Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy


Abstract
1. Introduction
- Definition of ICD together with a brief consideration of other types of cell death resulting from cancer therapy;
- Induction of ICD in cancer by chemotherapy and radiation therapy;
- The major types and key involvement of damage-associated molecular patterns (DAMPs) in triggering ICD;
- Evidence implicating the existence, involvement, and therapeutic benefit of ICD in the clinical setting of cancer chemotherapy and radiotherapy;
- Host defense- and tumor-related factors, which influence the antitumor efficacy of ICD;
- Combinatorial strategies based on ICD together with other types of oncoimmunotherapies;
- Novel inducers of ICD, as well as putative pharmacological adjuvant strategies that may augment ICD;
- Identification of predictive biomarkers, preferably systemic, but also in situ, which can be used to monitor the persistence and therapeutic efficacy of ICD.
2. Immunogenic Cell Death (ICD)
2.1. Types of Cell Death Relevant to Anticancer Treatment
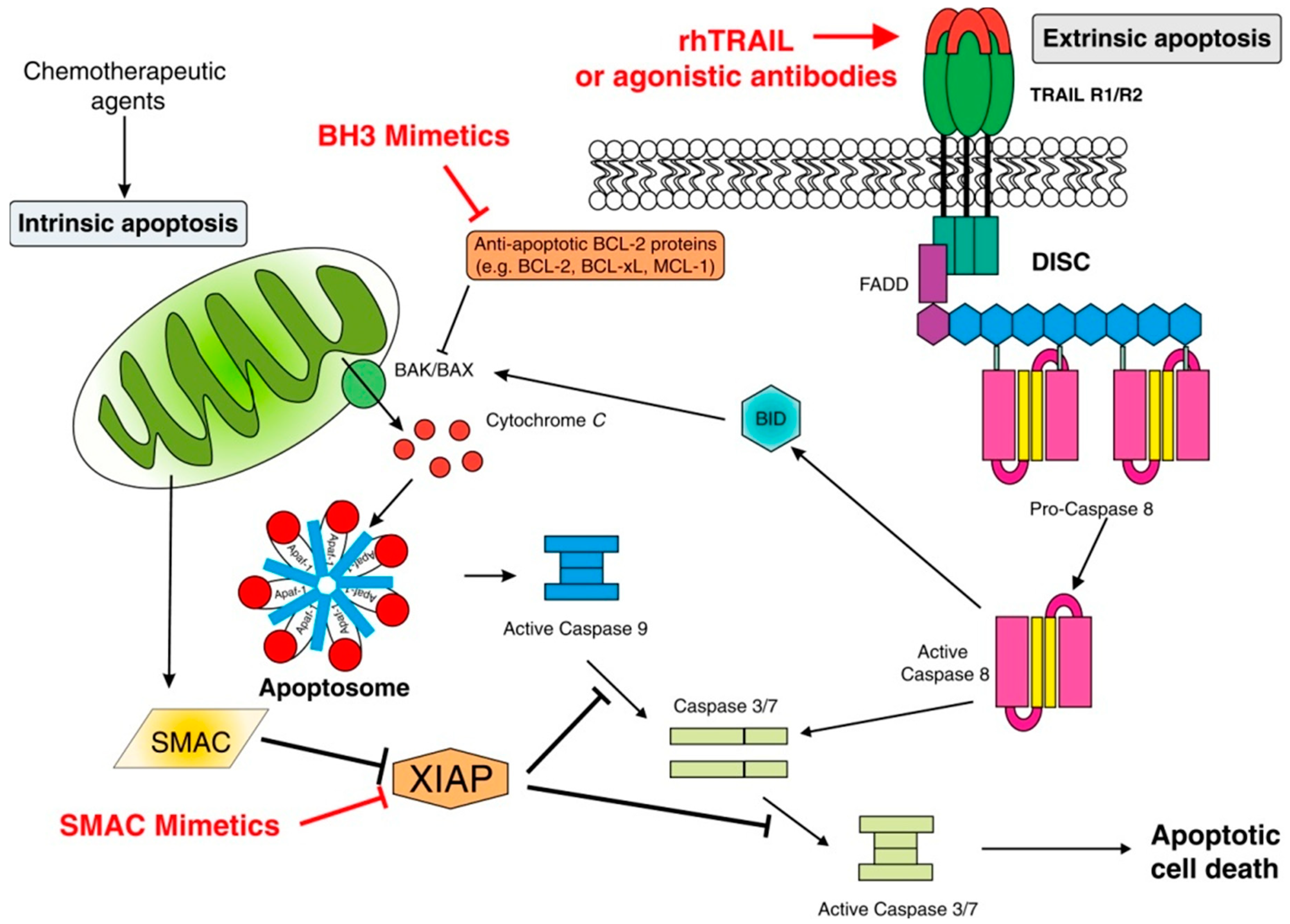
2.1.1. Intrinsic Apoptosis
2.1.2. Extrinsic Apoptosis
2.1.3. Necroptosis
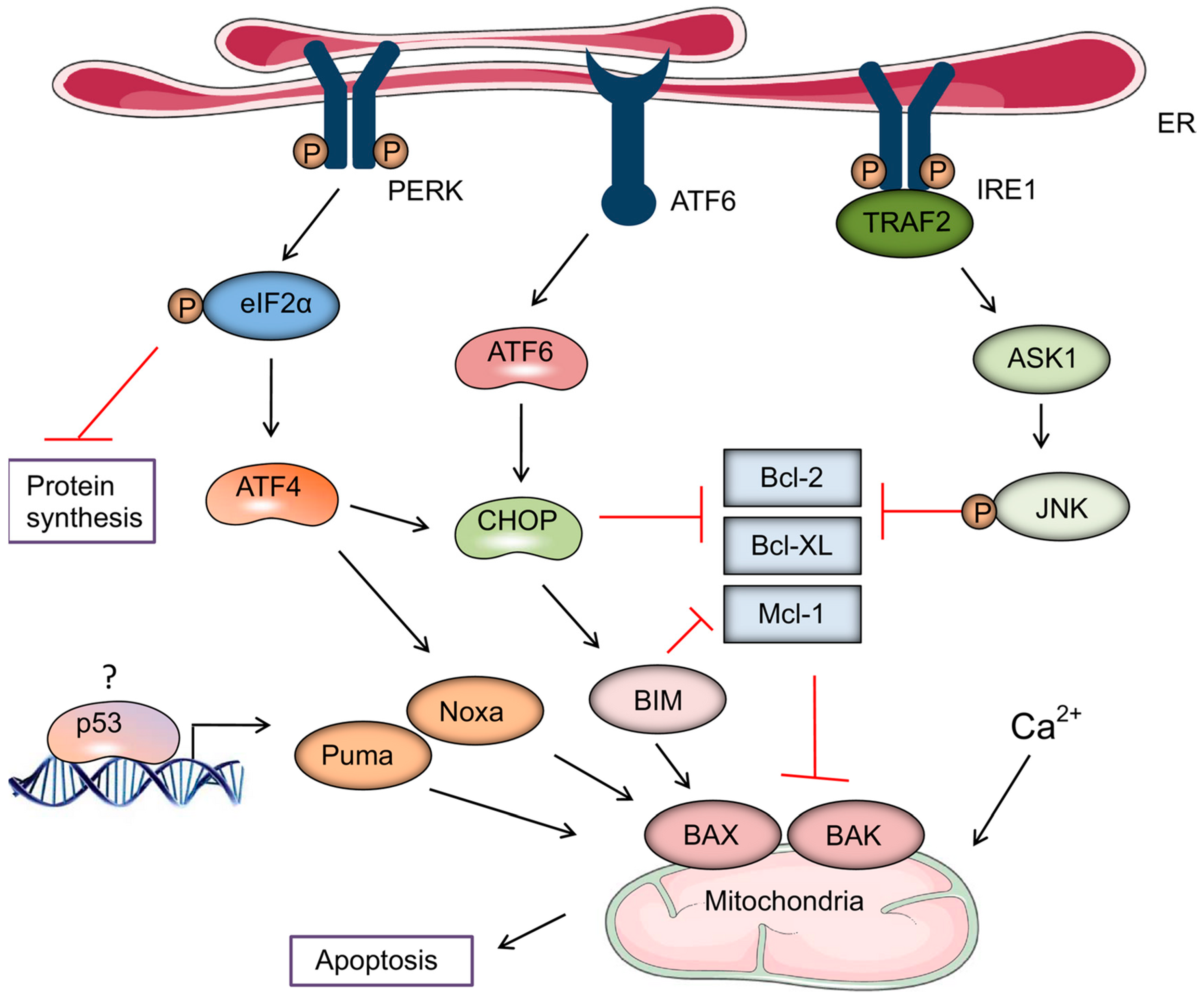
3. Role of Endoplasmic Reticulum (ER) Stress in the Induction of ICD
4. ICD in Cancer Chemotherapy and Radiation Therapy
5. Types and Key Involvement of Damage-Associated Molecular Patterns (DAMPs) in Triggering ICD
5.1. Calreticulin
5.2. Heat Shock Proteins (HSPs)
5.3. High Mobility Group Box 1
5.4. Adenosine 5-triphosphate (ATP)
5.5. Spliceosome-Associated Protein 130 (SAP130)
5.6. Defensins and S100 Proteins
6. Effects of DAMPs Released during Cell Death on Activation of Innate and Adaptive Immune Mechanisms
7. Evidence in Support of the Existence and Efficacy of ICD in the Therapeutic Setting
