BGFE: A Deep Learning Model for ncRNA-Protein Interaction Predictions Based on Improved Sequence Information


Abstract
1. Introduction
2. Results
2.1. Performance Evaluation
2.2. Comparison between Three Base Models and Final Integration Model BGFE
2.3. Comparison of Prediction with BGFE and Other Methods
3. Discussion
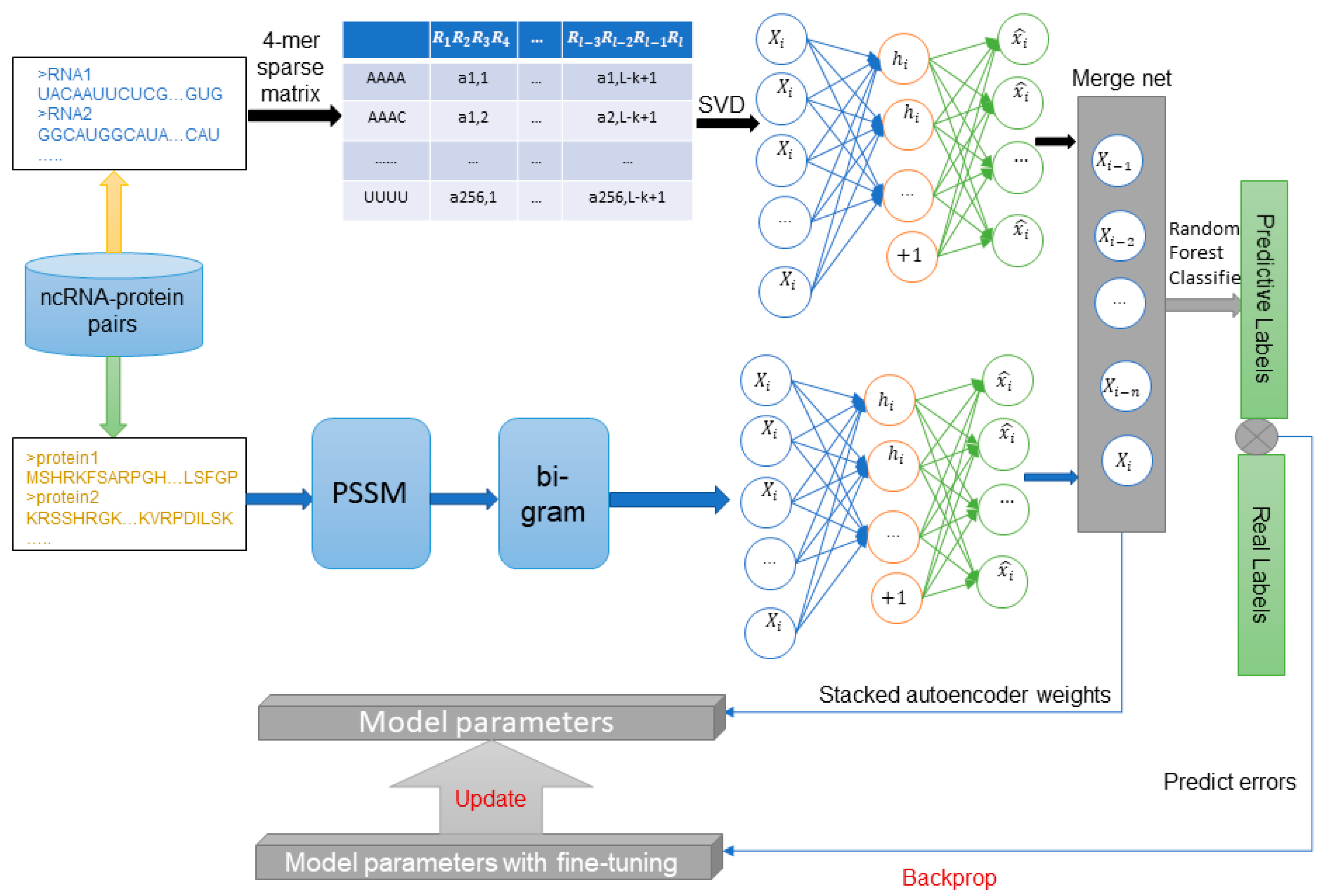
4. Materials and Methods
4.1. Datasets
4.2. Position Specific Scoring Matrix
4.3. Bi-gram Feature Extraction of PSSM
4.4. Representation of ncRNA Sequences Using K-mers Sparse Matrix and SVD
4.5. Stacked Auto-Encoder and Fine Tuning
4.6. Stacked Ensembling
4.7. Prediction Methods and Evaluation Criteria
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-H.; Lei, Y.-K.; Zhu, L.; Xia, J.; Wang, B. Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinform. 2013, 14, S10. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Q.; You, Z.-H.; Li, X.; Ming, Z.; Chen, X. PSPEL: In silico prediction of self-interacting proteins from amino acids sequences using ensemble learning. IEEE/ACM Trans. Comput. Biol. Bioinform. (TCBB) 2017, 14, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, M.; Agostini, F.; Masin, M.; Tartaglia, G.G. Predicting protein associations with long noncoding RNAs. Nat. Methods 2011, 8, 444. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Fan, Y.X.; Yan, J.; Shen, H.B. IPMiner: Hidden ncRNA-protein interaction sequential pattern mining with stacked autoencoder for accurate computational prediction. BMC Genomics 2016, 17, 582. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Wang, L.; You, Z.-H.; Huang, D.-S.; Zhou, F. Combining High Speed ELM Learning with a Deep Convolutional Neural Network Feature Encoding for Predicting Protein-RNA Interactions. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- SenGupta, D.J.; Zhang, B.; Kraemer, B.; Pochart, P.; Fields, S.; Wickens, M. A three-hybrid system to detect RNA-protein interactions in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 8496–8501. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.B. RNA–protein interactions. Curr. Opin. Struct. Biol. 2002, 12, 283–288. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Li, A.; Wang, M. A bipartite network-based method for prediction of long non-coding RNA–protein interactions. Genomics Proteomics Bioinform. 2016, 14, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Gawronski, A.R.; Uhl, M.; Zhang, Y.; Lin, Y.Y.; Niknafs, Y.S.; Ramnarine, V.R.; Malik, R.; Feng, F.; Chinnaiyan, A.M.; Collins, C.C.; et al. MechRNA: Prediction of lncRNA mechanisms from RNA–RNA and RNA–protein interactions. Bioinformatics 2018, 34, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Suresh, V.; Liu, L.; Adjeroh, D.; Zhou, X. RPI-Pred: Predicting ncRNA-protein interaction using sequence and structural information. Nucleic Acids Res. 2015, 43, 1370. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Kazan, H.; Chan, E.T.; Peña Castillo, L.; Chaudhry, S.; Talukder, S.; Blencowe, B.J.; Morris, Q.; Hughes, T.R. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 2009, 27, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Friedrich, S.; Kurgan, L. A comprehensive comparative review of sequence-based predictors of DNA- and RNA-binding residues. Briefings Bioinform. 2016, 17, 88. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.-C.; You, Z.-H.; Huang, D.-S.; Li, X.; Jiang, T.-H.; Li, L.-P. A Deep Learning Framework for Robust and Accurate Prediction of ncRNA-Protein Interactions Using Evolutionary Information. Mol. Ther.-Nucleic Acids 2018, 11, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; You, Z.-H.; Yan, X.; Xia, S.-X.; Liu, F.; Li, L.; Zhang, W.; Zhou, Y. Using Two-dimensional Principal Component Analysis and Rotation Forest for Prediction of Protein-Protein Interactions. Sci. Rep. 2018, 8, 12874. [Google Scholar] [CrossRef] [PubMed]
- Muppirala, U.K.; Honavar, V.G.; Dobbs, D. Predicting RNA-Protein Interactions Using Only Sequence Information. BMC Bioinform. 2011, 12, 489. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Liu, Z.P.; Huang, Q.; Wang, Y.; Xu, D.; Zhang, X.S.; Chen, R.; Chen, L. De novo prediction of RNA-protein interactions from sequence information. Mol. Biosyst. 2013, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank, 1999–. Int. Tables Crystallogr. 2000, 67, 675–684. [Google Scholar]
- Zahiri, J.; Mohammad-Noori, M.; Ebrahimpour, R.; Saadat, S.; Bozorgmehr, J.H.; Goldberg, T.; Masoudi-Nejad, A. LocFuse: Human protein–protein interaction prediction via classifier fusion using protein localization information. Genomics 2014, 104, 496. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-P.; Wang, Y.-B.; You, Z.-H.; Li, Y.; An, J.-Y. PCLPred: A Bioinformatics Method for Predicting Protein–Protein Interactions by Combining Relevance Vector Machine Model with Low-Rank Matrix Approximation. Int. J. Mol. Sci. 2018, 19, 1029. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Zhou, M.; Luo, X.; Li, S. Highly Efficient Framework for Predicting Interactions Between Proteins. IEEE Trans. Cybern. 2017, 47, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-B.; You, Z.-H.; Li, L.-P.; Huang, D.-S.; Zhou, F.-F.; Yang, S. Improving Prediction of Self-interacting Proteins Using Stacked Sparse Auto-Encoder with PSSM profiles. Int. J. Biol. Sci. 2018, 14, 983–991. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-H.; Huang, Z.A.; Zhu, Z.; Yan, G.Y.; Li, Z.W.; Wen, Z.; Chen, X. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, e1005455. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U.P. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204. [Google Scholar] [CrossRef] [PubMed]
- Hayat, M.; Khan, A. Predicting membrane protein types by fusing composite protein sequence features into pseudo amino acid composition. J. Theor. Biol. 2011, 271, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; You, Z.H.; Xia, S.-X.; Chen, X.; Yan, X.; Zhou, Y.; Liu, F. An improved efficient rotation forest algorithm to predict the interactions among proteins. Soft Comput. 2018, 22, 3373–3381. [Google Scholar] [CrossRef]
- An, J.Y.; You, Z.H.; Chen, X.; Huang, D.S.; Li, Z.W.; Liu, G.; Wang, Y. Identification of self-interacting proteins by exploring evolutionary information embedded in PSI-BLAST-constructed position specific scoring matrix. Oncotarget 2016, 7, 82440–82449. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The Database of Interacting Proteins: 2004 Update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed]
- Chatraryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, K.K.; Sharma, A.; Lyons, J.; Dehzangi, A. A Tri-Gram Based Feature Extraction Technique Using Linear Probabilities of Position Specific Scoring Matrix for Protein Fold Recognition. IEEE Trans. Nanobiosci. 2013, 320, 41. [Google Scholar] [CrossRef] [PubMed]
- Bouchaffra, D.; Tan, J. Protein Fold Recognition using a Structural Hidden Markov Model. In Proceedings of the International Conference on Pattern Recognition, Hong Kong, China, 20–24 August 2006; pp. 186–189. [Google Scholar]
- Chen, Z.-H.; You, Z.-H.; Li, L.-P.; Wang, Y.-B.; Li, X. RP-FIRF: Prediction of Self-interacting Proteins Using Random Projection Classifier Combining with Finite Impulse Response Filter. In Proceedings of the International Conference on Intelligent Computing, Wuhan, China, 15–18 August 2018; pp. 232–240. [Google Scholar]
- Chmielnicki, W.; Stapor, K. A hybrid discriminative/generative approach to protein fold recognition. Neurocomputing 2012, 75, 194–198. [Google Scholar] [CrossRef]
- Chen, H.; Huang, Z. Medical Image Feature Extraction and Fusion Algorithm Based on K-SVD. In Proceedings of the Ninth International Conference on P2P, Parallel, Grid, Cloud and Internet Computing, Guangdong, China, 8–10 November 2014; pp. 333–337. [Google Scholar]
- Vincent, P.; Larochelle, H.; Lajoie, I.; Bengio, Y.; Manzagol, P.A. Stacked Denoising Autoencoders: Learning Useful Representations in a Deep Network with a Local Denoising Criterion. J. Mach. Learn. Res. 2010, 11, 3371–3408. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Su, S.Z.; Liu, Z.H.; Xu, S.P.; Li, S.Z.; Ji, R. Sparse auto-encoder based feature learning for human body detection in depth image. Signal. Process. 2015, 112, 43–52. [Google Scholar] [CrossRef]
- Dahl, G.E.; Sainath, T.N.; Hinton, G.E. Improving deep neural networks for LVCSR using rectified linear units and dropout. In Proceedings of the IEEE International Conference on Acoustics, Speech and Signal. Processing, Vancouver, BC, Canada, 26–31 May 2013; pp. 8609–8613. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Pan, X.Y.; Tian, Y.; Huang, Y.; Shen, H.B. Towards better accuracy for missing value estimation of epistatic miniarray profiling data by a novel ensemble approach. Genomics 2011, 97, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Töscher, A.; Jahrer, M. The BigChaos Solution to the Netflix Grand Prize. Netflix Prize Documentation, 2009. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Jeong, E.; Chung, I.-F.; Miyano, S. A neural network method for identification of RNA-interacting residues in protein. Genome Inform. 2004, 15, 105–116. [Google Scholar] [PubMed]
- Hansen, L.K.; Salamon, P. Neural network ensembles. IEEE Trans. Pattern Anal. Mach. Intell. 1990, 12, 993–1001. [Google Scholar] [CrossRef]
- Zhang, H. The Optimality of Naive Bayes. In Proceedings of the International Flairs Conference, Miami Beach, FL, USA, 12–14 May 2004. [Google Scholar]
- You, Z.H.; Li, X.; Chan, K.C. An Improved Sequence-Based Prediction Protocol for Protein-Protein Interactions Using Amino Acids Substitution Matrix and Rotation Forest Ensemble Classifiers; Elsevier Science Publishers B. V.: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Statnikov, A.; Wang, L.; Aliferis, C.F. A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinform. 2008, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bengio, Y.; Grandvalet, Y. No unbiased estimator of the variance of k-fold cross-validation. J. Mach. Learn. Res. 2004, 5, 1089–1105. [Google Scholar]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta (BBA) Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Accuracy | Sensitivity | Specificity | Precision | MCC |
