in vivo antitumor effect of the histone deacetylase inhibitor MS-275 in combination with interleukin 2 in a murine model of renal cell carcinoma. Clin. Cancer Res. 2007, 13, 4538–4546. [Google Scholar] [CrossRef] [PubMed]
- Pili, R.; Quinn, D.I.; Hammers, H.J.; Monk, P.; George, S.; Dorff, T.B.; Olencki, T.; Shen, L.; Orillion, A.; Lamonica, D.; et al. Immunomodulation by entinostat in renal cell carcinoma patients receiving high-dose interleukin 2: A multicenter, single-arm, phase I/II trial (NCI-CTEP# 7870). Clin. Cancer Res. 2017, 23, 7199–7208. [Google Scholar] [PubMed]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Thomas, S.; Pawlowska, N.; Bartelink, I.; Grabowsky, J.; Jahan, T.; Cripps, A.; Harb, A.; Leng, J.; Reinert, A.; et al. Inhibiting histone deacetylase as a means to reverse resistance to angiogenesis inhibitors: Phase I study of abexinostat plus pazopanib in advanced solid tumor malignancies. J. Clin. Oncol. 2017, 35, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Pawlowsk, N.; Grabowsky, J.; Harb, A.; Abri-Lavasani, K.; Thomas, S.; Munster, P.N. Abstract A173: Long-Term Responders to Epigenetic Modulators: Abexinostat and Pazopanib; AACR: Philadelphia, PA, USA, 2018. [Google Scholar]


{kind=link}
| HDAC Inhibitor | Synonym | Specificity | Clinical Status |
|---|---|---|---|
| Vorinostat | SAHA | pan-HDAC | FDA approved for cutaneous T-cell lymphoma (CTCL) |
| Belinostat | PXD101 | FDA approved for peripheral T-cell lymphoma (PTCL) | |
| Panobinostat | LBH589 | FDA approved for multiple myeloma | |
| Trichostatin A | TSA | Not tested | |
| Givinostat | ITF2357 | Phase II—in combination With Hydroxyurea in Polycythemia Vera (NCT00928707), phase II—chronic myeloproliferative neoplasms (NCT01761968), phase II/III—duchenne’s muscular dystrophy (NCT03373968) and phase II—juvenile idiopathic arthritis (NCT01261624) | |
| Resminostat | RAS2410, 4SC-201 | Phase II—in combination with sorafenib in advanced hepatocellular carcinoma (NCT00943449 and NCT02400788), phase I/II—advanced colorectal carcinoma (NCT01277406) and phase II—refractory Hodgkin’s lymphoma (NCT01037478) and phase II—sézary syndrome (NCT02953301) | |
| Quisinostat | JNJ-26481585 | Phase II—combination with chemotherapy in ovarian cancer (NCT02948075) and phase II—cutaneous T-cell lymphoma (NCT01486277) | |
| Abexinostat | PCI-24781 | Phase I/II—in combination With doxorubicin to treat sarcoma (NCT01027910), phase II—relapsed/refractory follicular lymphoma (NCT03600441) and phase III—combination with pazopanib in metastatic renal cell carcinoma (NCT03592472) | |
| Romidepsin | FK228, desipeptide | Class I | FDA approved for PTCL and CTCL |
| CHR-3996 | Phase I/II—EBV-associated lymphoid malignancies (NCT03397706) | ||
| Entinostat | MS-275 | Phase II—in combination with azacitidine in breast cancer, colorectal cancer, chronic myelomonocytic leukemia or acute myeloid leukemia and non-small cell lung cancer (NCT01349959, NCT01105377, NCT00313586, NCT00387465), phase II—metastatic melanoma (NCT00185302), phase II—refractory Hodgkin’s lymphoma (NCT00866333), phase II—non-small cell lung cancer (NCT00750698) and phase III—in combination with exemestane in ER/PR+ breast cancer (NCT02115282) | |
| Tacedinaline | CI994 | Phase III—in combination with gemcitabine in non-small cell lung cancer (NCT00005093), phase II—in combination with gemcitabine in pancreatic cancer (NCT00004861) and phase II—advanced myeloma (NCT00005624) | |
| Domatinostat | 4SC202 | Phase I/II—advanced melanoma, non-responders to anti-PD-1 therapy (NCT03278665) | |
| Ricolinostat | ACY-1215 | Class II | Phase I/II—with various combination in multiple myeloma (NCT01997840, NCT01583283 and NCT01323751) |
| Valproic acid | VPA | Classes I and IIa | FDA approved for seizures and manic-depressive disorders |
| Mocetinostat | MGCD0103 | Classes I and IV | Phase II—urothelial carcinoma (NCT02236195), phase II—combination with gemcitabine in metastatic leiomyosarcoma (NCT02303262), phase II—relapsed and refractory lymphoma (NCT00359086), phase II—refractory chronic lymphocytic leukemia (NCT00431873) and phase II—combination with myelodysplastic syndrome or acute myelogenous leukemia (NCT00324220) |
| Pracinostat | SB939 | Classes I, II and IV | Phase II—in combination with ruxolitinib in myelofibrosis (NCT02267278), phase II—myelodysplastic syndrome (NCT01993641), phase II—combination with azacitidine in myelodysplastic syndrome (NCT01873703), phase II—combination with azacitidine in acute myeloid leukemia (NCT01912274), phase II—in patients myelofibrosis (NCT01200498) and phase II—metastatic sarcomas (NCT01112384) |
| Sirtuin inhibitors | SIRT family | Not tested |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatla, H.R.; Muniraj, N.; Thevkar, P.; Yavvari, S.; Sukhavasi, S.; Makena, M.R. Regulation of Chemokines and Cytokines by Histone Deacetylases and an Update on Histone Decetylase Inhibitors in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1110. https://doi.org/10.3390/ijms20051110
Gatla HR, Muniraj N, Thevkar P, Yavvari S, Sukhavasi S, Makena MR. Regulation of Chemokines and Cytokines by Histone Deacetylases and an Update on Histone Decetylase Inhibitors in Human Diseases. International Journal of Molecular Sciences. 2019; 20(5):1110. https://doi.org/10.3390/ijms20051110
Chicago/Turabian StyleGatla, Himavanth Reddy, Nethaji Muniraj, Prashanth Thevkar, Siddhartha Yavvari, Sahithi Sukhavasi, and Monish Ram Makena. 2019. "Regulation of Chemokines and Cytokines by Histone Deacetylases and an Update on Histone Decetylase Inhibitors in Human Diseases" International Journal of Molecular Sciences 20, no. 5: 1110. https://doi.org/10.3390/ijms20051110
APA StyleGatla, H. R., Muniraj, N., Thevkar, P., Yavvari, S., Sukhavasi, S., & Makena, M. R. (2019). Regulation of Chemokines and Cytokines by Histone Deacetylases and an Update on Histone Decetylase Inhibitors in Human Diseases. International Journal of Molecular Sciences, 20(5), 1110. https://doi.org/10.3390/ijms20051110