8. Properties of Tumors and Host Defenses that Determine the Efficacy of ICD
8.1. Tumor-Related Factors Impacting on the Efficacy of ICD
8.1.1. Tumor Mutational Burden
8.1.2. Tumor Expression of PD-L1
8.1.3. Overexpression of CD47 and MHC Class I
8.1.4. Immunosuppressive Factors Released by Tumor Cells
- Adenosine: While extracellular ATP is considered to be a major effector of ICD, it is also a substrate for ectonucleotidase enzymes, most prominently CD39 and CD73 [98]. These enzymes are highly expressed on various cell types present in the tumor microenvironment, including tumor cells per se, structural cells, and cells of both the innate and adaptive immune systems, particularly Tregs [98]. These ectonucleotidases, in turn, convert ATP to adenosine, a potent endogenous, immunosuppressive agent. Interaction of adenosine with adenosine 2A (A2A) subtype receptors, which are linked to activation of the enzyme, adenylyl cyclase, results in the synthesis of broadly immunosuppressive 3’,5’-cyclic adenosine monophosphate (cAMP) [98];
- Prostaglandin E2 (PGE2): It is well-recognized that many different types of cancer (colorectal, prostate, lung, pancreatic, breast) overexpress the enzyme, inducible cyclooxygenase 2 (COX-2), resulting in excessive production of PGE2 [99]. Operating via EP2 and EP4 receptors, PGE2, like adenosine, causes activation of adenylyl cyclase and synthesis of cAMP [99];
- Indole-2,3-dioxygenase: This enzyme is expressed by both tumor cells and cells of the innate and adaptive immune systems. It metabolizes tryptophan to kynurenine, anthranilic acid, and 3-hydroxyanthranilic acid, all of which are immunosuppressive and contribute to the evasion of immune-mediated eradication of tumors [100,101].
