The HslV Protease from Leishmania major and Its Activation by C-terminal HslU Peptides

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Expression and Purification of Recombinant LmHslV
2.2. LmHslV Is a Dodecameric and Inactive Protease that Can Be Activated by Peptides Corresponding to the C-terminal End of LmHslU1 and U2
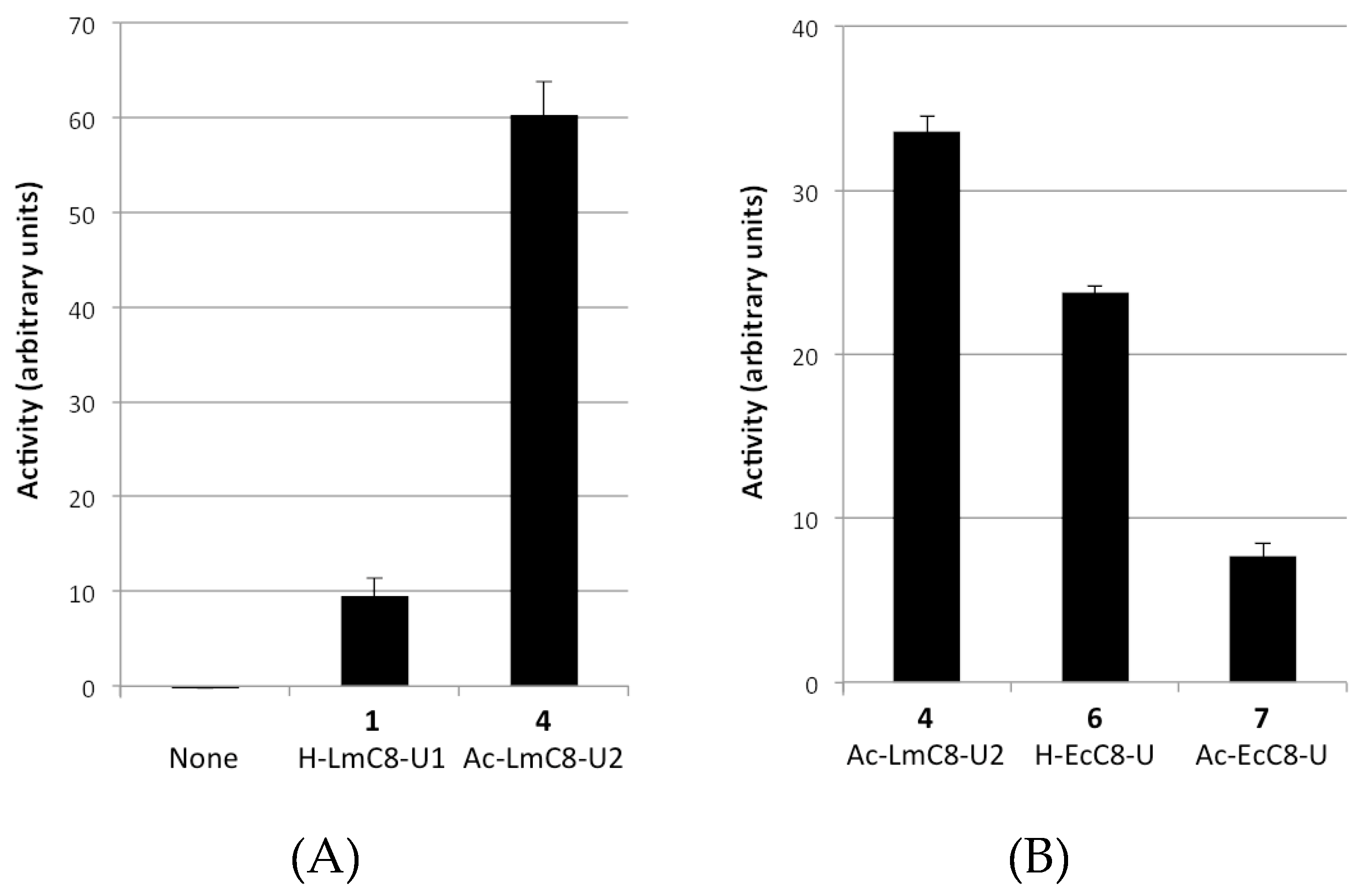
2.2.1. Peptides Derived from the C-terminal Ends of LmHslU1 and U2 Can Activate LmHslV
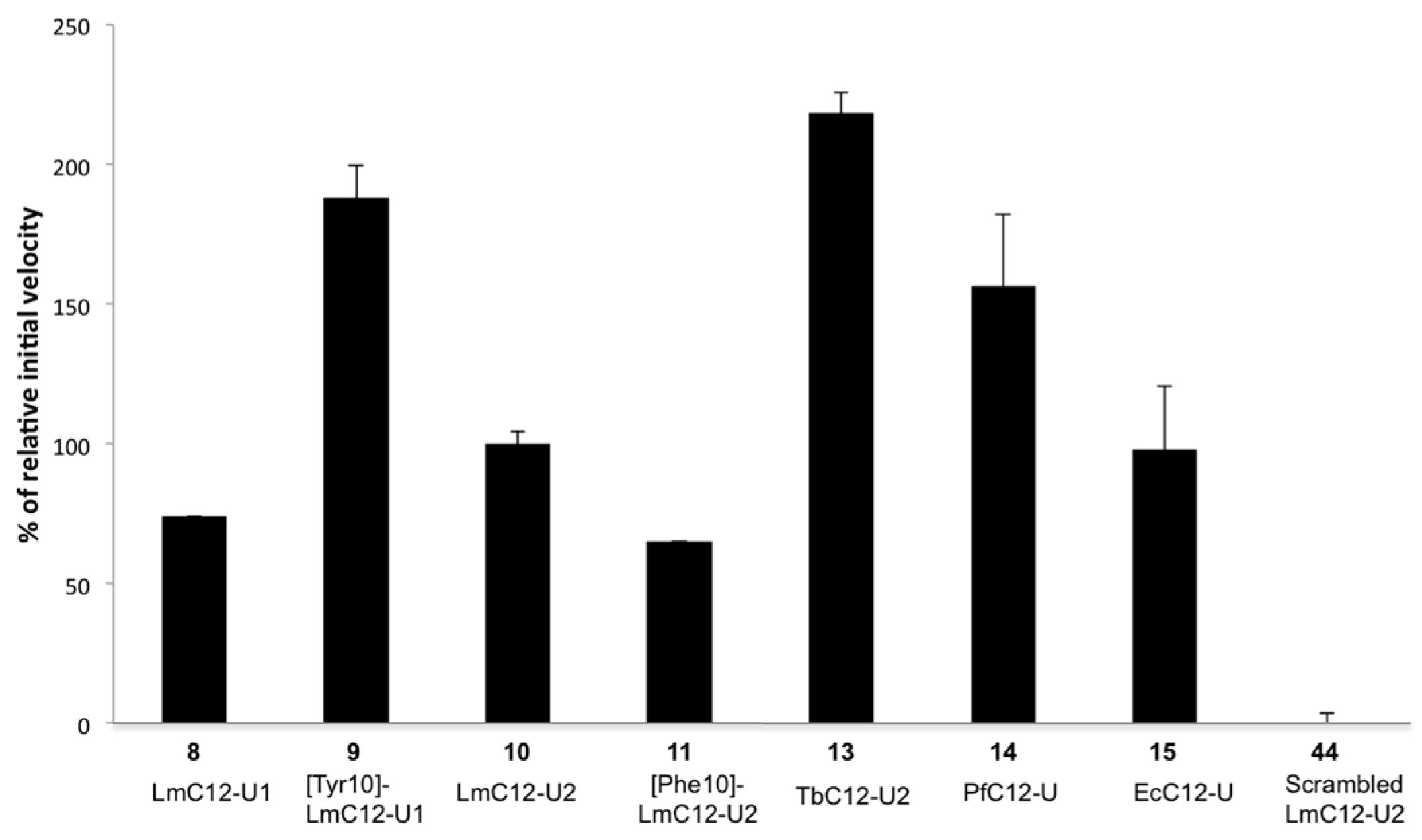
2.2.2. Sequence Requirements for Activation of LmHslV by C-terminal HslU Peptides
2.2.3. Structural Requirements for Activation of LmHslV by C-terminal LmHslU2 Peptides
2.3. LmHslV Activation by LmC12-U2 Is Accompanied by a Conformational Rearrangement of the Dodecamer
3. Discussion
3.1. Structural Requirements for Activation of LmHslV by C-terminal LmHslU2 Peptides
3.2. Specific Roles of HslU1 and HslU2
3.3. Conformational Rearrangement upon LmHslV Activation
3.4. Drugging HslVU
4. Materials and Methods
4.1. Material
4.2. Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAA | ATPases Associated with diverse cellular Activities |
| AMC | 7-amino-4-methylcoumarin |
| CD | Circular dichroism |
| DCM | Dichloromethane |
| DIEA | Diisopropylethylamine |
| DMF | Dimethylformamide |
| EM | Electronic microscopy |
| Fmoc | Fluorenylmethyloxycarbonyl |
| HBTU | N-[(1H-benzotriazol-1-yloxy) (dimethylamino)methylene]- N-methylmethanaminium hexafluorophosphate |
| NMP | N-methylpyrrolidone |
| O2Oc | [2-(2-aminoethoxy)ethoxy]acetyl -NH(CH2)2O(CH2)2OCH2CO- |
| Suc | Succinyl |
| TFE | Trifluoroethanol |
| TIS | Triisopropylsilane |
| Z | Benzyloxycarbonyl |
References
- Sauer, R.T.; Baker, T.A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2015, 41, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Missiakas, D.; Schwager, F.; Betton, J.M.; Georgopoulos, C.; Raina, S. Identification and characterization of HsIV HsIU (ClpQ ClpY) proteins involved in overall proteolysis of misfolded proteins in Escherichia coli. EMBO J. 1996, 15, 6899–6909. [Google Scholar] [CrossRef] [PubMed]
- Rohrwild, M.; Coux, O.; Huang, H.C.; Moerschell, R.P.; Yoo, S.J.; Seol, J.H.; Chung, C.H.; Goldberg, A.L. HslV-HslU: A novel ATP-dependent protease complex in Escherichia coli related to the eukaryotic proteasome. Proc. Natl. Acad. Sci. USA 1996, 93, 5808–5813. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.J.; Seol, J.H.; Shin, D.H.; Rohrwild, M.; Kang, M.S.; Tanaka, K.; Goldberg, A.L.; Chung, C.H. Purification and characterization of the heat shock proteins HslV and HslU that form a new ATP-dependent protease in Escherichia coli. J. Biol. Chem. 1996, 271, 14035–14040. [Google Scholar] [CrossRef] [PubMed]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar] [PubMed]