|---|---|---|---|---|---|
| RPI488 | 0.8868 | 0.9268 | 0.8354 | 0.9328 | 0.7743 |
| RPI1807 | 0.9600 | 0.9344 | 0.9989 | 0.9117 | 0.9217 |
| RPI2241 | 0.9130 | 0.8772 | 0.9660 | 0.8590 | 0.8335 |
| RPI488 | Accuracy | Sensitivity | Specificity | Precision | MCC |
|---|---|---|---|---|---|
| BGFE | 0.8868 | 0.9268 | 0.8354 | 0.9328 | 0.7743 |
| Raw feature | 0.8168 | 0.8083 | 0.8192 | 0.8104 | 0.6299 |
| Stacked auto-encoder | 0.8806 | 0.9243 | 0.8255 | 0.9351 | 0.7638 |
| Stacked auto-encoder without fine tuning | 0.8600 | 0.8848 | 0.8271 | 0.8850 | 0.7187 |
| RPI1807 | Accuracy | Sensitivity | Specificity | Precision | MCC |
|---|---|---|---|---|---|
| BGFE | 0.9600 | 0.9344 | 0.9989 | 0.9117 | 0.9217 |
| Raw feature | 0.9349 | 0.9508 | 0.9308 | 0.9400 | 0.8688 |
| Stacked auto-encoder | 0.9396 | 0.9029 | 0.9994 | 0.8651 | 0.8830 |
| Stacked auto-encoder without fine tuning | 0.9645 | 0.9672 | 0.9688 | 0.9590 | 0.9281 |
| RPI2241 | Accuracy | Sensitivity | Specificity | Precision | MCC |
|---|---|---|---|---|---|
| BGFE | 0.9130 | 0.8772 | 0.9660 | 0.8590 | 0.8335 |
| Raw feature | 0.6438 | 0.6525 | 0.6313 | 0.6565 | 0.2881 |
| Stacked auto-encoder | 0.9041 | 0.8895 | 0.9329 | 0.8747 | 0.8156 |
| Stacked auto-encoder without fine tuning | 0.6438 | 0.6517 | 0.6327 | 0.6551 | 0.2879 |
| RPI1807 | Accuracy | Sensitivity | Precision |
|---|---|---|---|
| BGFE | 0.9600 | 0.9344 | 0.9117 |
| RPI-Pred | 0.9300 | 0.9400 | 0.9400 |
| RPI2241 | Accuracy | Sensitivity | Precision |
| BGFE | 0.9130 | 0.8772 | 0.8590 |
| RPI-Pred | 0.8400 | 0.7800 | 0.8800 |
| Usha K Muppirala | 0.8960 | 0.9000 | 0.8900 |
| Ying Wang | 0.7400 | 0.9160 | 0.6990 |
| Dataset | Interaction Pairs | Number of Proteins | Number of RNAs |
|---|---|---|---|
| RPI488 1 | 243 | 25 | 247 |
| RPI1807 1 | 1807 | 1807 | 1078 |
| RPI2241 1 | 2241 | 2043 | 332 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, Z.-H.; Jia, L.-N.; Zhou, Y.; Li, L.-P.; Yi, H.-C. BGFE: A Deep Learning Model for ncRNA-Protein Interaction Predictions Based on Improved Sequence Information. Int. J. Mol. Sci. 2019, 20, 978. https://doi.org/10.3390/ijms20040978
Zhan Z-H, Jia L-N, Zhou Y, Li L-P, Yi H-C. BGFE: A Deep Learning Model for ncRNA-Protein Interaction Predictions Based on Improved Sequence Information. International Journal of Molecular Sciences. 2019; 20(4):978. https://doi.org/10.3390/ijms20040978
Chicago/Turabian StyleZhan, Zhao-Hui, Li-Na Jia, Yong Zhou, Li-Ping Li, and Hai-Cheng Yi. 2019. "BGFE: A Deep Learning Model for ncRNA-Protein Interaction Predictions Based on Improved Sequence Information" International Journal of Molecular Sciences 20, no. 4: 978. https://doi.org/10.3390/ijms20040978
APA StyleZhan, Z.-H., Jia, L.-N., Zhou, Y., Li, L.-P., & Yi, H.-C. (2019). BGFE: A Deep Learning Model for ncRNA-Protein Interaction Predictions Based on Improved Sequence Information. International Journal of Molecular Sciences, 20(4), 978. https://doi.org/10.3390/ijms20040978