8.2. Host-Associated Factors Which May Restrict the Efficacy of Immunogenic Cell Death
8.2.1. Chronic Infections
8.2.2. Smoking
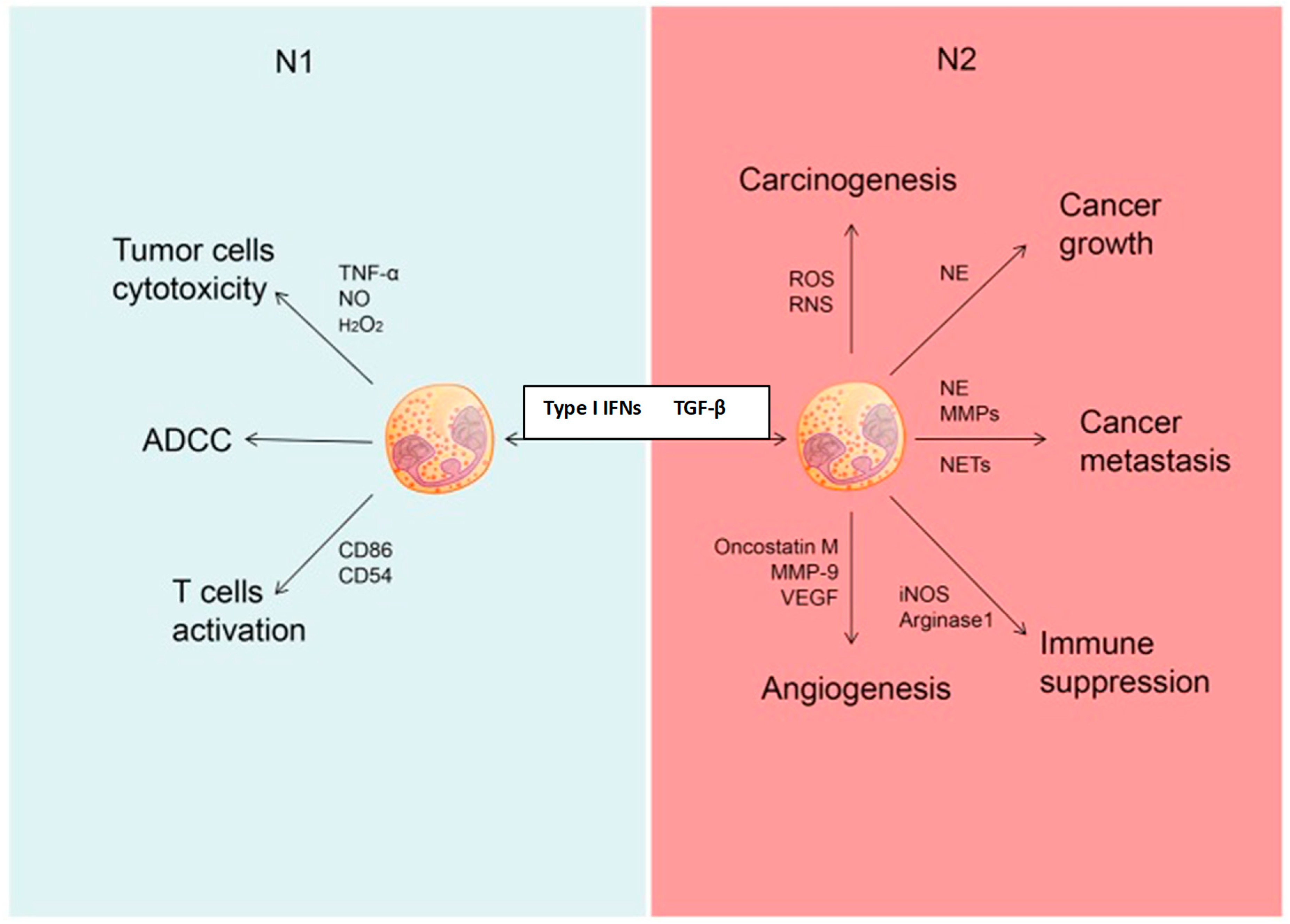
8.2.3. The Inflammatory Tumor Microenvironment
8.2.4. Immunosenescence
8.2.5. Obesity, Co-Morbidities, and Mental and Physical Stress
9. Strategies to Augment the Efficacy of ICD in Cancer Chemotherapy and Radiotherapy
9.1. Pretherapy Selection of Patients
- Type of malignancy;
- Normal circulating lymphocyte count and neutrophil:lymphocyte ratio, together with low numbers of immunosuppressive neutrophils;
- Low levels of circulating immunosuppressive cytokines, specifically IL-1Ra, IL-2R, IL-10, and TGF-β1;
- High tumor mutational burden, which may be performed at reasonable cost in the future [87];
- Digital image analysis of tumor bopsies to detect and enumerate types of infiltrating antitumor effector cells and suppressor cells; one such procedure gaining prominence is the “Immunoscore” [108];
- Presence of PD-L1 and/or CD47 on tumor cells and/or antigen-presenting cells, which may identify those patients likely to benefit from PD-1- and/or CD47-targeted immunotherapy; although extensive work has been done on PD-L1, CD47 remains to be validated in this context;
- Expression of IICP molecules on T cells in tumor biopsies, or the presence of systemic, soluble forms of these molecules;
- Specific epigenetic profiling termed “EPIMMUN”, which is based on the establishment of DNA methylation microarrays [109]. This strategy, albeit preliminary, has shown promise in identifying those patients with NSCLC who are likely to benefit from anti-PD-1 Mab therapy [109]. Together with the composition of the tumor immune cellular infiltrate, detection of the unmethylated status of the Treg transcription factor, forkhead box P1, was found to predict clinical response [109]. However, this strategy requires more intensive clinical evaluation and evidence of affordability.