- Sousa, M.C.; Trame, C.B.; Tsuruta, H.; Wilbanks, S.M.; Reddy, V.S.; McKay, D.B. Crystal and solution structures of an HslUV protease-chaperone complex. Cell 2000, 103, 633–643. [Google Scholar] [CrossRef]
- Wang, J.; Song, J.J.; Seong, I.S.; Franklin, M.C.; Kamtekar, S.; Eom, S.H.; Chung, C.H. Nucleotide-dependent conformational changes in a protease-associated ATPase HsIU. Structure 2001, 9, 1107–1116. [Google Scholar] [CrossRef]
- Yakamavich, J.A.; Baker, T.A.; Sauer, R.T. Asymmetric nucleotide transactions of the HslUV protease. J. Mol. Biol. 2008, 380, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, J.J.; Franklin, M.C.; Kamtekar, S.; Im, Y.J.; Rho, S.H.; Seong, I.S.; Lee, C.S.; Chung, C.H.; Eom, S.H. Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure 2001, 9, 177–184. [Google Scholar] [CrossRef]
- Park, E.; Lee, J.W.; Yoo, H.M.; Ha, B.H.; An, J.Y.; Jeon, Y.J.; Seol, J.H.; Eom, S.H.; Chung, C.H. Structural Alteration in the Pore Motif of the Bacterial 20S Proteasome Homolog HslV Leads to Uncontrolled Protein Degradation. J. Mol. Biol. 2013, 425, 2940–2954. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, M.; Hartmann, C.; Song, H.K.; Bourenkov, G.P.; Bartunik, H.D.; Huber, R. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature 2000, 403, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.C.; Kessler, B.M.; Overkleeft, H.S.; McKay, D.B. Crystal structure of HslUV complexed with a vinyl sulfone inhibitor: corroboration of a proposed mechanism of allosteric activation of HslV by HslU. J. Mol. Biol. 2002, 318, 779–785. [Google Scholar] [CrossRef]
- Ramachandran, R.; Hartmann, C.; Song, H.K.; Huber, R.; Bochtler, M. Functional interactions of HslV (ClpQ) with the ATPase HslU (ClpY). Proc. Natl. Acad. Sci. USA 2002, 99, 7396–7401. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.E.; Burland, V.; Plunkett, G., 3rd; Daniels, D.L.; Blattner, F.R. Sequence analysis of four new heat-shock genes constituting the hslTS/ibpAB and hslVU operons in Escherichia coli. Gene 1993, 134, 1–6. [Google Scholar] [CrossRef]
- Bochtler, M.; Ditzel, L.; Groll, M.; Huber, R. Crystal structure of heat shock locus V (HslV) from Escherichia coli. Proc. Natl. Acad. Sci. USA 1997, 94, 6070–6074. [Google Scholar] [CrossRef] [PubMed]