9.2. Combinatorial Immunotherapeutic Strategies
9.3. Small Molecule-Based Immunopharmacological Strategies
9.3.1. Repurposed Agents Which Act as Inducers/Enhancers of ICD
9.3.2. Cardiac Glycosides
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
9.3.3. Novel Small Molecule Inducers of ICD
Septacidin
LTX-315
CBP501
Dinaciclib
RT53
10. Small Molecule Immunostimulants for Cancer Immunotherapy Which Apparently Do Not Directly Induce ICD
10.1. Novel Agents
- CA-170: This is an inhibitory molecule, which targets PD-L1, as well as another IICP viz. VISTA (V-domain Ig suppressor of T cell activation [124];
- LYC-55716: This molecule is an agonist of the retinoic acid-related orphan receptor-γt (RORγt), a transcription factor intimately involved in promoting the proliferation, survival, and function of Th17 cells [125];
- Galunisertib: This agent is a potent antagonist of the tyrosine kinase domain of the TGF-β receptor type I with considerable immunotherapeutic potential [126];
- Indoximod: This agent is a prototype inhibitor of indoleamine-2,3-dioxygenase, which, together with several other similar agents, is currently under early clinical evaluation in various types of malignancy [123,127]; although a recent study reported lack of efficacy in melanoma [128], efficacy in other types of malignancy remains to be established;
- CB-1158: This agent is an inhibitor of arginase, an enzyme produced predominantly by myeloid suppressor cells, which mediates immunosuppressive activity via depletion of arginine [129];
10.2. Propranolol as a Repurposed Immunotherapeutic Agent for Cancer
11. Systemic Biomarkers Predictive of the Efficacy of ICD
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Iams, W.; Cruz, M.R.; Chandra, S.; Choi, J.; Giles, F. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J. Immunother Cancer. 2018, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Maio, M.; Scherpereel, A.; Calabrò, L.; Aerts, J.; Cedres Perez, S.; Bearz, A.; Nackaerts, K.; Fennell, D.A.; Kowalski, D.; Tsao, A.S.; et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): A multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. 2017, 18, 1261–1273. [Google Scholar] [CrossRef]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [PubMed]
- Radogna, F.; Diederich, M. Stress-induced cellular responses in immunogenic cell death: Implications for cancer immunotherapy. Biochem. Pharmacol. 2018, 153, 12–23. [Google Scholar] [CrossRef]
- Wang, Y.J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Vandecasteele, K.; Bachert, C.; Krysko, O.; Krysko, D.V. Immunogenic apoptotic cell death and anticancer immunity. Adv. Exp. Med. Biol. 2016, 930, 133–149. [Google Scholar] [CrossRef]
- Montico, B.; Nigro, A.; Casolaro, V.; Dal Col, J. Immunogenic apoptosis as a novel tool for anticancer vaccine development. Int. J. Mol. Sci. 2018, 19, 594. [Google Scholar] [CrossRef]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic immunotherapy: Dying the right way is a key to eliciting potent antitumor immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef] [PubMed]
- Krysko, O.; Aaes, T.L.; Kagan, V.E.; D’Herde, K.; Bachert, C.; Leybaert, L.; Vandenabeele, P.; Krysko, D.V. Necroptotic cell death in anti-cancer therapy. Immunol. Rev. 2017, 280, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.K.; Lake, R.A.; Marzo, A.L.; Scott, B.; Heath, W.R.; Collins, E.J.; Frelinger, J.A.; Robinson, B.W. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J. Immunol. 2003, 170, 4905–4913. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.K.; Robinson, B.W.; Lake, R.A. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res. 2003, 63, 4490–4496. [Google Scholar] [PubMed]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Pihán, P.; Carreras-Sureda, A.; Hetz, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017, 24, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Brumatti, G.; Salmanidis, M.; Ekert, P.G. Crossing paths: Interactions between the cell death machinery and growth factor survival signals. Cell Mol. Life Sci. 2010, 67, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer. 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA damage in stem cells. Mol. Cell. 2017, 66, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.L.; MacFarlane, M. Targeting cell death signalling in cancer: Minimising “Collateral damage”. Br. J. Cancer. 2016, 115, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [PubMed]
- Zaman, S.; Wang, R.; Gandhi, V. Targeting the apoptosis pathway in hematologic malignancies. Leuk. Lymphoma. 2014, 55, 1980–1992. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27, S2–S19. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Van de Walle, L.; Lamkanfi, M.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: An overview. Cell Death Differ. 2008, 15, 453–460. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef] [PubMed]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Su, D.; Zhang, J.; Ge, S.; Li, Y.; Wang, F.; Gravel, M.; Roulston, A.; Song, Q.; Xu, W.; et al. Improvement of pharmacokinetic profile of TRAIL via Trimer-Tag enhances its antitumor activity in vivo. Sci. Rep. 2017, 7, 8953, Erratum in: Sci Rep. 2018, 8, 5266. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell. 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Quade, B.; Wang, H.; Sun, L.; Wang, X.; Rizo, J. A plug release mechanism for membrane permeation by MLKL. Structure 2014, 22, 1489–1500. [Google Scholar] [CrossRef]
- Yang, H.; Ma, Y.; Chen, G.; Zhou, H.; Yamazaki, T.; Klein, C.; Pietrocola, F.; Vacchelli, E.; Souquere, S.; Sauvat, A.; et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016, 5, e1149673. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- Grootjans, S.; Van den Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zheng, W.; Rock, K.L. Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc. Natl. Acad. Sci. USA 2000, 97, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta. 2010, 1805, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Sistigu, A.; Yamazaki, T.; Vitale, I.; Zitvogel, L.; Kroemer, G. Autocrine signaling of type 1 interferons in successful anticancer chemotherapy. Oncoimmunology 2015, 4, e988042. [Google Scholar] [CrossRef] [PubMed]
- Iurescia, S.; Fioretti, D.; Rinaldi, M. Targeting cytosolic nucleic acid-sensing pathways for cancer immunotherapies. Front. Immunol. 2018, 9, 711. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.R.; Martin, S.; Garg, A.D.; Agostinis, P. The PERKs of damage-associated molecular patterns mediating cancer immunogenicity: From sensor to the plasma membrane and beyond. Semin. Cancer Biol. 2015, 33, 74–85. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. The emergence of phox-ER stress induced immunogenic apoptosis. Oncoimmunology 2012, 1, 786–788. [Google Scholar] [CrossRef]
- Kazama, H.; Ricci, J.E.; Herndon, J.M.; Hoppe, G.; Green, D.R.; Ferguson, T.A. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008, 29, 21–32. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Crippa, M.P.; Manfredi, A.A.; Mezzapelle, R.; Rovere Querini, P.; Venereau, E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev. 2017, 280, 74–82. [Google Scholar] [CrossRef]
- Roy, A.; Ganesh, G.; Sippola, H.; Bolin, S.; Sawesi, O.; Dagälv, A.; Schlenner, S.M.; Feyerabend, T.; Rodewald, H.R.; Kjellén, L.; et al. Mast cell chymase degrades the alarmins heat shock protein 70, biglycan, HMGB1, and interleukin-33 (IL-33) and limits danger-induced inflammation. J. Biol. Chem. 2014, 289, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Binder, R.J. Immunosurveillance of cancer and the heat shock protein-CD91 pathway. Cell. Immunol. 2018. [Epub ahead of print]. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Osman, R.; Tacnet-Delorme, P.; Kleman, J.P.; Millet, A.; Frachet, P. Calreticulin release at an early stage of death modulates the clearance by macrophages of apoptotic cells. Front. Immunol. 2017, 8, 1034. [Google Scholar] [CrossRef] [PubMed]
- Broere, F.; van der Zee, R.; van Eden, W. Heat shock proteins are no DAMPs, rather “DAMPERs”. Nat. Rev. Immunol. 2011, 11, 565. [Google Scholar] [CrossRef]
- Flechtner, J.B.; Cohane, K.P.; Mehta, S.; Slusarewicz, P.; Leonard, A.K.; Barber, B.H.; Levey, D.L.; Andjelic, S. High-affinity interactions between peptides and heat shock protein 70 augment CD8+ T lymphocyte immune responses. J. Immunol. 2006, 177, 1017–1027. [Google Scholar] [CrossRef]
- Salimu, J.; Spary, L.K.; Al-Taei, S.; Clayton, A.; Mason, M.D.; Staffurth, J.; Tabi, Z. Cross-presentation of the oncofetal tumor antigen 5T4 from irradiated prostate cancer cells—A key role for heat-shock protein 70 and receptor CD91. Cancer Immunol. Res. 2015, 3, 678–688. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Yanai, H.; Ban, T.; Taniguchi, T. High-mobility group box family of proteins: Ligand and sensor for innate immunity. Trends Immunol. 2012, 33, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Cambi, A.; Figdor, C. Necrosis: C-type lectins sense cell death. Curr. Biol. 2009, 19, R375–R378. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.H.; Torres-Hernandez, A.; Kalabin, A.; Whiteman, C.; Rokosh, R.; Ravirala, S.; Ochi, A.; Gutierrez, J.; Salyana, M.A.; Mani, V.R.; et al. Mincle Signaling Promotes Con A Hepatitis. J. Immunol. 2016, 197, 2816–2827. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.D.; Heijink, I.H.; ten Hacken, N.H.; Vandenabeele, P.; Krysko, D.V.; Nawijn, M.C.; van Oosterhout, A.J. DAMPs activating innate and adaptive immune responses in COPD. Mucosal. Immunol. 2014, 7, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Cappelletti, M.; Presicce, P.; Calcaterra, F.; Mavilio, D.; Della Bella, S. Bright expression of CD91 identifies highly activated human dendritic cells that can be expanded by defensins. Immunology 2015, 144, 661–667. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer. 2015, 15, 96–109. [Google Scholar] [CrossRef]
- Ferlazzo, G.; Morandi, B. Cross-Talks between Natural Killer Cells and Distinct Subsets of Dendritic Cells. Front. Immunol. 2014, 5, 159. [Google Scholar] [CrossRef]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Vandenberk, L.; Fang, S.; Fasche, T.; Van Eygen, S.; Maes, J.; Van Woensel, M.; Koks, C.; Vanthillo, N.; Graf, N.; et al. Pathogen response-like recruitment and activation of neutrophils by sterile immunogenic dying cells drives neutrophil-mediated residual cell killing. Cell Death Differ. 2017, 24, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiu, L.; Li, Z.; Wang, X.Y.; Yi, H. Understanding the multifaceted role of neutrophils in cancer and autoimmune diseases. Front. Immunol. 2018, 9, 2456. [Google Scholar] [CrossRef] [PubMed]
- Pylaeva, E.; Lang, S.; Jablonska, J. The essential role of type I interferons in differentiation and activation of tumor-associated neutrophils. Front. Immunol. 2016, 7, 629. [Google Scholar] [CrossRef] [PubMed]
- Grecian, R.; Whyte, M.K.B.; Walmsley, S.R. The role of neutrophils in cancer. Br. Med. Bull. 2018, 128, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Stoetzer, O.J.; Fersching, D.M.; Salat, C.; Steinkohl, O.; Gabka, C.J.; Hamann, U.; Braun, M.; Feller, A.M.; Heinemann, V.; Siegele, B.; et al. Circulating immunogenic cell death biomarkers HMGB1 and RAGE in breast cancer patients during neoadjuvant chemotherapy. Tumour Biol. 2013, 34, 81–90. [Google Scholar] [CrossRef]
- Fucikova, J.; Moserova, I.; Urbanova, L.; Bezu, L.; Kepp, O.; Cremer, I.; Salek, C.; Strnad, P.; Kroemer, G.; Galluzzi, L.; et al. Prognostic and predictive value of DAMPs and DAMP-associated processes in cancer. Front. Immunol. 2015, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Aoto, K.; Mimura, K.; Okayama, H.; Saito, M.; Chida, S.; Noda, M.; Nakajima, T.; Saito, K.; Abe, N.; Ohki, S.; et al. Immunogenic tumor cell death induced by chemotherapy in patients with breast cancer and esophageal squamous cell carcinoma. Oncol. Rep. 2018, 39, 151–159. [Google Scholar] [CrossRef]
- Laengle, J.; Stift, J.; Bilecz, A.; Wolf, B.; Beer, A.; Hegedus, B.; Stremitzer, S.; Starlinger, P.; Tamandl, D.; Pils, D.; et al. DNA damage predicts prognosis and treatment response in colorectal liver metastases superior to immunogenic cell death and T cells. Theranostics 2018, 8, 3198–3213. [Google Scholar] [CrossRef]