- Couvreur, B.; Wattiez, R.; Bollen, A.; Falmagne, P.; Le Ray, D.; Dujardin, J.-C. Eubacterial HslV and HslU subunits homologs in primordial eukaryotes. Mol. Biol. Evol. 2002, 19, 2110–2117. [Google Scholar] [CrossRef] [PubMed]
- Gille, C.; Goede, A.; Schlöetelburg, C.; Preissner, R.; Kloetzel, P.M.; Göbel, U.B.; Frömmel, C. A comprehensive view on proteasomal sequences: implications for the evolution of the proteasome. J. Mol. Biol. 2003, 326, 1437–1448. [Google Scholar] [CrossRef]
- Ruiz-González, M.X.; Marín, I. Proteasome-related HslU and HslV genes typical of eubacteria are widespread in eukaryotes. J. Mol. Evol. 2006, 63, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Bochtler, M.; Brandstetter, H.; Clausen, T.; Huber, R. Molecular machines for protein degradation. Chembiochem 2005, 6, 222–256. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lindsay, M.E.; Motyka, S.A.; Englund, P.T.; Wang, C.C. Identification of a bacterial-like HslVU protease in the mitochondria of Trypanosoma brucei and its role in mitochondrial DNA replication. PLoS Pathog. 2008, 4, e1000048. [Google Scholar] [CrossRef] [PubMed]
- Rathore, S.; Jain, S.; Sinha, D.; Gupta, M.; Asad, M.; Srivastava, A.; Narayanan, M.S.; Ramasamy, G.; Chauhan, V.S.; Gupta, D.; et al. Disruption of a mitochondrial protease machinery in Plasmodium falciparum is an intrinsic signal for parasite cell death. Cell Death Dis. 2011, 2, e231. [Google Scholar] [CrossRef] [PubMed]
- Chrobak, M.; Förster, S.; Meisel, S.; Pfefferkorn, R.; Förster, F.; Clos, J. Leishmania donovani HslV does not interact stably with HslU proteins. Int. J. Parasitol. 2012, 42, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Rathore, S.; Asad, M.; Hossain, M.E.; Sinha, D.; Datta, G.; Mohmmed, A. The prokaryotic ClpQ protease plays a key role in growth and development of mitochondria in Plasmodium falciparum. Cell. Microbiol. 2013, 15, 1660–1673. [Google Scholar] [PubMed]
- Mbang-Benet, D.-E.; Sterkers, Y.; Morelle, C.; Kebe, N.-M.; Crobu, L.; Portalès, P.; Coux, O.; Hernandez, J.-F.; Meghamla, S.; Pagès, M.; et al. The bacterial-like HslVU protease complex subunits are involved in the control of different cell cycle events in trypanosomatids. Acta Trop. 2014, 131, 22–31. [Google Scholar] [CrossRef] [PubMed]
- de Bettignies, G.; Coux, O. Proteasome inhibitors: Dozens of molecules and still counting. Biochimie 2010, 92, 1530–1545. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Li, D.; de Carvalho, L.P.S.; Deng, H.; Tao, H.; Vogt, G.; Wu, K.; Schneider, J.; Chidawanyika, T.; Warren, J.D.; et al. Inhibitors selective for mycobacterial versus human proteasomes. Nature 2009, 461, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; O’Donoghue, A.J.; van der Linden, W.A.; Xie, S.C.; Yoo, E.; Foe, I.T.; Tilley, L.; Craik, C.S.; da Fonseca, P.C.A.; Bogyo, M. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.J.N.; Myburgh, E.; Gao, M.-Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Rashid, Y.; Kamran Azim, M.; Saify, Z.S.; Khan, K.M.; Khan, R. Small molecule activators of proteasome-related HslV peptidase. Bioorg. Med. Chem. Lett. 2012, 22, 6089–6094. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.H.; Lee, S.Y.; Song, H.K. Structural and Biochemical Analyses of the Eukaryotic Heat Shock Locus V (HslV) from Trypanosoma brucei. J. Biol. Chem. 2013, 288, 23234–23243. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.H.; Song, H.K. Insights into the molecular evolution of HslU ATPase through biochemical and mutational analyses. PLoS ONE 2014, 9, e103027. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, G.; Gupta, D.; Mohmmed, A.; Chauhan, V.S. Characterization and localization of Plasmodium falciparum homolog of prokaryotic ClpQ/HslV protease. Mol. Biochem. Parasitol. 2007, 152, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Seong, I.S.; Kang, M.S.; Choi, M.K.; Lee, J.W.; Koh, O.J.; Wang, J.; Eom, S.H.; Chung, C.H. The C-terminal tails of HslU ATPase act as a molecular switch for activation of HslV peptidase. J. Biol. Chem. 2002, 277, 25976–25982. [Google Scholar] [CrossRef] [PubMed]
- Seol, J.H.; Yoo, S.J.; Shin, D.H.; Shim, Y.K.; Kang, M.S.; Goldberg, A.L.; Chung, C.H. The heat-shock protein HslVU from Escherichia coli is a protein-activated ATPase as well as an ATP-dependent proteinase. Eur. J. Biochem. 1997, 247, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.C.; Illangasekare, M.; Woody, R.W. Circular dichroism studies of distorted alpha-helices, twisted beta-sheets, and beta turns. Biophys. Chem. 1988, 31, 77–86. [Google Scholar] [CrossRef]
- Miranda, A.; Koerber, S.C.; Gulyas, J.; Lahrichi, S.L.; Craig, A.G.; Corrigan, A.; Hagler, A.; Rivier, C.; Vale, W.; Rivier, J. Conformationally restricted competitive antagonists of human/rat corticotropin-releasing factor. J. Med. Chem. 1994, 37, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.; Fasman, G.D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 1969, 8, 4108–4116. [Google Scholar] [CrossRef] [PubMed]
- Terskikh, A.V.; Le Doussal, J.M.; Crameri, R.; Fisch, I.; Mach, J.P.; Kajava, A.V. “Peptabody”: A new type of high avidity binding protein. Proc. Natl. Acad. Sci. USA 1997, 94, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. A corrected quaternary arrangement of the peptidase HslV and atpase HslU in a cocrystal structure. J. Struct. Biol. 2001, 134, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Song, H.K.; Bochtler, M.; Azim, M.K.; Hartmann, C.; Huber, R.; Ramachandran, R. Isolation and characterization of the prokaryotic proteasome homolog HslVU (ClpQY) from Thermotoga maritima and the crystal structure of HslV. Biophys. Chem. 2003, 100, 437–452. [Google Scholar] [CrossRef]
- Shi, L.; Kay, L.E. Tracing an allosteric pathway regulating the activity of the HslV protease. PNAS 2014, 111, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Peng, L.; Baldwin, P.R.; Mann, D.S.; Jiang, W.; Rees, I.; Ludtke, S.J. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007, 157, 38–46. [Google Scholar] [CrossRef] [PubMed]
- van Heel, M.; Harauz, G.; Orlova, E.V.; Schmidt, R.; Schatz, M. A new generation of the IMAGIC image processing system. J. Struct. Biol. 1996, 116, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bron, P.; Giudice, E.; Rolland, J.-P.; Buey, R.M.; Barbier, P.; Díaz, J.F.; Peyrot, V.; Thomas, D.; Garnier, C. Apo-Hsp90 coexists in two open conformational states in solution. Biol. Cell 2008, 100, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016, 193, 1–12. [Google Scholar] [CrossRef] [PubMed]
- van Heel, M.; Gowen, B.; Matadeen, R.; Orlova, E.V.; Finn, R.; Pape, T.; Cohen, D.; Stark, H.; Schmidt, R.; Schatz, M.; et al. Single-particle electron cryo-microscopy: towards atomic resolution. Q. Rev. Biophys. 2000, 33, 307–369. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure |
|---|---|
| C-ter HslU: Octamers | |
| LmHslU1 | |
| (1) H-LmC8-U1 | H-Val-Asp-Ile-Lys-Lys-Phe-Ile-Leu-OH |
| (2) Ac-LmC8-U1 | Ac-Val-Asp-Ile-Lys-Lys-Phe-Ile-Leu-OH |
| LmHslU2 | |
| (3) H-LmC8-U2 | H-Val-Asn-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (4) Ac-LmC8-U2 | Ac-Val-Asn-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (5) Dimer of LmC8-U2 | 1,2,3-Triazole-1,4 = [CH2-CH2CO-(O2Oc)3-Ala-Val-Asn-Leu-Ala-Lys-Tyr-Leu-Leu-OH]2 |
| EcHslU | |
| (6) H-EcC8-U | H-Glu-Asp-Leu-Ser-Arg-Phe-Ile-Leu-OH |
| (7) Ac-EcC8-U | Ac-Glu-Asp-Leu-Ser-Arg-Phe-Ile-Leu-OH |
| C-ter HslU: Dodecamers | |
| LmHslU1 | |
| (8) LmC12-U1 | H-arg-O2Oc-Met-Val-Lys-Lys-Val-Asp-Ile-Lys-Lys-Phe-Ile-Leu-OH |
| (9) [Tyr10]-LmC12-U1 | H-arg-O2Oc-Met-Val-Lys-Lys-Val-Asp-Ile-Lys-Lys-Tyr-Ile-Leu-OH |
| LmHslU2 | |
| (10) LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (11) [Phe10]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-Lys-Phe-Leu-Leu-OH |