- Ladoire, S.; Enot, D.; Andre, F.; Zitvogel, L.; Kroemer, G. Immunogenic cell death-related biomarkers: Impact on the survival of breast cancer patients after adjuvant chemotherapy. Oncoimmunology 2015, 5, e1082706. [Google Scholar] [CrossRef]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Martinez Monge, R.; Cerwenka, A.; Ajona, D.; Melero, I.; Lecanda, F. Radiation effects on antitumor immune responses: Current perspectives and challenges. Ther. Adv. Med. Oncol. 2018, 10, 1758834017742575. [Google Scholar] [CrossRef] [PubMed]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A., III; Yarchoan, M.; Lee, V.; Laheru, D.A.; Jaffee, E.M. Strategies for increasing pancreatic tumor immunogenicity. Clin. Cancer Res. 2017, 23, 1656–1669. [Google Scholar] [CrossRef]
- Haanen, J.B. Immunotherapy of melanoma. EJC Suppl. 2013, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Greil, R.; Hutterer, E.; Hartmann, T.N.; Pleyer, L. Reactivation of dormant anti-tumor immunity - a clinical perspective of therapeutic immune checkpoint modulation. Cell Commun. Signal. 2017, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Lyu, G.Y.; Yeh, Y.H.; Yeh, Y.C.; Wang, Y.C. Mutation load estimation model as a predictor of the response to cancer immunotherapy. NPJ Genom. Med. 2018, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Derer, A.; Spiljar, M.; Bäumler, M.; Hecht, M.; Fietkau, R.; Frey, B.; Gaipl, U.S. Chemoradiation increases PD-L1 expression in certain melanoma and glioblastoma cells. Front. Immunol. 2016, 7, 610. [Google Scholar] [CrossRef]
- Ock, C.Y.; Kim, S.; Keam, B.; Kim, S.; Ahn, Y.O.; Chung, E.J.; Kim, J.H.; Kim, T.M.; Kwon, S.K.; Jeon, Y.K.; et al. Changes in programmed death-ligand 1 expression during cisplatin treatment in patients with head and neck squamous cell carcinoma. Oncotarget 2017, 8, 97920–97927. [Google Scholar] [CrossRef]
- Lim, Y.J.; Koh, J.; Kim, S.; Jeon, S.R.; Chie, E.K.; Kim, K.; Kang, G.H.; Han, S.W.; Kim, T.Y.; Jeong, S.Y.; et al. Chemoradiation-induced alteration of programmed death-ligand 1 and CD8+ tumor-infiltrating lymphocytes identified patients with poor prognosis in rectal cancer: A matched comparison analysis. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 1216–1224. [Google Scholar] [CrossRef]
- Tang, H.; Liang, Y.; Anders, R.A.; Taube, J.M.; Qiu, X.; Mulgaonkar, A.; Liu, X.; Harrington, S.M.; Guo, J.; Xin, Y.; et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Investig. 2018, 128, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Hettich, M.; Lahoti, J.; Prasad, S.; Niedermann, G. Checkpoint antibodies but not T cell-recruiting diabodies effectively synergize with TIL-inducing γ-irradiation. Cancer Res. 2016, 76, 4673–4683. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Rückert, M.; Weber, J.; Mayr, X.; Derer, A.; Lotter, M.; Bert, C.; Rödel, F.; Fietkau, R.; Gaipl, U.S. Hypofractionated irradiation has immune stimulatory potential and induces a timely restricted infiltration of immune cells in colon cancer tumors. Front. Immunol. 2017, 8, 231. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.R.; Liu, L. “Eating” Cancer cells by blocking CD47 signaling: Cancer therapy by targeting the innate immune checkpoint. Cancer Transl. Med. 2017, 3, 200–208. [Google Scholar] [CrossRef]
- Barkal, A.A.; Weiskopf, K.; Kao, K.S.; Gordon, S.R.; Rosental, B.; Yiu, Y.Y.; George, B.M.; Markovic, M.; Ring, N.G.; Tsai, J.M.; et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 2018, 19, 76–84. [Google Scholar] [CrossRef]
- Thepmalee, C.; Panya, A.; Junking, M.; Chieochansin, T.; Yenchitsomanus, P.T. Inhibition of IL-10 and TGF-β receptors on dendritic cells enhances activation of effector T-cells to kill cholangiocarcinoma cells. Hum. Vaccin. Immunother. 2018, 14, 1423–1431. [Google Scholar] [CrossRef]
- Gemperle, C.; Schmid, M.; Herova, M.; Marti-Jaun, J.; Wuest, S.J.; Loretz, C.; Hersberger, M. Regulation of the formyl peptide receptor 1 (FPR1) gene in primary human macrophages. PLoS ONE 2012, 7, e50195. [Google Scholar] [CrossRef]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer. 2018, 6, 57. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef]
- Löb, S.; Königsrainer, A.; Rammensee, H.G.; Opelz, G.; Terness, P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: Can we see the wood for the trees? Nat. Rev. Cancer 2009, 9, 445–452. [Google Scholar] [CrossRef]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef] [PubMed]
- Wieland, D.; Hofmann, M.; Thimme, R. Overcoming CD8+ T-cell exhaustion in viral hepatitis: Lessons from the mouse model and clinical perspectives. Dig. Dis. 2017, 35, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Okoye, A.A.; Picker, L.J. CD4(+) T-cell depletion in HIV infection: Mechanisms of immunological failure. Immunol. Rev. 2013, 254, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Arnson, Y.; Shoenfeld, Y.; Amital, H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J. Autoimmun. 2010, 34, J258–J265. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.; Anderson, R. Cigarette smoking and mechanisms of susceptibility to infections of the respiratory tract and other organ systems. J. Infect. 2013, 67, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Baniyash, M. Chronic inflammation, immunosuppression and cancer: New insights and outlook. Semin. Cancer Biol. 2006, 16, 80–88. [Google Scholar] [CrossRef]
- Bottazzi, B.; Riboli, E.; Mantovani, A. Aging, inflammation and cancer. Semin. Immunol. 2018. [Epub ahead of print]. [Google Scholar] [CrossRef]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Duruisseaux, M.; Martínez-Cardús, A.; Calleja-Cervantes, M.E.; Moran, S.; Castro de Moura, M.; Davalos, V.; Piñeyro, D.; Sanchez-Cespedes, M.; Girard, N.; Brevet, M.; et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: A multicentre, retrospective analysis. Lancet Respir. Med. 2018, 6, 771–781. [Google Scholar] [CrossRef]
- Sukkurwala, A.Q.; Adjemian, S.; Senovilla, L.; Michaud, M.; Spaggiari, S.; Vacchelli, E.; Baracco, E.E.; Galluzzi, L.; Zitvogel, L.; Kepp, O.; et al. Screening of novel immunogenic cell death inducers within the NCI Mechanistic Diversity Set. Oncoimmunology 2014, 3, e28473. [Google Scholar] [CrossRef]
- Haux, J. Digitoxin is a potential anticancer agent for several types of cancer. Med. Hypotheses. 1999, 53, 543–548. [Google Scholar] [CrossRef]
- Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Shen, S.; Yamazaki, T.; Sukkurwala, A.Q.; Michaud, M.; Mignot, G.; et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci. Transl. Med. 2012, 4, 143ra99. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Sukkurwala, A.Q.; Michaud, M.; Galluzzi, L.; Zitvogel, L.; et al. Anticancer activity of cardiac glycosides: At the frontier between cell-autonomous and immunological effects. Oncoimmunology 2012, 1, 1640–1642. [Google Scholar] [CrossRef] [PubMed]
- Van Rensburg, C.E.; Anderson, R.; O’Sullivan, J.F. Riminophenazine compounds: Pharmacology and anti-neoplastic potential. Crit. Rev. Oncol. Hematol. 1997, 25, 55–67. [Google Scholar] [CrossRef]
- Van Rensburg, C.E.; van Staden, A.M.; Anderson, R. The riminophenazine agents clofazimine and B669 inhibit the proliferation of cancer cell lines in vitro by phospholipase A2-mediated oxidative and nonoxidative mechanisms. Cancer Res. 1993, 53, 318–323. [Google Scholar] [PubMed]
- Fletcher, R.E.; eibowitz, B.; Wang, Y.; Concha-Benavente, F.; Ferris, R.; Schoen, R.; Yu, J.; Zhang, L. Nonsteroidal anti-inflammatory drugs induce ER stress- and BID-dependent immunogenic cell death to suppress colorectal tumorigenesis [abstract]. In Proceedings of the American Association for Cancer Research Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018; AACR: Philadelphia, PA, USA. [Google Scholar] [CrossRef]
- Brunsvig, P.; Aamdal, S.; Kolstad, A.; Haaskjold, O.I.; Miller, R.M.; Rekdal, O.; Nicolaisen, B.; Olsen, W.M. A phase I study with LTX-315, an immunogenic cell death inducer, in patients with transdermally accessible tumors. J. Clin. Oncol. 2014, 32, 3067. Available online: http://ascopubs.org/doi/abs/10.1200/jco.2014.32.15_suppl.3067 (accessed on 23 October 2018). [CrossRef]
- Spicer, J.; Marabelle, A.; Baurain, J.F.; Awada, A.; Kristeleit, R.; Jøssang, D.E.; Jebsen, N.L.; Loirat, D.; Brunsvig, P.F.; Armstrong, A.; et al. A Phase I/II study of the oncolytic peptide LTX-315 combined with checkpoint inhibition generates de novo T-cell responses and clinical benefit in patients with advanced solid tumors. Poster presentation at the ASCO (American Society of Clinical Oncology) Annual Meeting, McCormick Place, Chicago, IL, USA, 1–5 June 2018; Available online: http://www.lytixbiopharma.com/uploads/posters/lytix-asco_2018-final_screen_file180529.pdf (accessed on 23 October 2018).
- Sakakibara, K.; Sato, T.; Kufe, D.W.; VonHoff, D.D.; Kawabe, T. CBP501 induces immunogenic tumor cell death and CD8 T cell infiltration into tumors in combination with platinum, and increases the efficacy of immune checkpoint inhibitors against tumors in mice. Oncotarget 2017, 8, 78277–78288. [Google Scholar] [CrossRef] [PubMed]
- NIH—U.S. National Library of Medicine. CBP501, Cisplatin and Nivolumab in advanced refractory tumors. ClinicalTrials; NCT03113188; Last Update: 17 November 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03113188 (accessed on 23 October 2018).
- Hossain, D.M.S.; Javaid, S.; Cai, M.; Zhang, C.; Sawant, A.; Hinton, M.; Sathe, M.; Grein, J.; Blumenschein, W.; Pinheiro, E.M.; et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J. Clin. Invest. 2018, 128, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Pasquereau-Kotula, E.; Habault, J.; Kroemer, G.; Poyet, J.L. The anticancer peptide RT53 induces immunogenic cell death. PLoS ONE 2018, 13, e0201220. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Yuan, W.E.; Su, J.; Liu, Y.; Chen, J. Recent advances in small molecule based cancer immunotherapy. Eur. J. Med. Chem. 2018, 157, 582–598. [Google Scholar] [CrossRef]
- Powderly, J.; Patel, M.R.; Lee, J.J.; Brody, J.; Meric-Bernstam, F.; Hamilton, E.; Ponce Aix, S.; Garcia-Corbacho, J.; Bang, Y.J.; Ahn, M.J.; et al. CA-170, a first in class oral small molecule dual inhibitor of immune checkpoints PD-L1 and VISTA, demonstrates tumor growth inhibition in pre-clinical models and promotes T cell activation in Phase 1 study. Ann. Oncol. 2017, 28. Abstract no. 1141PD, Abstract Book of the 42nd ESMO Congress (ESMO 2017) 8–12 September 2017, Madrid, Spain. Available online: https://doi.org/10.1093/annonc/mdx376.007 (accessed on 23 October 2018).
- Weems, G.A.; Hu, X.; Liu, X.; Li, H.; Bogdan, M.; Gao, Y.; Fox, B.; Wilkins, H.J.; Carter, L. LYC-55716: A novel small-molecule RORγ agonist immuno-oncology agent: Rationale for tumor selection and clinical evaluation of gastric and esophageal carcinoma in phase 2a expansion. J. Clin. Oncol. 2018, 36, 67. Available online: http://ascopubs.org/doi/abs/10.1200/JCO.2018.36.4_suppl.67 (accessed on 23 October 2018). [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.H.; Minton, S.E.; Han, H.S.; Ismail-Khan, R.; Neuger, A.; Khambati, F.; Noyes, D.; Lush, R.; Chiappori, A.A.; Roberts, J.D.; et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget 2016, 7, 22928–22938. [Google Scholar] [CrossRef] [PubMed]
- Komiya, T.; Huang, C.H. Updates in the clinical development of epacadostat and other indoleamine 2,3-dioxygenase 1 inhibitors (IDO1) for human cancers. Front. Oncol. 2018, 8, 423. [Google Scholar] [CrossRef] [PubMed]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer. 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Ho, P.Y.; Hotson, A.; Hill, C.; Piccione, E.C.; Hsieh, J.; Liu, L.; Buggy, J.J.; McCaffery, I.; Miller, R.A. A2AR antagonism with CPI-444 induces antitumor responses and augments efficacy to anti-PD-(L)1 and anti-CTLA-4 in preclinical models. Cancer Immunol. Res. 2018, 6, 1136–1149. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother. 2018. [Epub ahead of print]. [Google Scholar] [CrossRef] [PubMed]
- NIH—U.S. National Library of Medicine. A Phase 1 Clinical Study of AZD4635 and Durvalumab in Patients with Advanced Solid Malignancies. ClinicalTrials; NCT02740985; Last Updated on September 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT02740985 (accessed on 24 October 2018).