| (12) Dimer of LmC12-U2 | 1,2,3-Triazole-1,4 = [CH2CH2CO-(O2Oc)3-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-Lys-Tyr-Leu-Leu-OH]2 |
| C-ter HslU from other origins | |
| (13) TbC12-U2 | H-arg-O2Oc-Leu-Met-Asn-Asn-Ile-Asp-Leu-Ala-Lys-Tyr-Ile-Leu-OH |
| (14) PfC12-U | H-arg-O2Oc-Phe-Ile-Lys-Gln-Tyr-Asp-Leu-Lys-Lys-Tyr-Ile-Ile-OH |
| (15) EcC12-U | H-arg-O2Oc-Leu-Val-Ala-Asp-Glu-Asp-Leu-Ser-Arg-Phe-Ile-Leu-OH |
| Ala-scan (C-ter LmHslU2) | |
| (16) [Ala5]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Ala-Asn-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (17) [Ala6]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Ala-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (18) [Ala7]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Ala-Ala-Lys-Tyr-Leu-Leu-OH |
| (19) [Ala9]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-Ala-Tyr-Leu-Leu-OH |
| (20) [Ala10]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-Lys-Ala-Leu-Leu-OH |
| (21) [Ala11]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-Lys-Tyr-Ala-Leu-OH |
| (22) [Ala12]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-Lys-Tyr-Leu-Ala-OH |
| D-scan (C-ter LmHslU2) | |
| (23) [leu7]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (24) [ala8]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-ala-Lys-Tyr-Leu-Leu-OH |
| (25) [lys9]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-lys-Tyr-Leu-Leu-OH |
| (26) [tyr10]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-Lys-tyr-Leu-Leu-OH |
| (27) [leu11]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-Lys-Tyr-leu-Leu-OH |
| (28) [leu12]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-Asn-Val-Asn-Leu-Ala-Lys-Tyr-Leu-leu-OH |
| Truncated peptides (C-ter LmHslU2) | |
| (29) LmC9-U2 | H-arg-O2OC-Asn-Val-Asn-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (30) LmC8-U2 | H-arg-O2Oc-Val-Asn-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (31) LmC7-U2 | H-arg-O2Oc-Asn-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (32) Ac-LmC7-U2 | Ac-Asn-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (33) LmC6-U2 | H-arg-O2Oc-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (34) Ac-LmC6-U2 | Ac-Leu-Ala-Lys-Tyr-Leu-Leu-OH |
| (35) LmC4-U2 | H-arg-O2Oc-Lys-Tyr-Leu-Leu-OH |
| (36) Ac-LmC4-U2 | Ac-Lys-Tyr-Leu-Leu-OH |
| Cyclic peptides (C-ter LmHslU2) | |
| (37) cyclo[Asp4, Lys8]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-c[Asp-Val-Asn-Leu-Lys]-Lys-Tyr-Leu-Leu-OH |
| (38) cyclo[asp4, Lys8]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-c[asp-Val-Asn-Leu-Lys]-Lys-Tyr-Leu-Leu-OH |