- Theron, A.J.; Steel, H.C.; Tintinger, G.R.; Feldman, C.; Anderson, R. Can the anti-inflammatory activities of β2-agonists be harnessed in the clinical setting? Drug Des. Devel. Ther. 2013, 7, 1387–1398. [Google Scholar] [CrossRef]
- Jean Wrobel, L.; Bod, L.; Lengagne, R.; Kato, M.; Prévost-Blondel, A.; Le Gal, F.A. Propranolol induces a favourable shift of anti-tumor immunity in a murine spontaneous model of melanoma. Oncotarget 2016, 7, 77825–77837. [Google Scholar] [CrossRef]
- Bucsek, M.J.; Qiao, G.; MacDonald, C.R.; Giridharan, T.; Evans, L.; Niedzwecki, B.; Liu, H.; Kokolus, K.M.; Eng, J.W.; Messmer, M.N.; et al. β-adrenergic signaling in mice housed at standard temperatures suppresses an effector phenotype in CD8+ T cells and undermines checkpoint inhibitor therapy. Cancer Res. 2017, 77, 5639–5651. [Google Scholar] [CrossRef]
- Kokolus, K.M.; Zhang, Y.; Sivik, J.M.; Schmeck, C.; Zhu, J.; Repasky, E.A.; Drabick, J.J.; Schell, T.D. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology 2017, 7, e1405205. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed]
- NIH—U.S. National Library of Medicine. Detection of Circulating Biomarkers of Immunogenic Cell Death (ICD). ClinicalTrials; NCT02921854; Last updated: 17 August 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT02921854 (accessed on 14 January 2019).
- Smart Patients. Detection of Circulating Biomarkers of Immunogenic Cell Death. Available online: https://www.smartpatients.com/trials/NCT02921854 (accessed on 14 January 2019).
- Kim, J.H.; Kim, E.; Lee, M.Y. Exosomes as diagnostic biomarkers in cancer. Mol. Cell Toxicol. 2018, 14, 113–122. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mechanism | Consequence |
|---|---|
| Low mutational load [86,87] * | Decreased immunogenicity and immune evasion |
| Expression of PD-L1 on tumor cells, as well as on tumor-infiltrating macrophages [91] | Decreased immunogenicity and immune evasion |
| Upregulation of expression of CD47 on tumor cells [94] | Interacts with SIRPα on dendritic cells to suppress “eat me” signals |
| Expression of MHC class I molecules by tumor cells [95] | Interact with LILRB1 on macrophages, suppressing phagocytic activity |
| Release of various types of immunosuppressive factor by tumor cells [83,96,97,98,99,100,101] | IL-10 and TGF-β1 Adenosine Prostaglandin E2 Indole-2,3-dioxygenase |
| Strategy | Comment |
|---|---|
| Type of malignancy, e.g., NSCLC, melanoma [83,84,85] * | Tend to have higher levels of immunogenicity |
| Absence of lymphopenia and high neutrophil:lymphocyte ratios | Nonspecific indicators of poor antitumor immune responses |
| Measurement of systemic immunosuppressive cytokines [83,96,97] | IL-1Ra, IL-2R, IL-10, and TGF-β1 are of particular significance in this context |
| Measurement of tumor mutational load [86,87] | A high mutational load is predictive of good immunogenicity |
| Computerized image analysis of tumor biopsies [108] | Enables immunological profiling of the tumor microenvironment |
| Detection of the presence of PD-L1 and/or CD47 and MHC 1 on tumor cells [91,94,95] | Mab-targeting of these immunosuppressive proteins may promote immune eradication |
| Specific epigenetic profiling (“EPIMMUN”) to predict responsiveness to PD-1-targeted MAbs in NSCLC [109] | Based on detection of the unmethylated status of the Treg transcription factor, Forkhead box P1 |
| Novel | Re-Purposed |
|---|---|
| Septacidin: an N-acyl-amino acid antibiotic [110] * | Cardiac glycosides: Inhibit Na+/K+-ATPase [110,111,112,113] Clofazimine: The prototype riminophenazine, cationic amphiphilic agent with membrane-disruptive activity, but untested in induction of ICD [114,115] Nonsteroidal anti-inflammatory drugs: Induce endoplasmic reticulum stress associated with death receptor signaling and BID activation [116] |
| LTX-315: a cationic amphiphilic, membrane-disruptive, chemically-modified 9-mer peptide [117,118] | |
| CBP501: a calmodulin-binding peptide that promotes uptake of cisplatin into tumor cells [119,120] | |
| Dinaciclib: a cyclin-dependent kinase inhibitor [121] | |
| RT53: a cationic amphiphilic, membrane-disruptive peptide [122] |
| Agent | Immunostimulatory Mechanism |
|---|---|
| CA-170 | Targets PD-L1 and VISTA [124] * |
| LYC-55716 | Agonist of RORγt [125] |
| Galunisertib | Antagonist of the tyrosine kinase domain of TGF-β receptor type 1 [126] |
| Indoximod | Prototype inhibitor of indoleamine-2,3-dioxygenase [123,127,128] |
| CB-1158 | Granulocyte arginase inhibitor [129] |
| CPI-444 and AZD4635 | Antagonists of the T cell A2a receptor [130,131,132] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rapoport, B.L.; Anderson, R. Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy. Int. J. Mol. Sci. 2019, 20, 959. https://doi.org/10.3390/ijms20040959
Rapoport BL, Anderson R. Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy. International Journal of Molecular Sciences. 2019; 20(4):959. https://doi.org/10.3390/ijms20040959
Chicago/Turabian StyleRapoport, Bernardo L., and Ronald Anderson. 2019. "Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy" International Journal of Molecular Sciences 20, no. 4: 959. https://doi.org/10.3390/ijms20040959
APA StyleRapoport, B. L., & Anderson, R. (2019). Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy. International Journal of Molecular Sciences, 20(4), 959. https://doi.org/10.3390/ijms20040959