| (39) cyclo[Asp4, lys8]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-c[Asp-Val-Asn-Leu-lys]-Lys-Tyr-Leu-Leu-OH |
| (40) cyclo[asp4, lys8]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-c[asp-Val-Asn-Leu-lys]-Lys-Tyr-Leu-Leu-OH |
| (41) cyclo[Asp4, Orn8]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-c[Asp-Val-Asn-Leu-Orn]-Lys-Tyr-Leu-Leu-OH |
| (42) cyclo[Asp4, Dap8]-LmC12-U2 | H-arg-O2Oc-Leu-Gln-Lys-c[Asp-Val-Asn-Leu-Dap]-Lys-Tyr-Leu-Leu-OH |
| (43) cyclo[Asp4, (1) Lys8]-LmC9-U2 | H-arg-O2Oc-c[Asp-Val-Asn-Leu-Lys]-Lys-Tyr-Leu-Leu-OH |
| Scrambled LmC12-U2 | |
| (44) Scr-LmC12-U2 | H-arg-O2Oc-Asn-Leu-Tyr-Lys-Leu-Val-Gln-Leu-Leu-Asn-Lys-Ala-OH |
| Compound | θmre (deg.cm2.dmol−1) at 222 nm (Helicity%) | |||
|---|---|---|---|---|
| N | Name | 0% TFE | 25% TFE | 50% TFE |
| 10 | LmC12-U2 | −1186 (3) | −3727 (10) | −4328 (12) |
| 29 | LmC9-U2 | −1711 (5) | −4130 (11) | −4733 (13) |
| 30 | LmC8-U2 | −1377 (4) | −3519 (10) | −3592 (10) |
| 37 | c[Asp4,Lys8]-LmC12-U2 | −1626 (5) | −6143 (17) | −7628 (21) |
| 43 | c[Asp4,Lys8]-LmC9-U2 | −2225 (6) | −5026 (14) | −5140 (14) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kebe, N.M.; Samanta, K.; Singh, P.; Lai-Kee-Him, J.; Apicella, V.; Payrot, N.; Lauraire, N.; Legrand, B.; Lisowski, V.; Mbang-Benet, D.-E.; et al. The HslV Protease from Leishmania major and Its Activation by C-terminal HslU Peptides. Int. J. Mol. Sci. 2019, 20, 1021. https://doi.org/10.3390/ijms20051021
Kebe NM, Samanta K, Singh P, Lai-Kee-Him J, Apicella V, Payrot N, Lauraire N, Legrand B, Lisowski V, Mbang-Benet D-E, et al. The HslV Protease from Leishmania major and Its Activation by C-terminal HslU Peptides. International Journal of Molecular Sciences. 2019; 20(5):1021. https://doi.org/10.3390/ijms20051021
Chicago/Turabian StyleKebe, Ndeye Mathy, Krishnananda Samanta, Priyanka Singh, Joséphine Lai-Kee-Him, Viviana Apicella, Nadine Payrot, Noémie Lauraire, Baptiste Legrand, Vincent Lisowski, Diane-Ethna Mbang-Benet, and et al. 2019. "The HslV Protease from Leishmania major and Its Activation by C-terminal HslU Peptides" International Journal of Molecular Sciences 20, no. 5: 1021. https://doi.org/10.3390/ijms20051021
APA StyleKebe, N. M., Samanta, K., Singh, P., Lai-Kee-Him, J., Apicella, V., Payrot, N., Lauraire, N., Legrand, B., Lisowski, V., Mbang-Benet, D.-E., Pages, M., Bastien, P., Kajava, A. V., Bron, P., Hernandez, J.-F., & Coux, O. (2019). The HslV Protease from Leishmania major and Its Activation by C-terminal HslU Peptides. International Journal of Molecular Sciences, 20(5), 1021. https://doi.org/10.3390/ijms20051021